
The Progress of the Interfacial Diffusion between Virgin and Aged Asphalt Based on Molecular Dynamics Simulation: A Review

Abstract
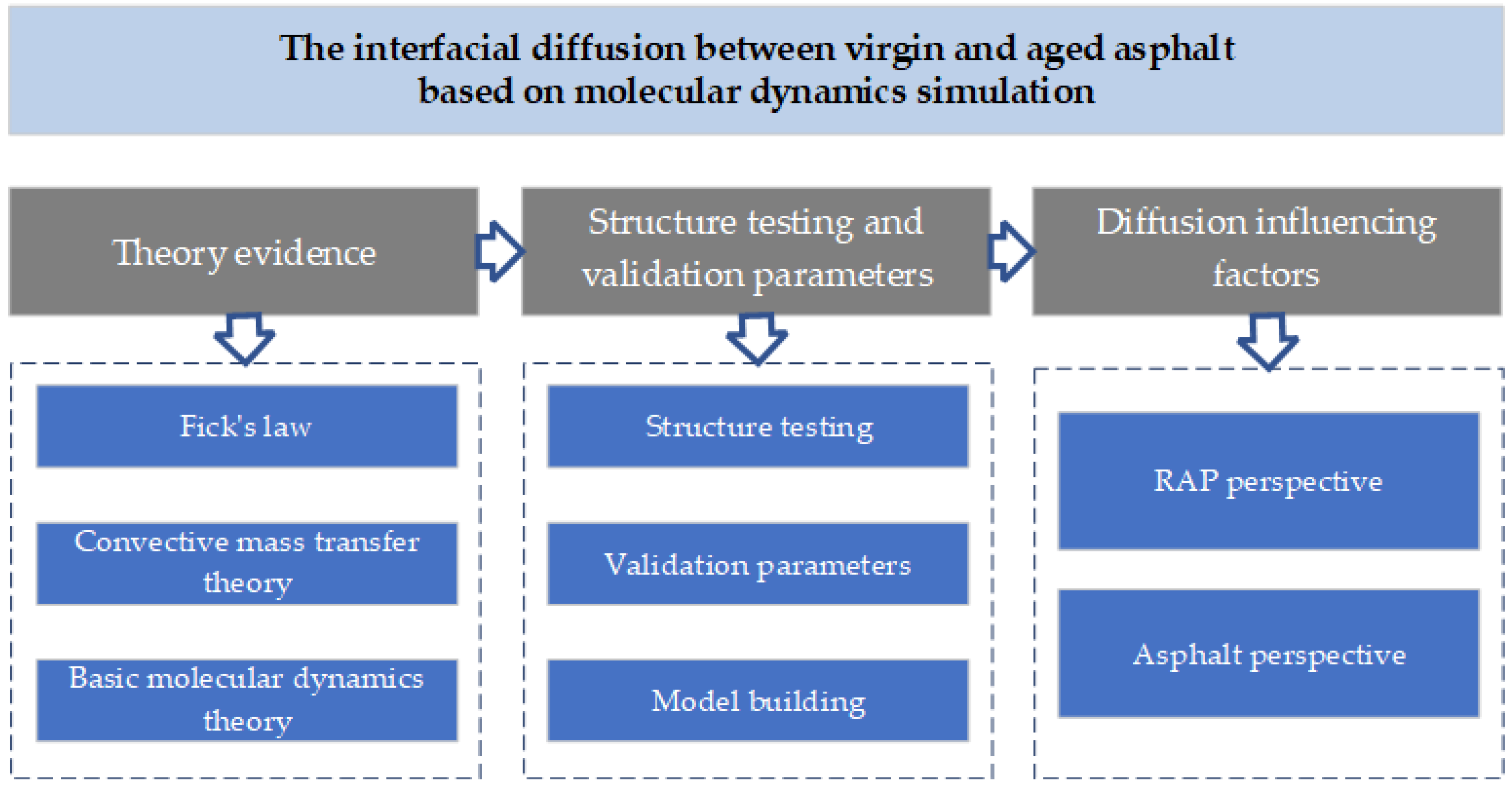
:1. Introduction
2. Theory Evidence
2.1. Asphalt Diffusion Theory
2.1.1. Fick’s Law and Composite Theory
2.1.2. Convective Mass Transfer Theory
2.2. Basic Theory of Molecular Dynamics
3. Structure Testing and Validation Parameters
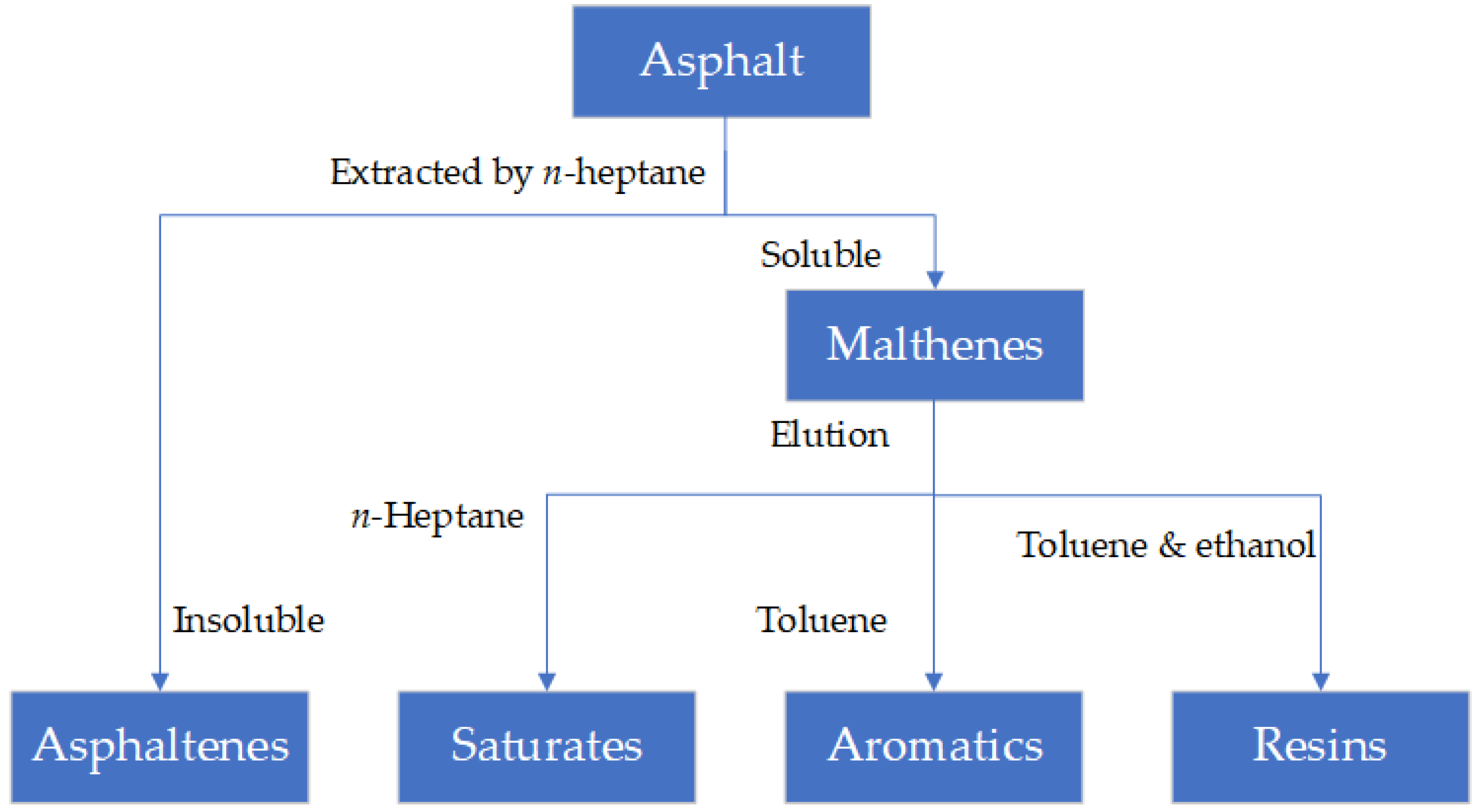
3.1. Main Methods for Determining Molecular Structure
3.1.1. Alumina Chromatographic Column Method
3.1.2. Thin-Layer Chromatography with Flame Ionization Detection (TCL-FID)
3.2. Establishment and Validation of a Virgin and Aged Asphalt Interface Model
- Representative molecular mixed phases could approach the characteristics of real mixed phases;
- Each component of virgin and aged asphalt should comply with structural validation tests;
- The simulation calculation accuracy of assembly models should be improved.
