
Infantile-Onset Isolated Neurohypophyseal Langerhans Cell Histiocytosis with Central Diabetes Insipidus: A Case Report



Abstract
1. Introduction
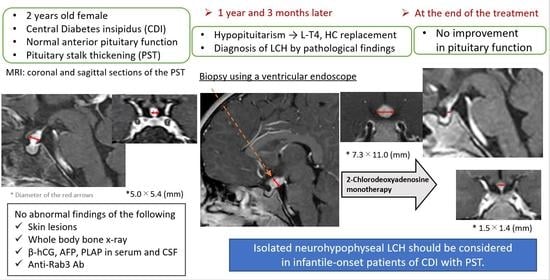
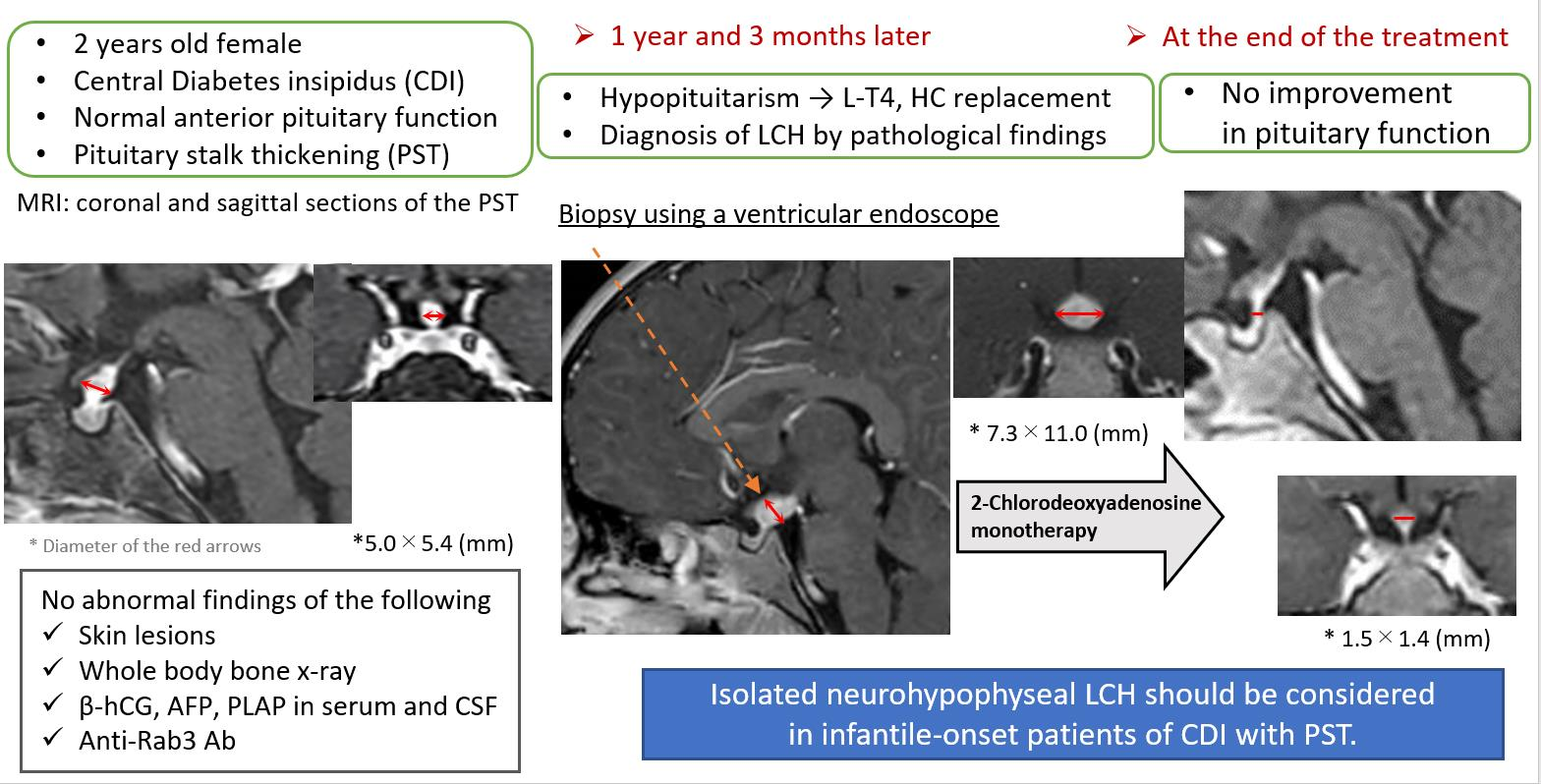
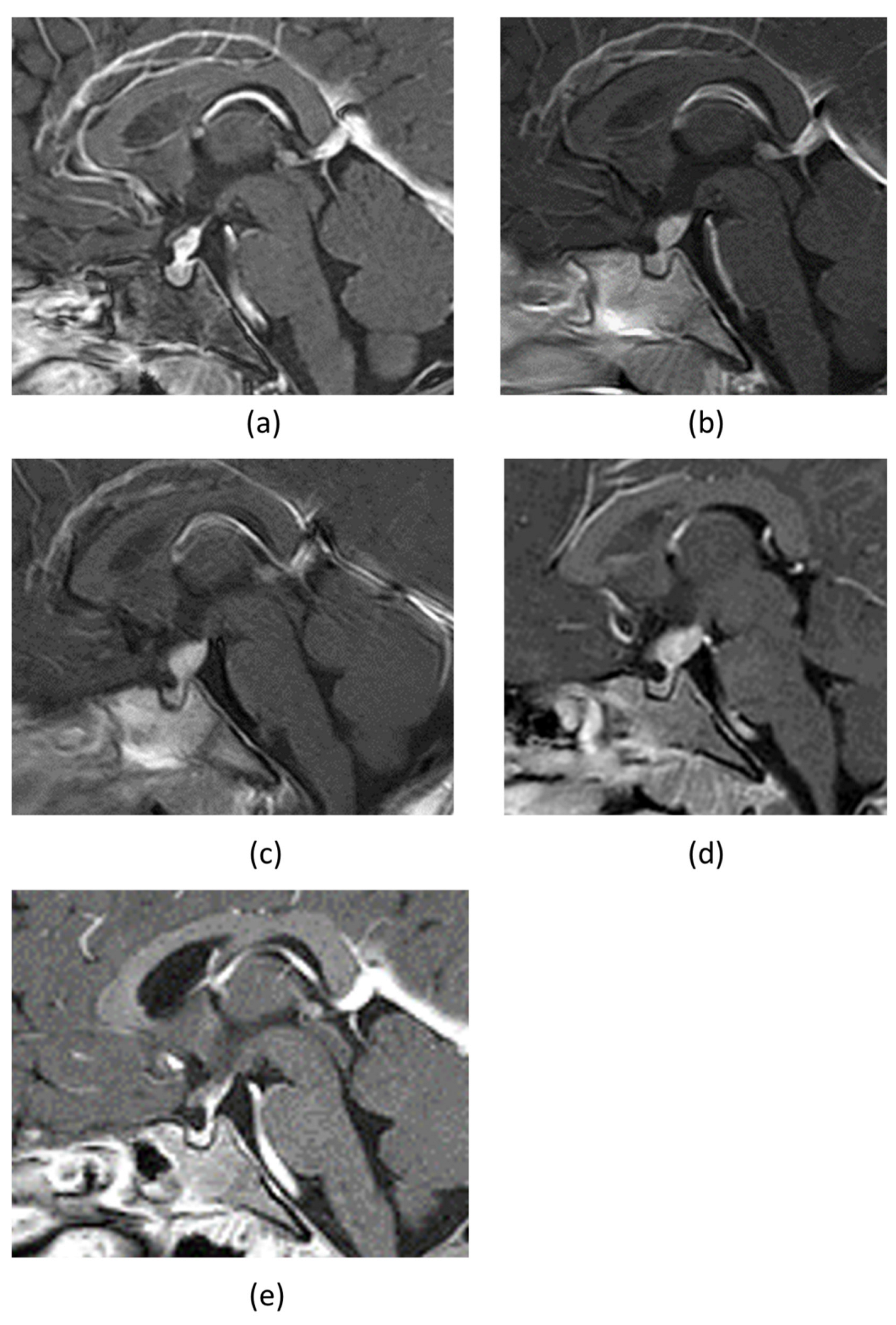
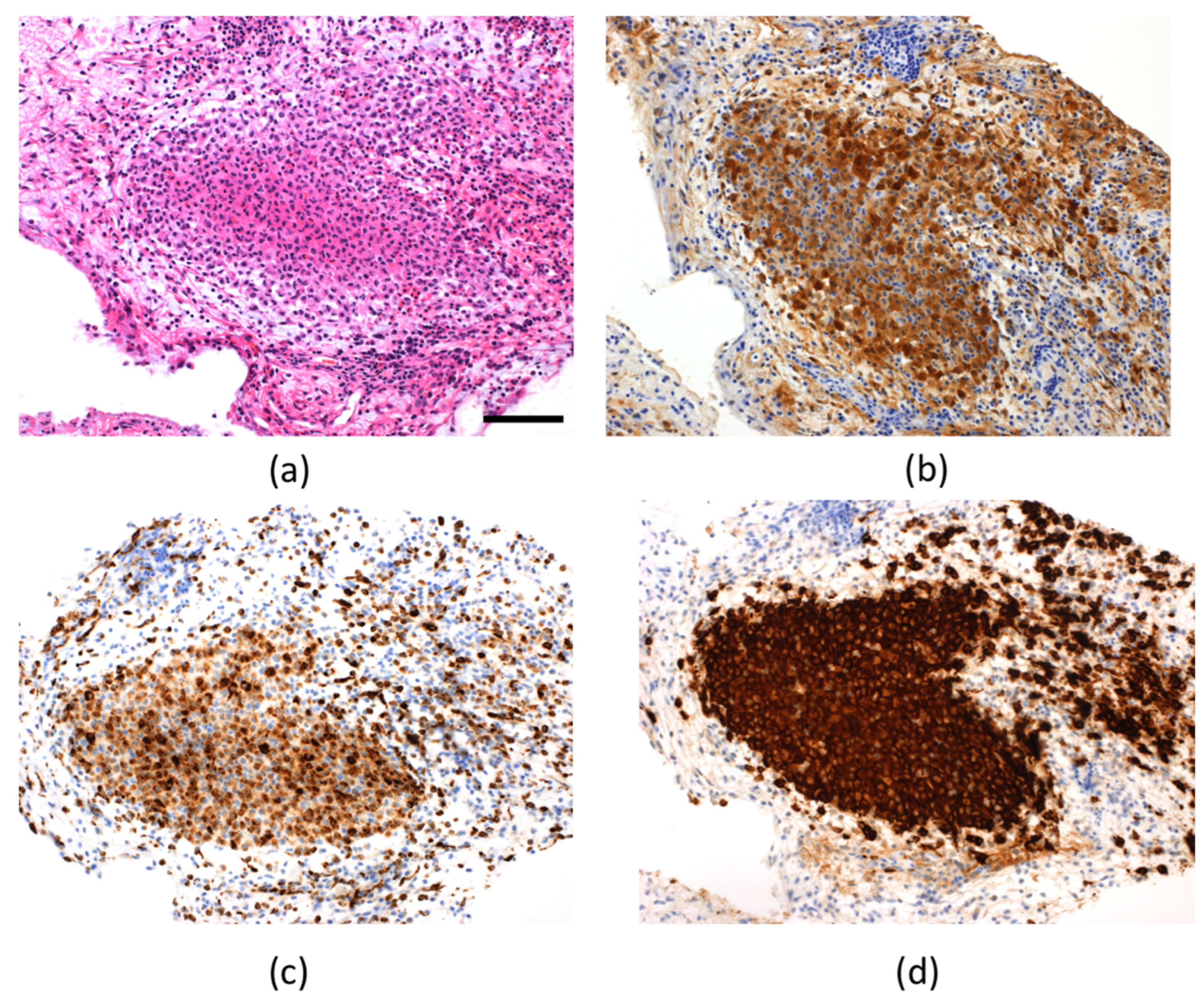
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maghnie, M.; Cosi, G.; Genovese, E.; Manca-Bitti, M.L.; Cohen, A.; Zecca, S.; Tinelli, C.; Gallucci, M.; Bernasconi, B.; Severi, F.; et al. Central diabetes insipidus in children and young adults. N. Engl. J. Med. 2000, 343, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Turcu, A.F.; Erickson, B.J.; Lin, E.; Guadalix, S.; Schwartz, K.; Scheithauer, B.W.; Atkinson, J.L.D.; Young, W.F., Jr. Pituitary stalk lesions: The Mayo Clinic experience. J. Clin. Endocrinol. Metab. 2013, 98, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Huo, Z.; Lu, T.; Liang, Z.; Ping, F.; Shen, J.; Lu, J.; Ma, W.; Zhao, D.; Zhong, D. Clinicopathological features and BRAF V600E mutations in patients with isolated hypothalamic-pituitary Langerhans cell histiocytosis. Diagn. Pathol. 2016, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Rao, J.; Li, C. Isolated Langerhans cell histiocytosis in the hypothalamic-pituitary region: A case report. BMC Endocr. Disord. 2019, 19, 143. [Google Scholar] [CrossRef]
- Yeh, E.A.; Greenberg, J.; Abla, O.; Longoni, G.; Diamond, E.; Hermiston, M.; Tran, B.; Rodriguez-Galindo, C.; Allen, C.E.; McClain, K.L. Evaluation and treatment of Langerhans cell histiocytosis patients with central nervous system abnormalities: Current views and new vistas. Pediatric Blood Cancer 2018, 65, e26784. [Google Scholar] [CrossRef]
- Robison, N.J.; Prabhu, S.P.; Sun, P.; Chi, S.N.; Kieran, M.W.; Manley, P.E.; Cohen, L.E.; Goumnerova, L.; Smith, E.R.; Scott, R.M.; et al. Predictors of neoplastic disease in children with isolated pituitary stalk thickening. Pediatric Blood Cancer 2013, 60, 1630–1635. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kim, P.H.; Jung, A.Y.; Choi, J.H.; Cho, Y.A.; Yoon, H.M.; Lee, J.S. Neoplastic Etiology and Natural Course of Pituitary Stalk Thickening. J. Clin. Endocrinol. Metab. 2022, 107, 563–574. [Google Scholar] [CrossRef]
- Aihara, Y.; Watanabe, S.; Amano, K.; Komatsu, K.; Chiba, K.; Imanaka, K.; Hori, T.; Ohba, T.; Dairoku, H.; Okada, Y.; et al. Placental alkaline phosphatase levels in cerebrospinal fluid can have a decisive role in the differential diagnosis of intracranial germ cell tumors. J. Neurosurg. 2018, 131, 687–694. [Google Scholar] [CrossRef]
- Iwama, S.; Sugimura, Y.; Kiyota, A.; Kato, T.; Enomoto, A.; Suzuki, H.; Iwata, N.; Takeuchi, S.; Nakashima, K.; Takagi, H.; et al. Rabphilin-3A as a Targeted Autoantigen in Lymphocytic Infundibulo-neurohypophysitis. J. Clin. Endocrinol. Metab. 2015, 100, E946–E954. [Google Scholar] [CrossRef]
- Rodriguez-Galindo, C.; Allen, C.E. Langerhans cell histiocytosis. Blood 2020, 135, 1319–1331. [Google Scholar] [CrossRef]
- Cerbone, M.; Visser, J.; Bulwer, C.; Ederies, A.; Vallabhaneni, K.; Ball, S.; Kamaly-Asl, I.; Grossman, A.; Gleeson, H.; Korbonits, M.; et al. Management of children and young people with idiopathic pituitary stalk thickening, central diabetes insipidus, or both: A national clinical practice consensus guideline. Lancet Child Adolesc. Health 2021, 5, 662–676. [Google Scholar] [CrossRef]