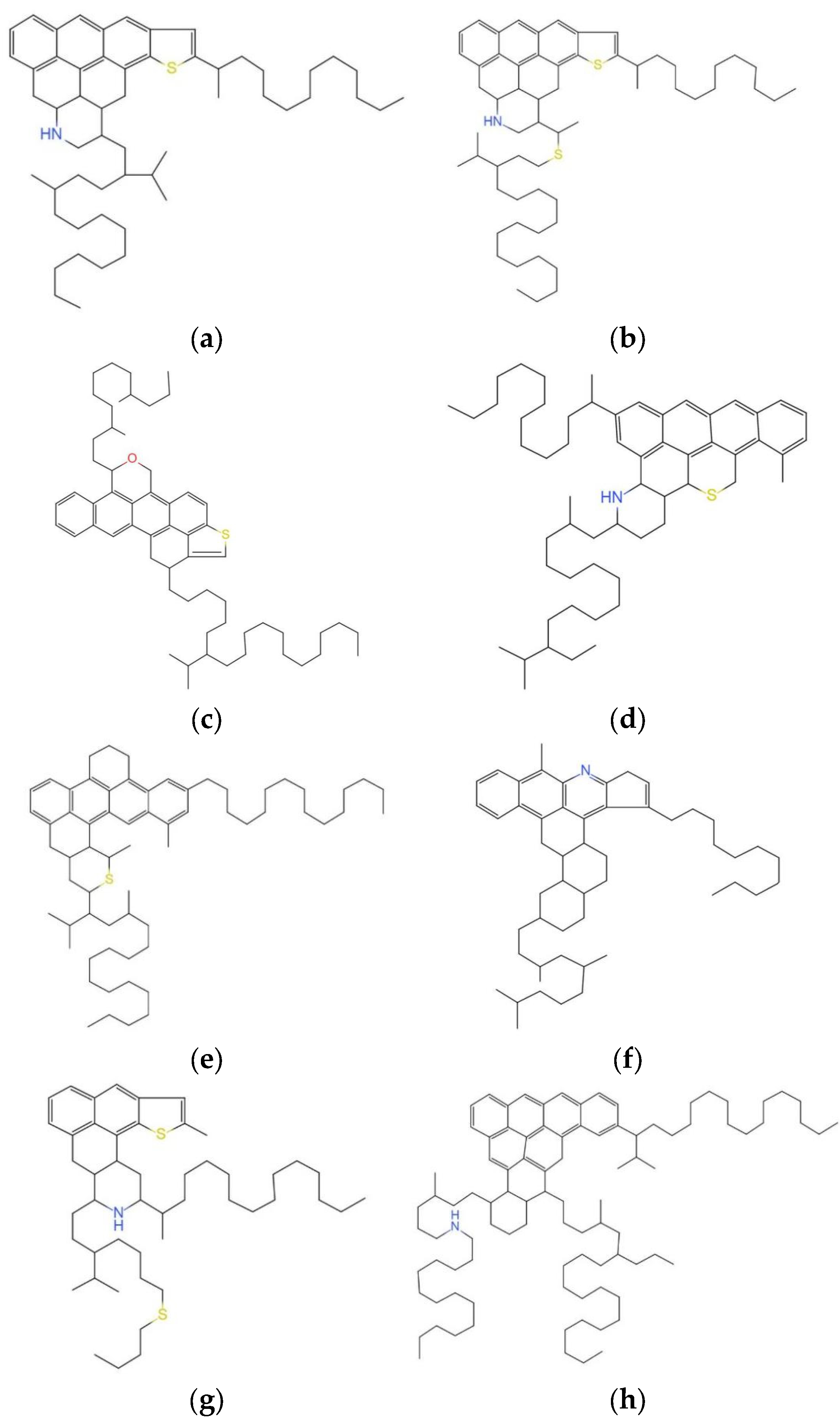
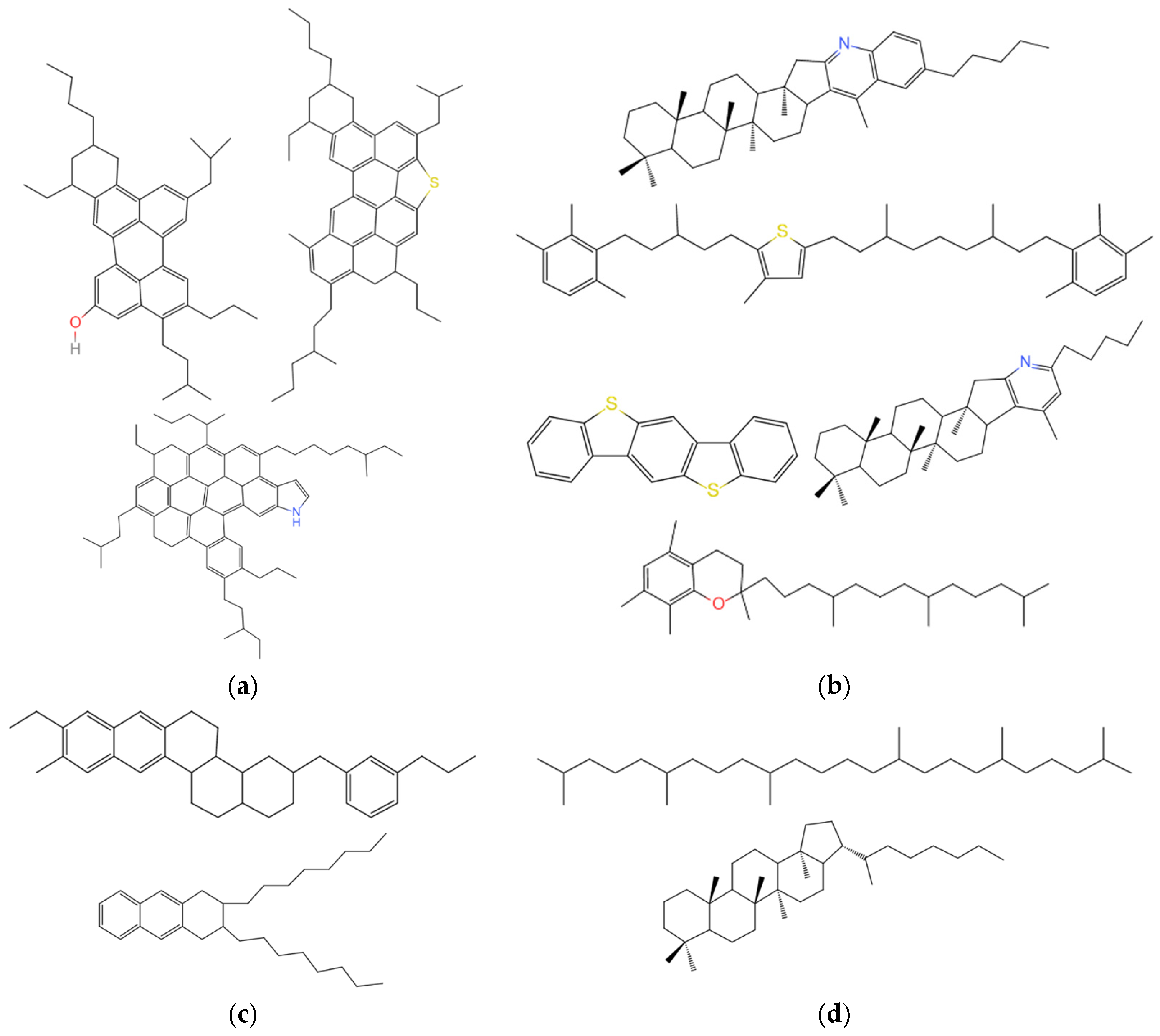
3.2.1. Molecular Structure of Virgin Asphalt
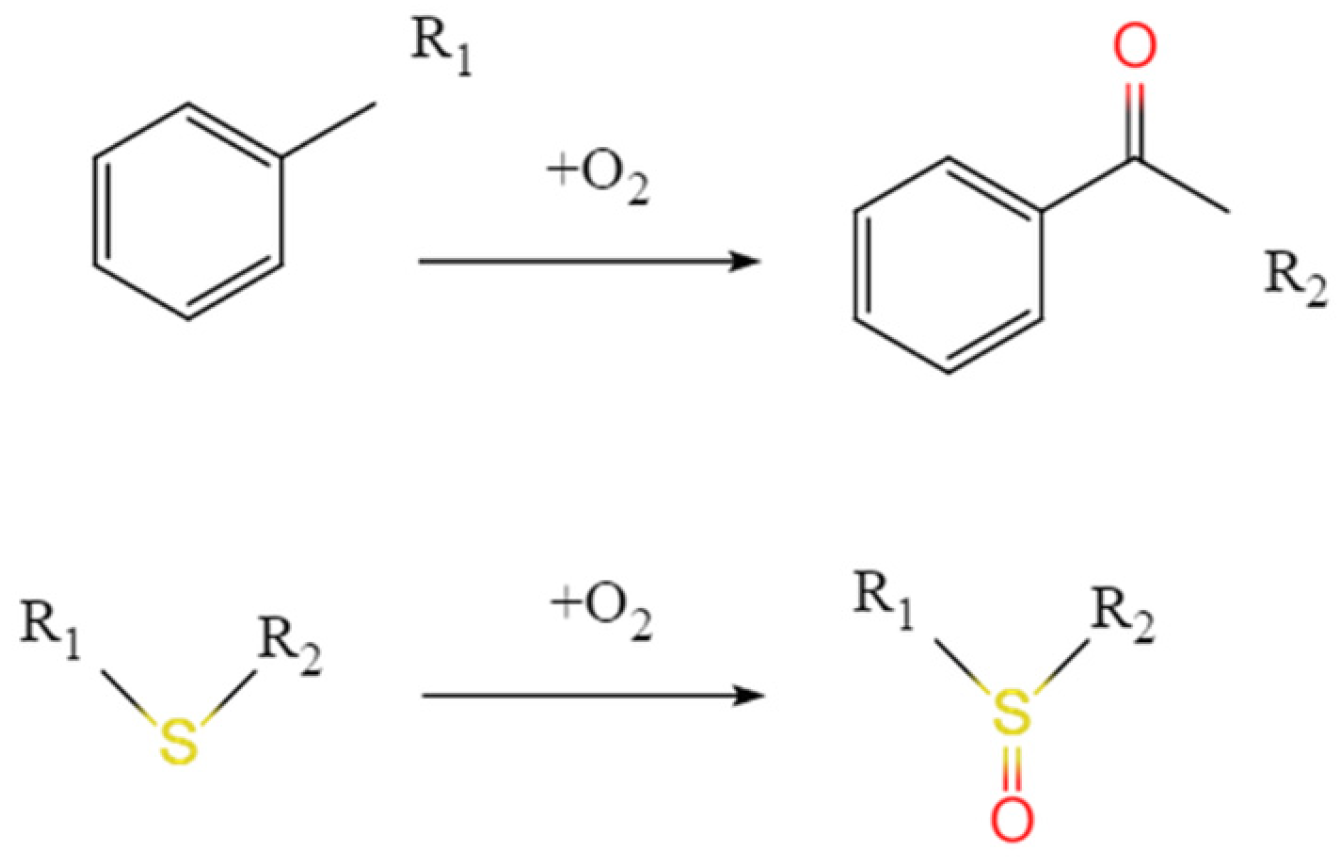
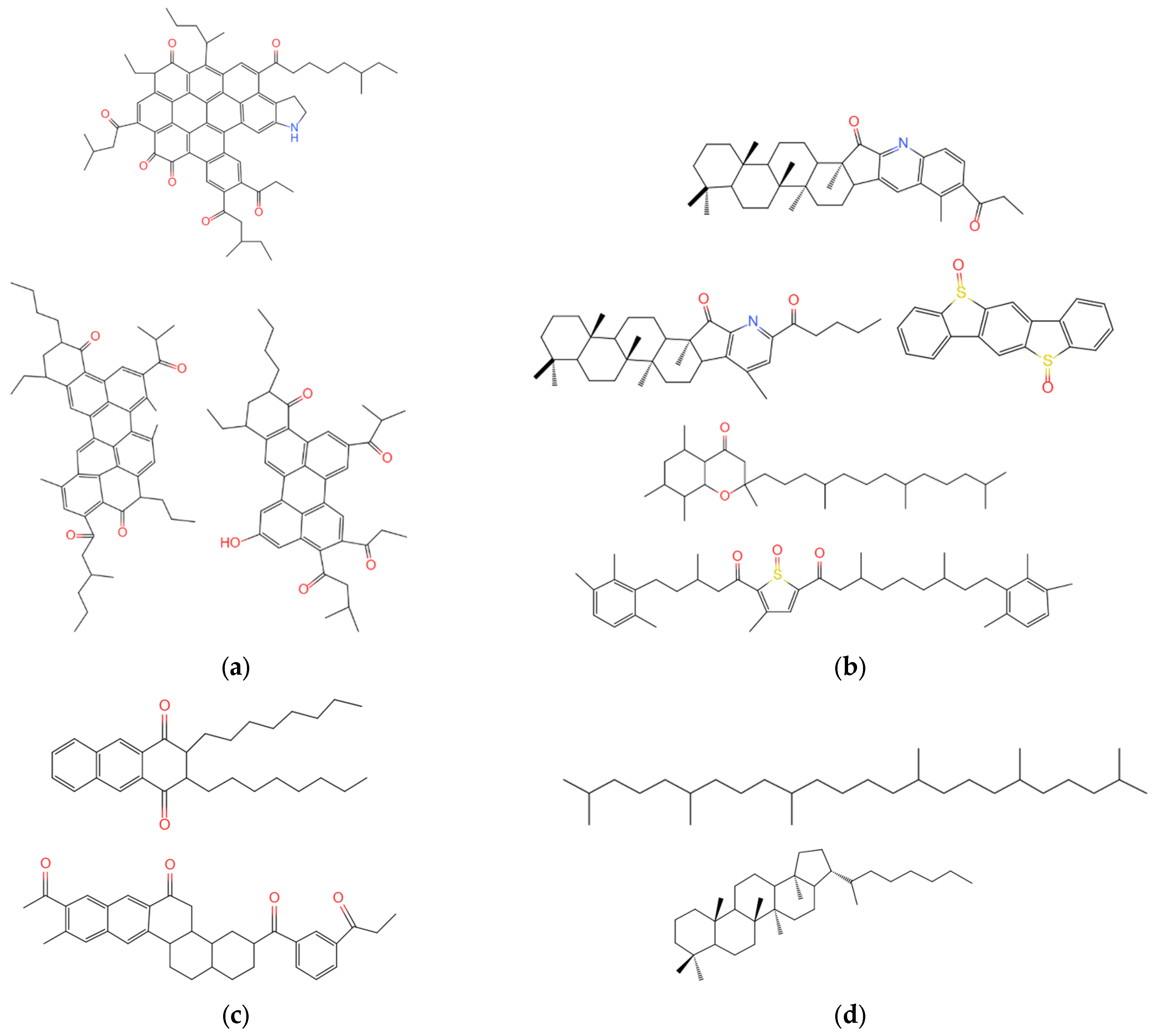
3.2.2. Molecular Structure of Aged Asphalt
3.3. Molecular Structure Validation Methods
3.3.1. Density
3.3.2. Viscosity
- WLF equation [67]:
- Rouse model [68]:
- Reverse non-equilibrium molecular dynamics (rNEMD) [69]:
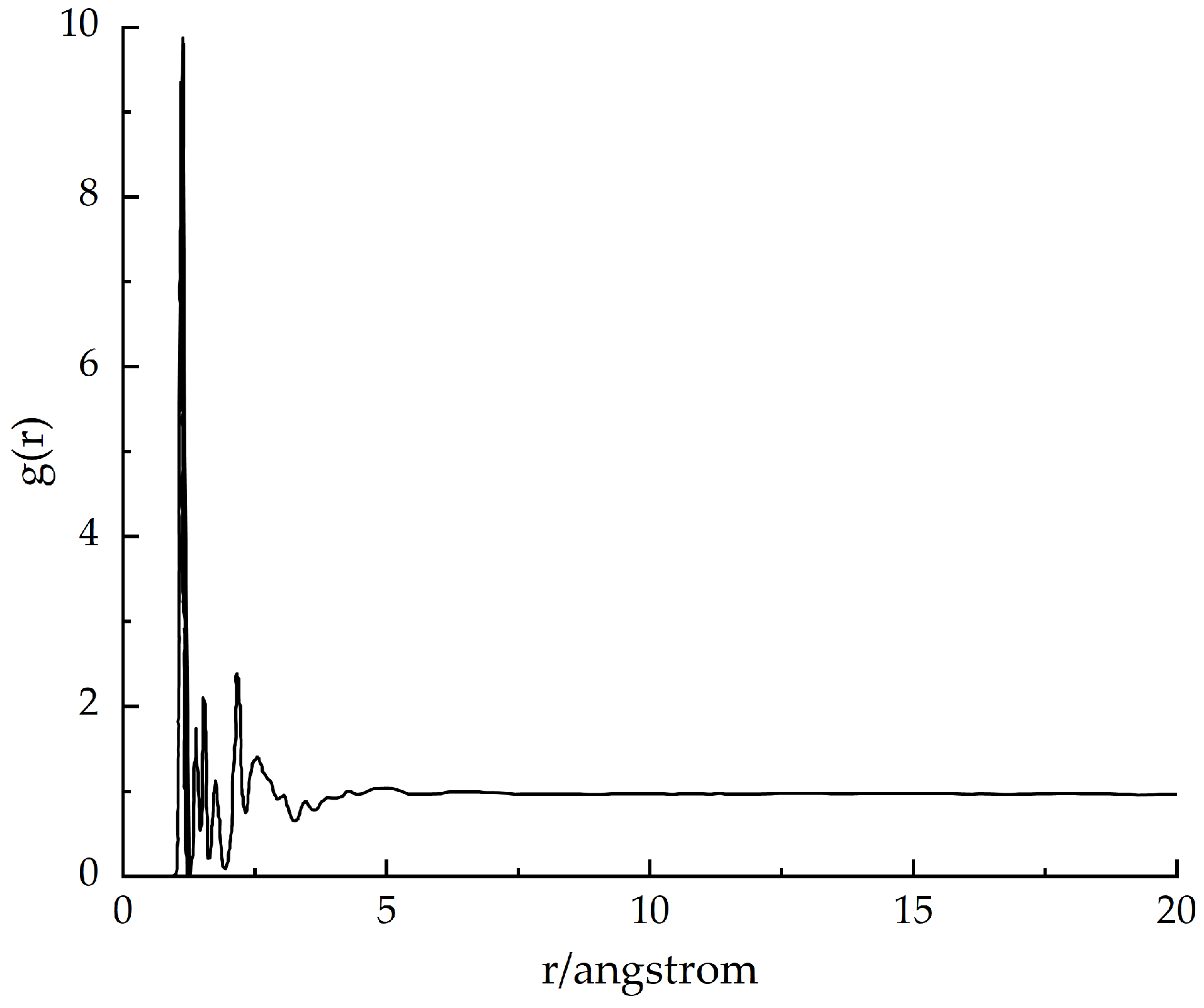
3.3.3. Radial Distribution Function (RDF)
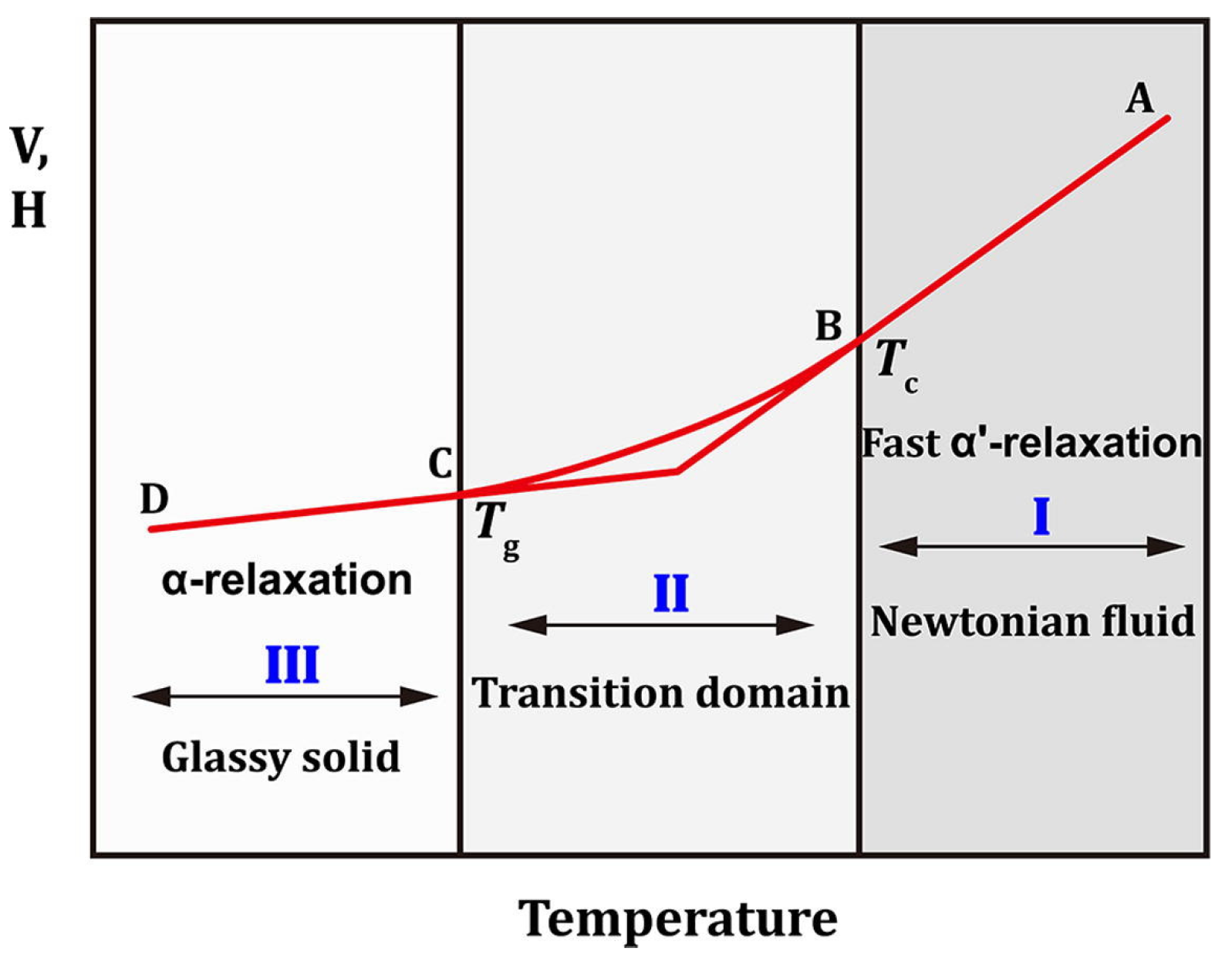
3.3.4. Glass Transition Temperature
3.3.5. Solubility Parameter
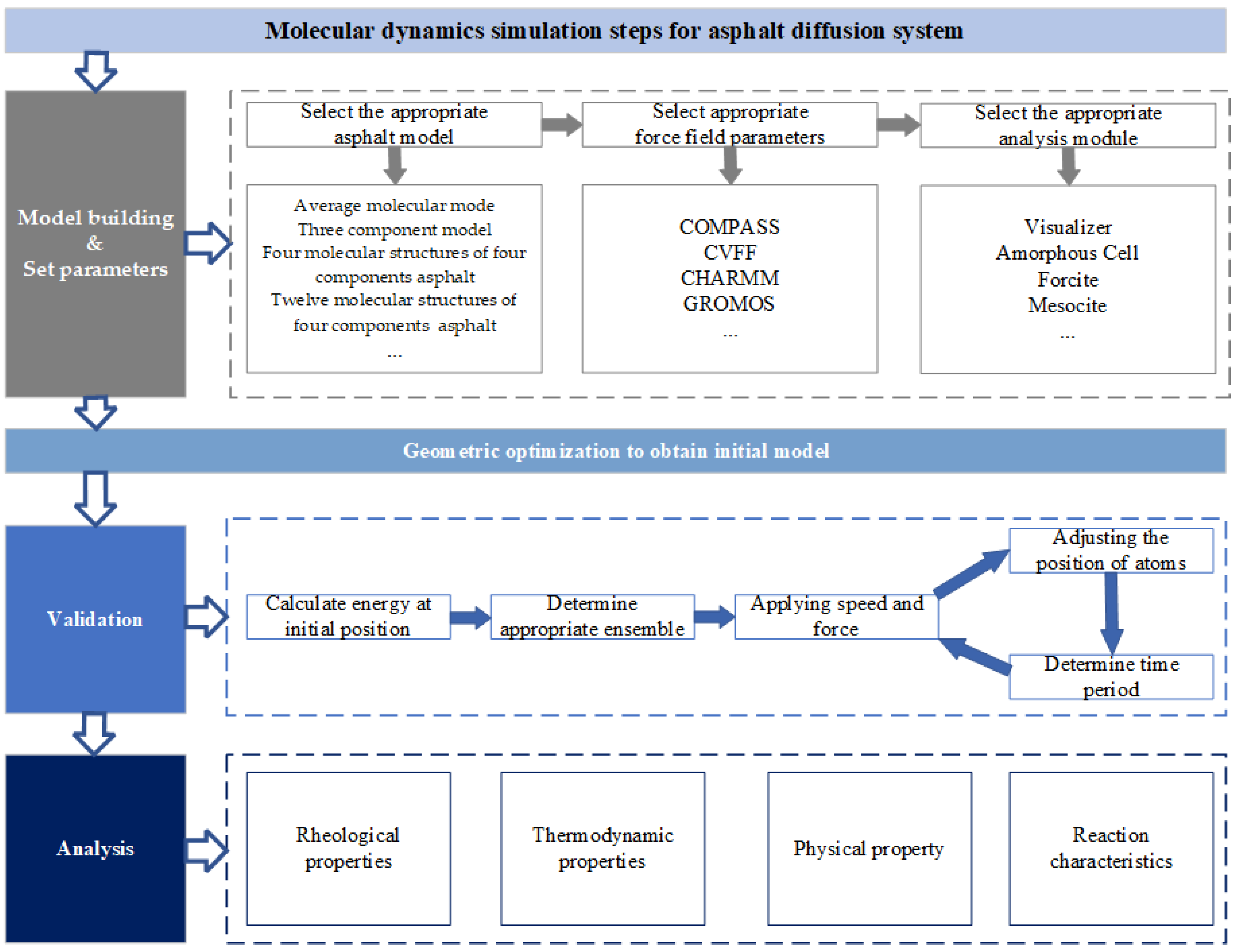
3.4. Module and Parameter Selection
4. Diffusion Influencing Factors
4.1. RAP Perspective
4.2. Asphalt Perspective
5. Conclusions and Perspective
- The paper summarizes the main theories of interface diffusion and the basic characteristics and application scenarios of each module in software, proposing the basic steps of molecular dynamics simulation. Two methods for measurement of the molecular structure of asphalt are summarized and compared. The alumina chromatographic column method is widely used and could be suitable for most measurement situations. The TCL-FID method needs to be promoted and used according to unified standards.