- Morimoto, A.; Oh, Y.; Shioda, Y.; Kudo, K.; Imamura, T. Recent advances in Langerhans cell 169 histiocytosis. Pediatric Int. 2014, 56, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Grois, N.; Pötschger, U.; Prosch, H.; Minkov, M.; Arico, M.; Braier, J.; Henter, J.; Janka-Schaub, G.; Ladisch, S.; Ritter, J.; et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatric Blood Cancer 2006, 46, 228–233. [Google Scholar] [CrossRef]
- Catford, S.; Wang, Y.Y.; Wong, R. Pituitary stalk lesions: Systematic review and clinical guidance. Clin. Endocrinol. 2016, 85, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Dhall, G.; Finlay, J.L.; Dunkel, I.J.; Ettinger, L.J.; Kellie, S.J.; Allen, J.C.; Egeler, R.M.; Arceci, R.J. Analysis of outcome for patients with mass lesions of the central nervous system due to Langerhans cell histiocytosis treated with 2-chlorodeoxyadenosine. Pediatric Blood Cancer 2008, 50, 72–79. [Google Scholar] [CrossRef]
- Okamoto, M.; Yamaguchi, S.; Ishi, Y.; Motegi, H.; Mori, T.; Hashimoto, T.; Terashita, Y.; Hirabayashi, S.; Sugiyama, M.; Iguchi, A.; et al. Diagnostic Capability of Cerebrospinal Fluid-Placental Alkaline Phosphatase Value in Intracranial Germ Cell Tumor. Oncology 2021, 99, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Morota, K.; Tadokoro, H.; Sawano, K.; Watanabe, K.; Iwata, N.; Fujisawa, H.; Suzuki, A.; Sugimura, Y.; Nagasaki, K. A 7-year-old boy with central diabetes insipidus presenting with thickened pituitary stalk and anti-rabphilin-3A antibody positivity. J. Pediatric Endocrinol. Metab. 2022, 35, 687–690. [Google Scholar] [CrossRef]
- Jessop, S.; Crudgington, D.; London, K.; Kellie, S.; Howman-Giles, R. FDG PET-CT in pediatric Langerhans cell histiocytosis. Pediatric Blood Cancer 2020, 67, e28034. [Google Scholar] [CrossRef]
- Marchand, I.; Barkaoui, M.A.; Garel, C.; Polak, M.; Donadieu, J. Central diabetes insipidus as the inaugural manifestation of Langerhans cell histiocytosis: Natural history and medical evaluation of 26 children and adolescents. J. Clin. Endocrinol. Metab. 2011, 96, e1352–e1360. [Google Scholar] [CrossRef]
- Isoo, A.; Ueki, K.; Ishida, T.; Yoshikawa, T.; Fujimaki, T.; Suzuki, I.; Sasaki, T.; Kirino, T. Langerhans cell histiocytosis limited to the pituitary-hypothalamic axis--two case reports. Neurol. Med. Chir. 2000, 40, 532–535. [Google Scholar] [CrossRef][Green Version]
- Nagasaki, K.; Tsumanuma, I.; Yoneoka, Y.; Ogawa, Y.; Kikuchi, T.; Uchiyama, M. Spontaneous regression of isolated neurohypophyseal langerhans cell histiocytosis with diabetes insipidus. Endocr. J. 2009, 56, 721–725. [Google Scholar] [CrossRef]
- Horn, E.M.; Coons, S.W.; Spetzler, R.F.; Rekate, H.L. Isolated Langerhans cell histiocytosis of the infundibulum presenting with fulminant diabetes insipidus. Child’s Nerv. Syst. 2006, 22, 542–544. [Google Scholar] [CrossRef] [PubMed]
- Kurtulmus, N.; Mert, M.; Tanakol, R.; Yarman, S. The pituitary gland in patients with Langerhans cell histiocytosis: A clinical and radiological evaluation. Endocrine 2015, 48, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Nellan, A.; Bodlak, A.; Mirsky, D.M.; Levy, J.M.; Garrington, T.P.; Foreman, N.K.; Gilani, A.; Hayashi, M. ddPCR Analysis Reveals BRAF V600E Mutations Are Infrequent in Isolated Pituitary Langerhans Cell Histiocytosis Patients. J. Neuropathol. Exp. Neurol. 2020, 79, 1313–1319. [Google Scholar] [CrossRef]