- The structures of virgin and aged asphalt molecules are summarized. The twelve molecular structures of four-component models have improved the accuracy of simulation, and the simulation parameters are close to those of real asphalt. Under the control of calculation speed, the model could effectively represent the molecular structure of real asphalt. Multi-parameter and multi-indicator validation could reduce the negative impact caused by different parameter fluctuations. The COMPASS series of force fields could effectively simulate the complex structures of various molecules and predict thermodynamic performance, being suitable for interface problems. The AC module and NPT ensemble and NVT are selected based on actual problems.
- This paper analyzes the current influencing factors affecting mixed diffusion systems. From a RAP perspective, DOB is related to the mixing temperature. DOB increases by approximately 7% every 15 °C. When the RAP content is 20%, the effect of the content on partial mixed systems is not significant. From the perspective of asphalt, different types of asphalt have a crucial impact on the degree of diffusion. When the heating temperature is 130 °C and the heating time is 60 min, the DOB of 90# asphalt increases by about 10% compared to 70# asphalt. In addition, there is a good logarithmic relationship between DSR and the fusion rate of simulation. The diffusion effect of temperature on asphaltene is higher than that of resin and aromatics. The coefficient of variation differs by about 10%.
- In current research, molecular dynamics simulations have limitations on the time scale. Asphalt pavement can involve long-term problems. It is not possible to directly simulate the long-term aging effect. Finally, some studies have used more types of molecular models in aggregate interfaces. However, various types mean the expansion of the entire model, which influences computational efficiency and spatial structure authenticity. So, in the modeling of virgin and aged asphalt, the molecular structure components of asphalt should be expanded, and a complete, unified, and realistic asphalt molecular model dataset should be formed from different sources and aging methods. The optimization of parameters such as the interaction potential function and initial conditions should be taken into account. Sensitivity analysis and validation of parameters should be taken seriously. The simulation of adding different additives such as modifiers and rejuvenators to the system should be considered. Additionally, it is recommended to combine macro-scale tests and molecular levels to clarify the mechanisms of low temperature and salt damage.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Amorphous Cell |
| AFM | Atomic Force Microscope |
| CED | Cohesive Energy Density |
| CHARMM | Chemistry at HARvard Macromolecular Mechanics |
| COMPASS | Condensed-Phase Optimized Molecular Potentials for Atomistic Simulation Studies |
| CT | Computed Tomography |
| CVFF | Consistent Valence Force Field |
| DOA | Degree of Activity |
| DOB | Degree of Blending |
| DSC | Differential Scanning Calorimetry |
| DSR | Dynamics Shear Rheometry |
| FTIR | Fourier Transform Infrared Spectrometry |
| GAFF | Generation Amber Force Field |
| GPC | Gel Permeation Chromatography |
| GPU | Graphic Process Unit |
| GROMOS | Groningen Molecular Simulation |
| ICO | Index of Carbonyl |
| ISO | Index of Sulfoxide |
| J-OCTA | J-Open Computational Tool for Advanced Material Technology |
| LAMMPS | Large-scale Atomic/Molecular Massively Parallel Simulator |
| NVT, NVE, NPT, NPH | N: the Number of Moles; V: Volume; P: Pressure; T: Temperature; E: Energy; H: Enthalpy |
| OPLS | Optimized Potentials for Liquid Simulations |
| PCFF | Polymer Consistent Family of Force Field |
| RAP | Reclaimed Asphalt Pavement |
| RDF | Radial Distribution Function |
| rNEMD | Reverse Non-Equilibrium Molecular Dynamics |
| RTF | RAP Film Thickness |
| RTFOT | Rolling Thin Film Oven Test |
| SARA | Saturates, Asphaltene, Resin, and Aromatics |
| SE method | Stokes–Einstein method |
| TCL-FID | Thin-layer Chromatography with Flame Ionization Detection |
| WLF | Williams–Landel–Ferry |
References
- Ministry of Transport of the People’s Republic of China. Statistical Bulletin on the Development of the Transportation Industry in 2022. China Water Transp. 2023, 29–33. [Google Scholar] [CrossRef]
- Xu, W.Y.; Li, W.; Ji, Y.C. Mechanical Behavior Investigation of Reclaimed Asphalt Aggregate Concrete in a Cold Region. Materials 2021, 14, 4101. [Google Scholar] [CrossRef] [PubMed]
- Shirodkar, P.; Mehta, Y.; Nolan, A.; Sonpal, K.; Norton, A.; Tomlinson, C.; Dubois, E.; Sullivan, P.; Sauber, R. A study to determine the degree of partial blending of reclaimed asphalt pavement (RAP) binder for high RAP hot mix asphalt. Constr. Build. Mater. 2011, 25, 150–155. [Google Scholar] [CrossRef]
- Wang, Z.J.; Zhang, H.X.; Liang, Q.Y.; Yan, F.F. Quantitative Evaluation of Emulsified Asphalt Mortar on RAP Coating Degree. China J. Highw. Transp. 2021, 34, 111–124. [Google Scholar] [CrossRef]
- Guo, P.; Xie, F.; Meng, J.; Meng, X.; Wei, L.; Xu, J.; Feng, Y. Review on the Interface Blending Behavior of Virgin Asphalt and Aged Asphalt During Asphalt Reclaiming. Mater. Rep. 2020, 34, 13100–13108. [Google Scholar] [CrossRef]
- Wang, J.; Qin, Y.C.; Xu, J.; Liu, L.P. In-situ characterization of microscale mechanical properties of interface between cement emulsified asphalt and aged asphalt. J. Southeast Univ. Nat. Sci. Ed. 2022, 52, 288–298. [Google Scholar] [CrossRef]
- Guo, P.; Lu, C.H.; Xie, Z.; Wang, D.W.; Gong, H.R.; Liu, F. Study on Interfacial Fusion Characteristics of Virgin and Aged Asphalt of Warm Mix Recycled Mixture at Micro Scale. China J. Highw. Transp. 2021, 34, 89–97. [Google Scholar] [CrossRef]
- Sun, J.C.; Zhang, W.; Wang, Z.; Qi, Y. Quantitative Characterization of Fusion Degree of New and Old Asphalt Based on Infrared Spectrometer. Sci. Tech. Eng. 2022, 22, 4143–4151. [Google Scholar] [CrossRef]
- Chen, L.; Chen, H.B.; Li, P.; Hu, P. Quantitative Evaluation on Interfacial Diffusion Behavior of Asphalt with High Percentage of RAP. J. Build. Mater. 2021, 24, 811–819. [Google Scholar] [CrossRef]
- Zou, G.L.; Zhang, J.J.; Li, Y.Y.; Lin, Z.P. Quantitative characterize binder blending and diffusion in recycled asphalt mixture: An environmental-friendly solution using wooden cube and 3D fluorescence image technology. J. Clean. Prod. 2021, 293, 12. [Google Scholar] [CrossRef]
- Xu, G.J.; Wang, H. Study of cohesion and adhesion properties of asphalt concrete with molecular dynamics simulation. Comput. Mater. Sci. 2016, 112, 161–169. [Google Scholar] [CrossRef]
- Xu, G.J.; Wang, H. Diffusion and interaction mechanism of rejuvenating agent with virgin and recycled asphalt binder: A molecular dynamics study. Mol. Simul. 2018, 44, 1433–1443. [Google Scholar] [CrossRef]
- Ding, H.Y.; Wang, H.N.; Qu, X.; Varveri, A.; Gao, J.F.; You, Z.P. Towards an understanding of diffusion mechanism of bio-rejuvenators in aged asphalt binder through molecular dynamics simulation. J. Clean. Prod. 2021, 299, 15. [Google Scholar] [CrossRef]
- Cui, B.Y.; Gu, X.Y.; Hu, D.L.; Dong, Q. A multiphysics evaluation of the rejuvenator effects on aged asphalt using molecular dynamics simulations. J. Clean. Prod. 2020, 259, 14. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Clarendon Press: London, UK, 1956; pp. 1–10. [Google Scholar]
- He, Y.; Alavi, Z.; Harvey, J.; Jones, D. Evaluating Diffusion and Aging Mechanisms in Blending of New and Age-Hardened Binders During Mixing and Paving. Transp. Res. Rec. 2016, 2574, 64–73. [Google Scholar] [CrossRef]
- Karlsson, R.; Isacsson, U. Application of FTIR-ATR to characterization of bitumen rejuvenator diffusion. J. Mater. Civ. Eng. 2003, 15, 157–165. [Google Scholar] [CrossRef]
- Jin, L. Study on the Diffusion Behavior of Asphalt Rejuvenator and Influence Factors. Ph.D. Thesis, China University of Petroleum, Beijing, China, 2010. [Google Scholar]
- Gao, F. Diffusion Mechanism and Macro-Micro Characteristics of Fresh-RAP Binders. Master’s Thesis, Harbin Institute of Technology, Harbin, China, 2018. [Google Scholar]
- Davison, R.R.; Bullin, J.A.; Glover, C.J.; Chaffin, J.M.; Peterson, G.D. Verification of an Asphalt Aging Test and Development of Superior Recycling Agents and Asphalts; Final Report; Texas Transportation Institute: College Station, TX, USA, 1994; 358p.