- Haupt, R.; Minkov, M.; Astigarraga, I.; Schäfer, E.; Nanduri, V.; Jubran, R.; Egeler, R.M.; Janka, G.; Micic, D.; Rodriguez-Galindo, C.; et al. Langerhans cell histiocytosis (LCH): Guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatric Blood Cancer 2013, 60, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Héritier, S.; Emile, J.F.; Barkaoui, M.A.; Thomas, C.; Fraitag, S.; Boudjemaa, S.; Renaud, F.; Moreau, A.; Peuchmaur, M.; Chassagne-Clément, C.; et al. BRAF Mutation Correlates With High-Risk Langerhans Cell Histiocytosis and Increased Resistance to First-Line Therapy. J. Clin. Oncol. 2016, 34, 3023–3030. [Google Scholar] [CrossRef]
- Nann, D.; Schneckenburger, P.; Steinhilber, J.; Metzler, G.; Beschorner, R.; Schwarze, C.P.; Lang, P.; Handgretinger, R.; Fend, F.; Ebinger, M.; et al. Pediatric Langerhans cell histiocytosis: The impact of mutational profile on clinical progression and late sequelae. Ann. Hematol. 2019, 98, 1617–1626. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Loading Agents | Time | 0 | 15 | 30 | 60 | 90 | 120 | Assessment c | |
|---|---|---|---|---|---|---|---|---|---|
| Hormones | |||||||||
| Arginine | GH a (ng/mL) | 1.32 | - | 3.91 | 6.18 | 3.04 | 1.50 | Normal | |
| Insulin | Glucose b (mg/dL) | 55 | 29 | 35 | 43 | 45 | 69 | ||
| Insulin | GH b (ng/mL) | 0.2 | 0.2 | 0.2 | 0.9 | 0.8 | 1.7 | Low | |
| Insulin | ACTH b (pg/mL) | 18.2 | 20.6 | 31.2 | 29.8 | 24.2 | 32.8 | ||
| Insulin | Cortisol b (µg/dL) | 7.1 | 10.2 | 13.1 | 15.6 | 16 | 14.9 | Low | |