- Xu, M.; Zhang, Y.Z. Study of rejuvenators dynamic diffusion behavior into aged asphalt and its effects. Constr. Build. Mater. 2020, 261, 14. [Google Scholar] [CrossRef]
- Wei, W.; Guo, P.; Tang, B. Review of the Research on Diffusion Efficiency of Virgin-Aged Asphalt in Recycled Asphalt Mixture. Mater. Rep. 2017, 31, 109–114. [Google Scholar] [CrossRef]
- Yang, Y.W.; Ma, T. Proposed Testing Procedure for Estimation of Effective Recycling Ratio of Aged Asphalt in Hot Recycling Technical Conditions. J. Build. Mater. 2011, 14, 418–422. [Google Scholar] [CrossRef]
- Tao, X. Research on Mechanism of Asphalt Film Transfer Based on Convective Mass Transfer Theory. J. Highw. Transp. Res. Dev. Chin. Ed. 2010, 27, 21–24. [Google Scholar] [CrossRef]
- Huang, F.; Xia, C.; Chen, M. Film-Penetration Method to Determining the Convective Mass Transfer Coefficient. J. Nanjing Norm. Univ. Eng. Technol. Ed. 2007, 7, 26–29. [Google Scholar] [CrossRef]
- Wen, Y.; Zhu, R.; Zhou, F.; Wang, C. An Overivew on Molecular Dynamics Simulation. Adv. Mech. 2003, 33, 65–73. [Google Scholar] [CrossRef]
- Llovell, F.; Pamies, J.C.; Vega, L.F. Thermodynamic properties of Lennard-Jones chain molecules: Renormalization-group corrections to a modified statistical associating fluid theory. J. Chem. Phys. 2004, 121, 10715–10724. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.F. The Colloidal Aspect of a Macrostructure of Petroleum Asphalt. Fuel Sci. Technol. Int. 1992, 10, 723–733. [Google Scholar] [CrossRef]
- Rahmani, S.; McCaffrey, W.C.; Dettman, H.D.; Gray, M.R. Coking kinetics of asphaltenes as a function of chemical structure. Energy Fuels 2003, 17, 1048–1056. [Google Scholar] [CrossRef]
- Zhang, C.C.; Xu, T.; Shi, H.Q.; Wang, L.L. Physicochemical and pyrolysis properties of SARA fractions separated from asphalt binder. J. Therm. Anal. Calorim. 2015, 122, 241–249. [Google Scholar] [CrossRef]
- Feng, Z.G.; Zhang, J.B.; Li, X.J.; Yu, J.Y. Determination of the four generic fractions of aged bitumen by thin-layer chromatography with flame ionization detection. Chin. J. Chromatogr. 2015, 33, 195–200. [Google Scholar] [CrossRef]
- Zou, Y.H.; Chen, S.M.; Chen, W.S. Test methods study of bar thin layer chromatograph in measuring four components of bitumen. Pet. Asph. 2009, 23, 17–21. [Google Scholar] [CrossRef]
- Yang, B.; Zheng, L.; Zhang, K.; Cui, Z.; Wang, X.; Li, X. Study on SARA Composition of Crude Oil by TLC/FID. Chem. Eng. Oil Gas 2011, 40, 201–203+222+101. [Google Scholar]
- Du, G.; Yang, H.; Gu, J.; Lin, Y. TCL/FID Determination of the Family Compositon of Petroleum Asphalt Using PLS. PTCA Part B Chem. Anal. 2004, 12, 715–718. [Google Scholar] [CrossRef]
- Jennings, P.W.; Pribanic, J.A.S.; Desando, M.A.; Raub, M.F.; Moats, R.A.; Smith, J.A.; Mendes, T.M.; McGrane, M.T.; Fanconi, B.M.; Vanderhart, D.L.; et al. Binder Characterization and Evaluation by Nuclear Magnetic Resonance Spectroscopy; National Academy of Sciences: Washington, DC, USA, 1993. [Google Scholar]
- Pauli, A.T.; Grimes, W.; Huang, S.C.H.; Robertson, R.E. Surface energy studies of SHRP asphalts by AFM. Abstr. Pap. Am. Chem. Soc. 2003, 225, U422. [Google Scholar]
- Storm, D.A.; Edwards, J.C.; Decanio, S.J.; Sheu, E.Y. Molecular Representations of Ratawi and Alaska North Slope Asphaltenes based on Liquid-State and Solid-State NMR. Energy Fuels 1994, 8, 561–566. [Google Scholar] [CrossRef]
- Murgich, J.; Abanero, J.A.; Strausz, O.P. Molecular Recognition in Aggregates Formed by Asphaltene and Resin Molecules from the Athabasca Oil Sand. Energy Fuels 1999, 13, 278–286. [Google Scholar] [CrossRef]
- Murgich, J.; Rodríguez; Aray, Y. Molecular Recognition and Molecular Mechanics of Micelles of Some Model Asphaltenes and Resins. Energy Fuels 1996, 10, 68–76. [Google Scholar] [CrossRef]
- Artok, L.; Su, Y.; Hirose, Y.; Hosokawa, M.; Murata, S.; Nomura, M. Structure and Reactivity of Petroleum-Derived Asphaltene. Energy Fuels 1999, 13, 287–296. [Google Scholar] [CrossRef]
- Groenzin, H.; Mullins, O.C. Molecular size and structure of asphaltenes from various sources. Energy Fuels 2000, 14, 677–684. [Google Scholar] [CrossRef]
- Takanohashi, T.; Sato, S.; Saito, I.; Tanaka, R. Molecular Dynamics Simulation of the Heat-Induced Relaxation of Asphaltene Aggregates. Energy Fuels 2003, 17, 135–139. [Google Scholar] [CrossRef]
- Mullins, O.C. The Modified Yen Model. Energy Fuels 2010, 24, 2179–2207. [Google Scholar] [CrossRef]
- Dickie, J.P.; Yen, T.F. Macrostructures of the asphaltic fractions by various instrumental methods. Anal. Chem. 1967, 39, 1847–1852. [Google Scholar] [CrossRef]
- Zhang, L.; Greenfield, M.L. Molecular Orientation in Model Asphalts Using Molecular Simulation. Energy Fuels 2007, 21, 1102–1111. [Google Scholar] [CrossRef]
- Zhang, L.Q.; Greenfield, M.L. Analyzing properties of model asphalts using molecular simulation. Energy Fuels 2007, 21, 1712–1716. [Google Scholar] [CrossRef]
- Zhang, L.Q.; Greenfield, M.L. Rotational relaxation times of individual compounds within simulations of molecular asphalt models. J. Chem. Phys. 2010, 132, 10. [Google Scholar] [CrossRef]
- Qu, X.; Liu, Q.; Guo, M.; Wang, D.W.; Oeser, M. Study on the effect of aging on physical properties of asphalt binder from a microscale perspective. Constr. Build. Mater. 2018, 187, 718–729. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. Chemical compositions of improved model asphalt systems for molecular simulations. Fuel 2014, 115, 347–356. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. Viscosity, relaxation time, and dynamics within a model asphalt of larger molecules. J. Chem. Phys. 2014, 140, 10. [Google Scholar] [CrossRef]
- Li, D.D.; Greenfield, M.L. High Internal Energies of Proposed Asphaltene Structures. Energy Fuels 2011, 25, 3698–3705. [Google Scholar] [CrossRef]
- Tarefder, R.A.; Arisa, I. Molecular Dynamic Simulations for Determining Change in Thermodynamic Properties of Asphaltene and Resin Because of Aging. Energy Fuels 2011, 25, 2211–2222. [Google Scholar] [CrossRef]
- Pan, J.L.; Tarefder, R.A. Investigation of asphalt aging behaviour due to oxidation using molecular dynamics simulation. Mol. Simul. 2016, 42, 667–678. [Google Scholar] [CrossRef]
- Tang, W.; Guo, Y.J.; Ly, Y.J.; Chen, H. Influence of Biological Rejuvenator on Molecular Agglomeration Behavior of Aged Asphalt. J. Chongqing Jiaotong Univ. Nat. Sci. 2022, 41, 92–97. [Google Scholar] [CrossRef]
- Das, P.K.; Balieu, R.; Kringos, N.; Birgisson, B. On the oxidative ageing mechanism and its effect on asphalt mixtures morphology. Mater. Struct. 2015, 48, 3113–3127. [Google Scholar] [CrossRef]
- Pan, J.L.; Tarefder, R.A.; Hossain, M.I. Study of Moisture Impact on Asphalt Before and After Oxidation Using Molecular Dynamics Simulations. Transp. Res. Rec. 2016, 2574, 38–47. [Google Scholar] [CrossRef]
- Sun, D.Q.; Yu, F.; Li, L.H.; Lin, T.B.; Zhu, X.Y. Effect of chemical composition and structure of asphalt binders on self-healing. Constr. Build. Mater. 2017, 133, 495–501. [Google Scholar] [CrossRef]