| TRH | TSH b (µIU/mL) | 2.6 | 11.1 | 15.5 | 15.6 | 15.3 | 14.6 | Delayed | |
| TRH | PRL b (ng/mL) | 28.1 | 57.6 | 72 | 57.1 | 63.3 | 45.2 | Excessive | |
| LHRH | LH b (mIU/mL) | <0.2 | <0.2 | 0.2 | <0.2 | <0.2 | <0.2 | Low | |
| LHRH | FSH b (mIU/mL) | 1 | 3.1 | 5 | 5.4 | 5.3 | 4.9 | Normal | |
| Author (Year) | Age (Years)/Sex | DI before Treatment | APD before Treatment | Sites of Involvement | Treatment | Prognosis | References |
|---|---|---|---|---|---|---|---|
| Marchand, I. et al., (2011) | 13/male | + | - | PS | Mass resection | Shrinkage | [19] |
| 16/female | + | + | PS | VBL, steroid/ Rx/2-CdA | Progression followed by regression | ||
| 8/male | + | + | PS | VBL | Shrinkage | ||
| 12/female | + | + | PS | VBL, steroid | Shrinkage | ||
| Isoo, A. et al., (2000) | 14/female | + | - | PS | Rx 20 Gy | Shrinkage | [20] |
| 9/female | + | n/A | PS, H | Rx 20 Gy | Shrinkage | ||
| Nagasaki, K. et al., (2009) | 13/female | + | - | PS | No Tx | Spontaneous shrinkage | [21] |
| Horn, E.M. et al., (2006) | 18/male | + | + | PS | Rx 21 Gy | n/A | [22] |
| Kurtulmus, N. et al., (2015) | 16/male | + | + | PS | Rx | Shrinkage | [23] |
| Huo, Z. et al., (2016) | 9/female | + | + | PS | Rx | Shrinkage | [3] |
| 12/female | + | + | PS, H | No Tx | Spontaneous shrinkage | ||
| 13/male | + | + | PS, H | Rx | Shrinkage | ||
| 15/male | + | - | PS, p | No Tx | No change | ||
| Zhou, W. et al., (2019) | 17/male | + | - | PS | No Tx | No recurrence | [4] |
| Nellan, A. et al., (2020) | 7/male | + | n/A | PS | VBL, prednisone | No recurrence | [24] |
| 4/female | + | n/A | PS | VBL, prednisone | No recurrence | ||