- Qin, Q.; Schabron, J.F.; Boysen, R.B.; Farrar, M.J. Field aging effect on chemistry and rheology of asphalt binders and rheological predictions for field aging. Fuel 2014, 121, 86–94. [Google Scholar] [CrossRef]
- Hu, C.; Zhou, Z.G.; Luo, Y.Y.; Yan, X. Effect of Acid Rain on Mechanical Properties and Aging Mechanism of Asphalt. J. Mater. Civ. Eng. 2023, 35, 17. [Google Scholar] [CrossRef]
- Cui, Y.N.; Li, X.S.; Zhang, S.Y. Diffusion Mechanism of Regenerant Aged Asphalt Based on Molecular Dynamics Simulation. J. Build. Mater. 2021, 24, 1105–1109. [Google Scholar] [CrossRef]
- Williams, M.L.; Landel, R.F.; Ferry, J.D. The Temperature Dependence of Relaxation Mechanisms in Amorphous Polymers and Other Glass-forming Liquids. J. Am. Chem. Soc. 1955, 77, 3701–3707. [Google Scholar] [CrossRef]
- Tan, Y.Q.; Li, G.N.; Shan, L.Y.; Lv, H.J.; Meng, A.X. Research progress of bitumen microstructures and components. J. Traffic Transp. Eng. 2020, 20, 1–17. [Google Scholar] [CrossRef]
- Liu, F.J.; Xin, C.; Guan, M.Y.; Guo, M. Research on Diffusion Behavior of Waste Edible Oil-Aged Asphalt Based on Molecular Dynamics Simulation. Highw. Eng. 2022, 47, 120–125+131. [Google Scholar] [CrossRef]
- Zheng, C.F.; Shan, C.; Liu, J.; Zhang, T.; Yang, X.; Lyu, D. Microscopic adhesion properties of asphalt-mineral aggregate interface in cold area based on molecular simulation technology. Constr. Build. Mater. 2021, 268, 13. [Google Scholar] [CrossRef]
- Mazza, M.G.; Giovambattista, N.; Stanley, H.E.; Starr, F.W. Connection of translational and rotational dynamical heterogeneities with the breakdown of the Stokes-Einstein and Stokes-Einstein-Debye relations in water. Phys. Rev. E 2007, 76, 031203. [Google Scholar] [CrossRef]
- Nevins, D.; Spera, F.J. Accurate computation of shear viscosity from equilibrium molecular dynamics simulations. Mol. Simul. 2007, 33, 1261–1266. [Google Scholar] [CrossRef]
- Luo, L.; Chu, L.J.; Fwa, T.F. Molecular dynamics analysis of oxidative aging effects on thermodynamic and interfacial bonding properties of asphalt mixtures. Constr. Build. Mater. 2021, 269, 12. [Google Scholar] [CrossRef]
- Zhou, X.X.; Zhang, G.F.; Liu, R.M.; Zheng, L. Molecular Simulations of Anti-aging Mechanisms on Nano-LDHs Modified Asphalt. In Proceedings of the Annual Meetings of Chinese-Society’s-Building-Materials, Professional Committees of Stone and Aggregate and Utilization of Solid Waste, Wuhan, China, 28 November–1 December 2013; pp. 198–202. [Google Scholar]
- Yao, H.; Dai, Q.L.; You, Z.P. Molecular dynamics simulation of physicochemical properties of the asphalt model. Fuel 2016, 164, 83–93. [Google Scholar] [CrossRef]
- Przygocki, W. Radial distribution function in polymers. In Proceedings of the 3rd International School on X-ray Investigations of Polymer Structures (XIPS), Bielsko Biala, Poland, 10–13 June 1996; pp. 92–102. [Google Scholar]
- Xia, L.; Xiao, J.J.; Fan, J.F.; Zhu, W.; Xiao, H.M. Molecular Dynamics Simulation of Mechanical Properties and Surface Interaction for Nitrate Plasticizer. Acta Chim. Sin. 2008, 66, 874–878. [Google Scholar] [CrossRef]
- Yang, J.; Guo, N.; Guo, X.; Wang, Z.; Fang, C.; Chu, Z. Adhesion of Foamed Asphalt-Aggregate Interface Based on Molecular Dynamics. Mater. Rep. 2021, 35, 138–144. [Google Scholar]
- Kang, Y.; Zhou, D.H.; Wu, Q.; Lang, R.; Shangguan, S.X.; Liao, Z.W.; Wei, N. Molecular dynamics study on the glass forming process of asphalt. Constr. Build. Mater. 2019, 214, 430–440. [Google Scholar] [CrossRef]
- Qiu, Y.J.; Su, T.; Zheng, P.F.; Ding, H.B. Physical Aging Mechanism of Asphalt Binder Based on Molecular Simulation. J. Build. Mater. 2020, 23, 1464–1470. [Google Scholar] [CrossRef]
- Tabatabaee, H.A.; Velasquez, R.; Bahia, H.U. Predicting low temperature physical hardening in asphalt binders. Constr. Build. Mater. 2012, 34, 162–169. [Google Scholar] [CrossRef]
- Tang, W.; Wang, J.S.; Lyu, Y.J. Study on self-healing behavior of asphalt binder based on molecular dynamics. J. Wuhan Univ. Sci. Technol. 2020, 43, 123–127. [Google Scholar] [CrossRef]
- Hansen, C.M. 50 Years with solubility parameters—Past and future. Prog. Org. Coat. 2004, 51, 77–84. [Google Scholar] [CrossRef]
- Zhu, J.Y.; He, Z.Y. Research of Compatiblity of Asphalt and Anti-stripping Agent Using Molecular Dynamics. J. Highw. Transp. Res. Dev. 2016, 33, 34–40. [Google Scholar] [CrossRef]
- Zhu, J.Y. Molecular Dynamic Simulation of Self-healing Behavior of Asphalt Binder. J. Build. Mater. 2018, 21, 433–439. [Google Scholar] [CrossRef]
- Cao, L.P.; Zhang, X.K.; Yang, C. Modification mechanism of iron tailings asphalt mixture by silane coupling agents based on molecular dynamics. J. Cent. South Univ. 2021, 52, 2276–2286. [Google Scholar] [CrossRef]
- Zhai, R.X.; Hao, P.W. Research on the impact of mineral type and bitumen ageing process on asphalt-mineral adhesion performance based on molecular dynamics simulation method. Road Mater. Pavement Des. 2021, 22, 2000–2013. [Google Scholar] [CrossRef]
- Du, Z.; Zhu, X.Y.; Zhang, Y.Q. Diffusive Dynamics and Structural Organization of Moisture in Asphaltic Materials Based on Molecular Dynamics Simulation. J. Mater. Civ. Eng. 2021, 33, 13. [Google Scholar] [CrossRef]
- Xu, M.; Yi, J.Y.; Feng, D.C.; Huang, Y.D.; Wang, D.S. Analysis of Adhesive Characteristics of Asphalt Based on Atomic Force Microscopy and Molecular Dynamics Simulation. ACS Appl. Mater. Interfaces 2016, 8, 12393–12403. [Google Scholar] [CrossRef]
- Wang, H.; Lin, E.; Xu, G. Molecular dynamics simulation of asphalt-aggregate interface adhesion strength with moisture effect. Int. J. Pavement Eng. 2017, 18, 414–423. [Google Scholar] [CrossRef]
- Guo, M.; Huang, Y.; Wang, L.; Yu, J.; Hou, Y. Using atomic force microscopy and molecular dynamics simulation to investigate the asphalt micro properties. Int. J. Pavement Res. Technol. 2018, 11, 321–326. [Google Scholar] [CrossRef]
- Bhasin, A.; Bommavaram, R.; Greenfield, M.L.; Little, D.N. Use of Molecular Dynamics to Investigate Self-Healing Mechanisms in Asphalt Binders. J. Mater. Civ. Eng. 2011, 23, 485–492. [Google Scholar] [CrossRef]
- Xu, G.J.; Wang, H. Molecular dynamics study of interfacial mechanical behavior between asphalt binder and mineral aggregate. Constr. Build. Mater. 2016, 121, 246–254. [Google Scholar] [CrossRef]
- Sun, W.; Wang, H. Moisture effect on nanostructure and adhesion energy of asphalt on aggregate surface: A molecular dynamics study. Appl. Surf. Sci. 2020, 510, 11. [Google Scholar] [CrossRef]
- Sun, D.Q.; Sun, Q.G.; Zhu, X.Y.; Ye, F.Y.; Xu, J.Y. Intrinsic temperature sensitive self-healing character of asphalt binders based on molecular dynamics simulations. Fuel 2018, 211, 609–620. [Google Scholar] [CrossRef]
- Ding, Y.J.; Huang, B.S.; Shu, X. Modeling Shear Viscosity of Asphalt through Nonequilibrium Molecular Dynamics Simulation. Transp. Res. Rec. 2018, 2672, 235–243. [Google Scholar] [CrossRef]
- Zhang, W.W.; Hu, M.M.; Liu, K.; Yu, L.; He, Z.Y. Study on Road Performance of Hot Regeneration Mixture Based on Interfacial Regeneration Fusion Degree. J. Wuhan Univ. Technol. 2021, 45, 552–557. [Google Scholar]
- Navaro, J.; Bruneau, D.; Drouadaine, I.; Pouteau, B.; Colin, J.; Dony, A. Analyzing the influence of manufacturing conditions of reclaimed asphalt concrete on the characteristics of the asphalt binder: Development of a gradual binder extraction method. Eur. Phys. J. Appl. Phys. 2012, 58, 14. [Google Scholar] [CrossRef]
- Pires, G.M.; Lo Presti, D.; Airey, G.D. A practical approach to estimate the degree of binder activity of reclaimed asphalt materials. Road Mater. Pavement Des. 2021, 22, 1093–1116. [Google Scholar] [CrossRef]
- Stimilli, A.; Virgili, A.; Canestrari, F. New method to estimate the “re-activated” binder amount in recycled hot-mix asphalt. Road Mater. Pavement Des. 2015, 16, 442–459. [Google Scholar] [CrossRef]
- Rinaldini, E.; Schuetz, P.; Partl, M.N.; Tebaldi, G.; Poulikakos, L.D. Investigating the blending of reclaimed asphalt with virgin materials using rheology, electron microscopy and computer tomography. Compos. Part B 2014, 67, 579–587. [Google Scholar] [CrossRef]
- Yu, S.; Shen, S.H.; Zhang, C.; Zhang, W.G.; Jia, X.Y. Evaluation of the Blending Effectiveness of Reclaimed Asphalt Pavement Binder. J. Mater. Civ. Eng. 2017, 29, 8. [Google Scholar] [CrossRef]
- Chen, L.; He, Z.Y.; Chen, H.B.; Wang, X.D.; Xiang, H. Rheological Characteristics and Molecular Dynamics Simulation of Interface Regeneration Between Virgin and Aged Asphalts. China J. Highw. Transp. 2019, 32, 25–33. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, C.; Wan, M.; Zhou, X.X.; Wang, Y.F.; Wu, S.P. Study of the Diffusion of Rejuvenators and Its Effect on Aged Bitumen Binder. Appl. Sci. 2017, 7, 397. [Google Scholar] [CrossRef]
- Ding, Y.J.; Huang, B.S.; Shu, X.; Zhang, Y.Z.; Woods, M.E. Use of molecular dynamics to investigate diffusion between virgin and aged asphalt binders. Fuel 2016, 174, 267–273. [Google Scholar] [CrossRef]
- Newcomb, D.E.; Nusser, B.J.; Kiggundu, B.M.; Zallen, D.M. Laboratory Study of the Effects of Recycling Modifiers on Aged Asphalt Cement. Transp. Res. Rec. 1984, 968, 66–77. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Modules | Characteristics | Application |
|---|---|---|
| Visualizer | Graphical interface, a part of model building | It mainly builds and adjusts visualized 3D model |
| Amorphous Cell | Build an amorphous cell model by Monte Carlo method | It mainly constructs polymer blend systems with different components, ratios, solutions, and interface models |
| Forcite | Fast energy calculation of systems, geometric optimization and energy minimization, analyze different ensembles | It was applied in organic materials, inorganic small molecules, high-polymer materials, commonly used in asphaltene and modifiers |
| Mesocite | Two methods: coarse-grained and dissipative particle molecular dynamics, simulating at the mesoscopic level | It was applied in polymer dilution and dissolution, surface adsorption, surface activity, and composite materials |
| Test Method | Principles | Result Source | Features |
|---|---|---|---|
| Alumina chromatographic column method [32] | The chromatographic column serves as a stationary phase, achieving separation based on the adsorption capacity of different polar components | The ratio of the actual mass of each component to the total mass of the test sample |
|
| TCL-FID [34] | Extended separation of different solvents combined with hydrogen flame ionization detection | The ratio of peak area to the total peak area of each component in the graph |
|
| Molecular Component | Molecular Formula | Atomicity | Molecular Weight |
|---|---|---|---|
| Asphaltene | C42H54O | 97 | 575.0 |
| C66H81N | 148 | 888.5 | |
| C54H62S | 114 | 707.2 | |
| Polar Aromatics | C40H59N | 100 | 554.0 |
| C40H60S | 101 | 573.1 | |
| C18H10S2 | 30 | 290.4 | |
| C36H57N | 94 | 573.9 | |
| C29H50O | 80 | 414.8 | |
| Naphthene Aromatics | C35H44 | 79 | 464.8 |
| C30H46 | 76 | 406.8 | |
| Saturates | C30H62 | 92 | 422.9 |
| C35H62 | 97 | 483.0 |
| Author | Asphalt Type | Molecular Structure | Platform | Force Field | Module Selection | Ensemble Selection |
|---|---|---|---|---|---|---|
| Li Derek (2013) [49] | Virgin | SARA-12 1 | Windows: LAMMPS Linux: Spartan | OPLS-AA | Monte Carlo | —— |
| Cao L. (2021) [80] | Virgin and modified | SARA-12 | Windows: Material Studio | COMPASS | Visualizer Amorphous Cell | NPT NVT |
| Ruixin Z. (2020) [81] | Aged | SARA-12 | Windows: Material Studio | COMPASSII | Forcite | NVT NPT |
| Zhao D. (2020) [82] | Virgin | SARA-12 | Windows: Material Studio | PCFF Dreiding COMPASSII | Visualizer Amorphous Cell | NVT NPT NVE |
| Xu M. (2015) [63] | Virgin and aged | SARA-12 | Windows: LAMMPS | CVFF | Monte Carlo | NVT NPT |
| Cui Y. (2020) [60] | Aged | SARA-12 | Windows: Material Studio | COMPASSII | Visualizer Amorphous Cell | NVT NPT |
| Qiu Y. (2020) [74] | Virgin | SARA-4 2 | Windows: Material Studio | COMPASS | Visualizer Amorphous Cell Forcite | NVT NPT |
| Xu M. (2016) [83] | Virgin and aged | SARA-12 | Windows: LAMMPS | COMPASS | Monte Carlo | —— |
| Wang H. (2015) [84] | Virgin | SARA-12 | Windows: Plimpton1995 | CVFF | Monte Carlo | NVT |
| Guo Meng (2017) [85] | Virgin | SARA-12 | Windows: Material Studio | COMPASS | Visualizer Amorphous Cell | NVT |
| Bahasin (2011) [86] | Virgin | Average | Windows: Material Studio | COMPASS | Visualizer Amorphous Cell | —— |
| Xu G. (2016) [87] | Virgin | SARA-4 | Windows: Material Studio | COMPASSII | Visualizer Amorphous Cell | NVT NPT |
| Sun W. (2020) [88] | Virgin | SARA-4 | Windows: Material Studio | COMPASSII | Visualizer Amorphous Cell | NVT NPT |
| Sun D. (2018) [89] | Virgin | Average | Windows: Material Studio | Universal COMPASSII | Visualizer Amorphous Cell | NPT |
| Ding Y. (2018) [90] | Virgin and modified | SAR-8 3 | Windows: LAMMPS | —— | NEMD method | NVT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Wang, C.; Yang, Y. The Progress of the Interfacial Diffusion between Virgin and Aged Asphalt Based on Molecular Dynamics Simulation: A Review. Processes 2023, 11, 3024. https://doi.org/10.3390/pr11103024
Yang Y, Wang C, Yang Y. The Progress of the Interfacial Diffusion between Virgin and Aged Asphalt Based on Molecular Dynamics Simulation: A Review. Processes. 2023; 11(10):3024. https://doi.org/10.3390/pr11103024
Chicago/Turabian StyleYang, Yanhai, Chonghua Wang, and Ye Yang. 2023. "The Progress of the Interfacial Diffusion between Virgin and Aged Asphalt Based on Molecular Dynamics Simulation: A Review" Processes 11, no. 10: 3024. https://doi.org/10.3390/pr11103024
APA StyleYang, Y., Wang, C., & Yang, Y. (2023). The Progress of the Interfacial Diffusion between Virgin and Aged Asphalt Based on Molecular Dynamics Simulation: A Review. Processes, 11(10), 3024. https://doi.org/10.3390/pr11103024