| 9/male | + | n/A | PS | VBL, prednisone | Recurrence | ||
| 10/male | + | n/A | PS | VBL, prednisone | No recurrence | ||
| 3/male | + | n/A | PS | VBL, prednisone | No recurrence | ||
| 2/female | + | n/A | PS | VBL, prednisone | No recurrence | ||
| 7/female | + | n/A | PS | VBL, prednisone | No recurrence | ||
| 8/female | + | n/A | PS | Rx | No recurrence | ||
| Present case | 2/female | + | + | PS | 2-CdA | Shrinkage |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tani, M.; Hiroshima, S.; Sato, H.; Sawano, K.; Ogawa, Y.; Imamura, M.; Oishi, M.; Nagasaki, K. Infantile-Onset Isolated Neurohypophyseal Langerhans Cell Histiocytosis with Central Diabetes Insipidus: A Case Report. Children 2022, 9, 716. https://doi.org/10.3390/children9050716
Tani M, Hiroshima S, Sato H, Sawano K, Ogawa Y, Imamura M, Oishi M, Nagasaki K. Infantile-Onset Isolated Neurohypophyseal Langerhans Cell Histiocytosis with Central Diabetes Insipidus: A Case Report. Children. 2022; 9(5):716. https://doi.org/10.3390/children9050716
Chicago/Turabian StyleTani, Mizuki, Shota Hiroshima, Hidetoshi Sato, Kentaro Sawano, Yohei Ogawa, Masaru Imamura, Makoto Oishi, and Keisuke Nagasaki. 2022. "Infantile-Onset Isolated Neurohypophyseal Langerhans Cell Histiocytosis with Central Diabetes Insipidus: A Case Report" Children 9, no. 5: 716. https://doi.org/10.3390/children9050716
APA StyleTani, M., Hiroshima, S., Sato, H., Sawano, K., Ogawa, Y., Imamura, M., Oishi, M., & Nagasaki, K. (2022). Infantile-Onset Isolated Neurohypophyseal Langerhans Cell Histiocytosis with Central Diabetes Insipidus: A Case Report. Children, 9(5), 716. https://doi.org/10.3390/children9050716