Management of Congenital Heart Disease: State of the Art—Part II—Cyanotic Heart Defects


Abstract
:1. Introduction
2. Tetralogy of Fallot
2.1. Conventional TOF
2.1.1. Corrective Surgery
2.1.2. Palliative Procedures
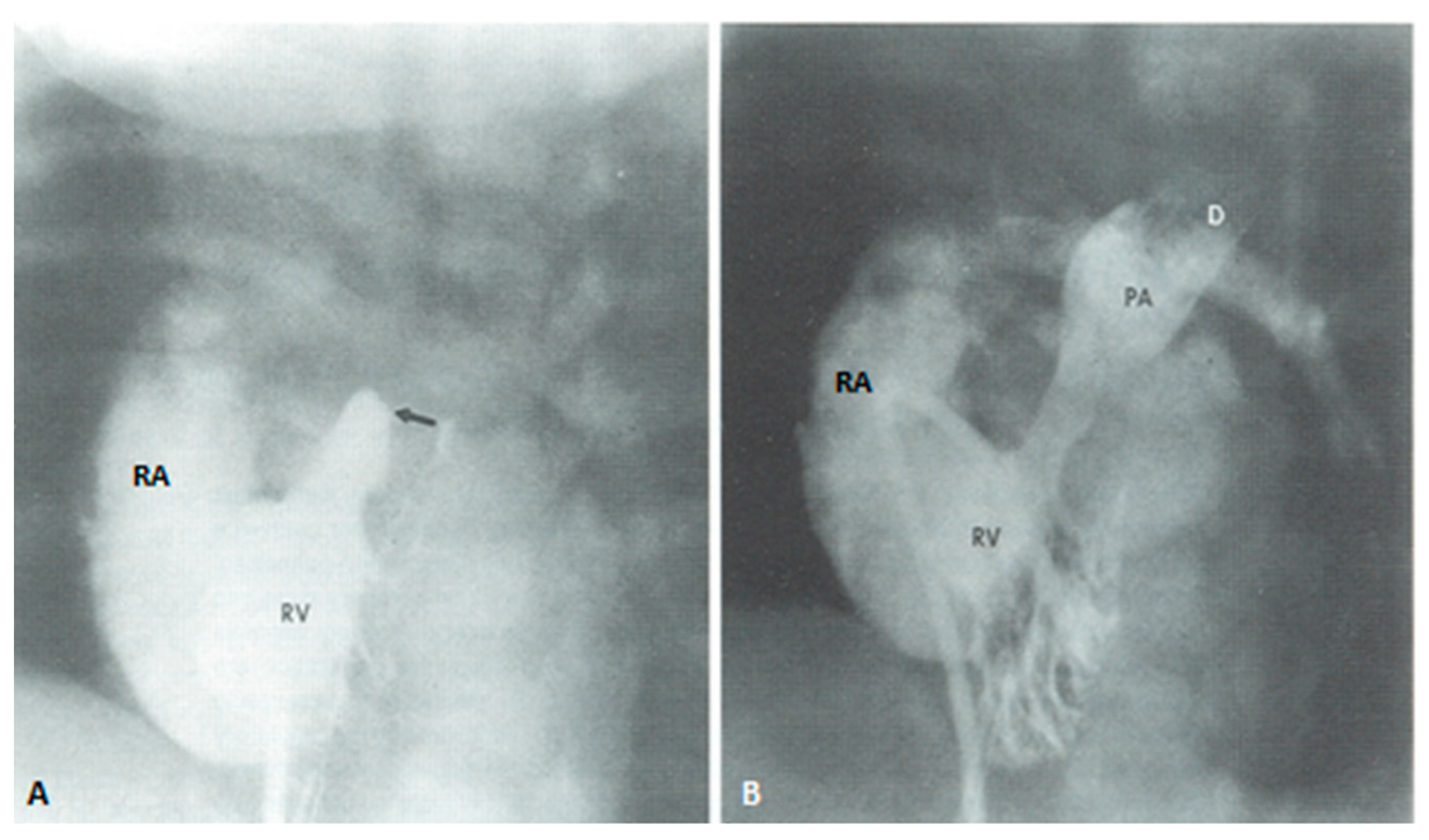
2.1.3. Management of Cyanotic Spells
2.2. TOF with Pulmonary Atresia
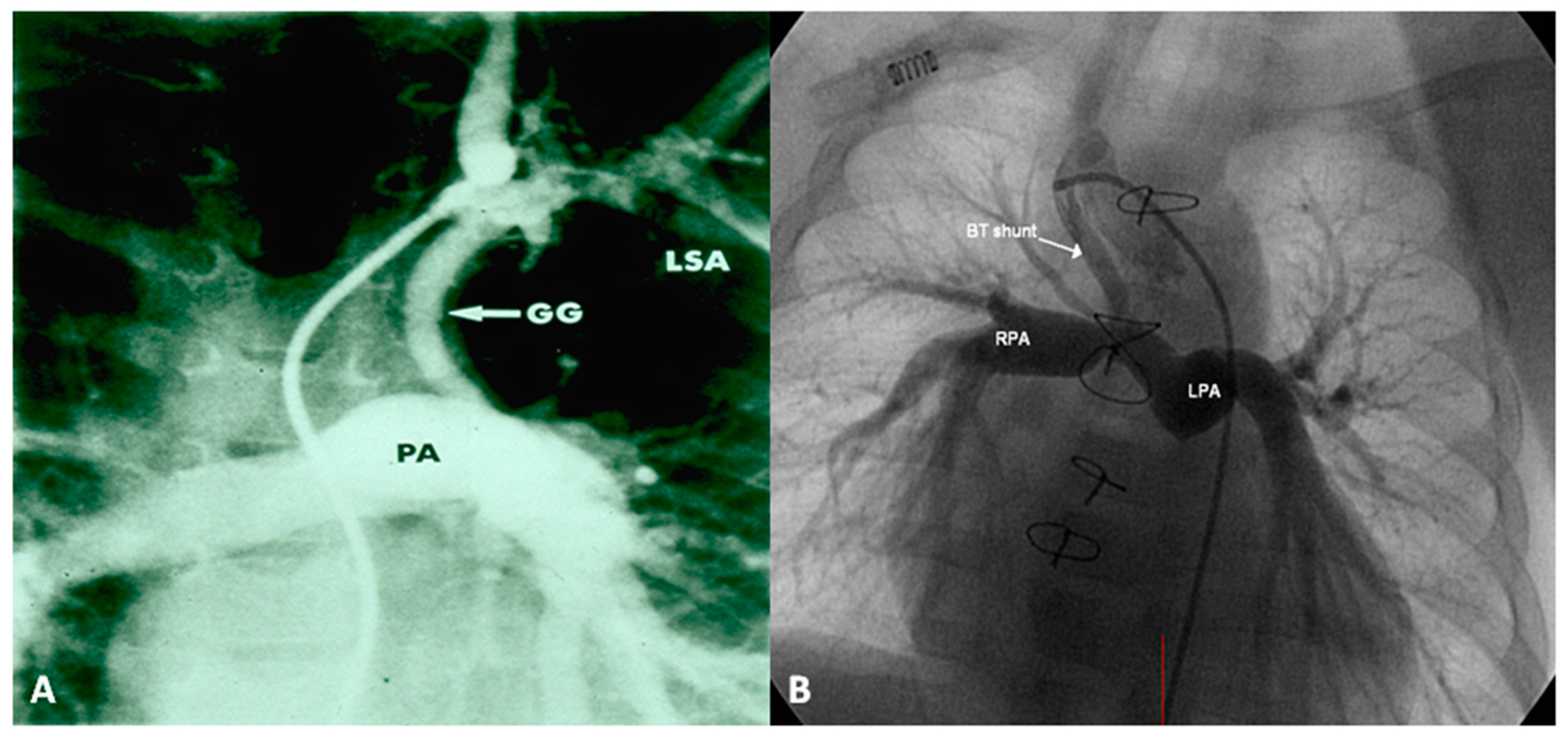
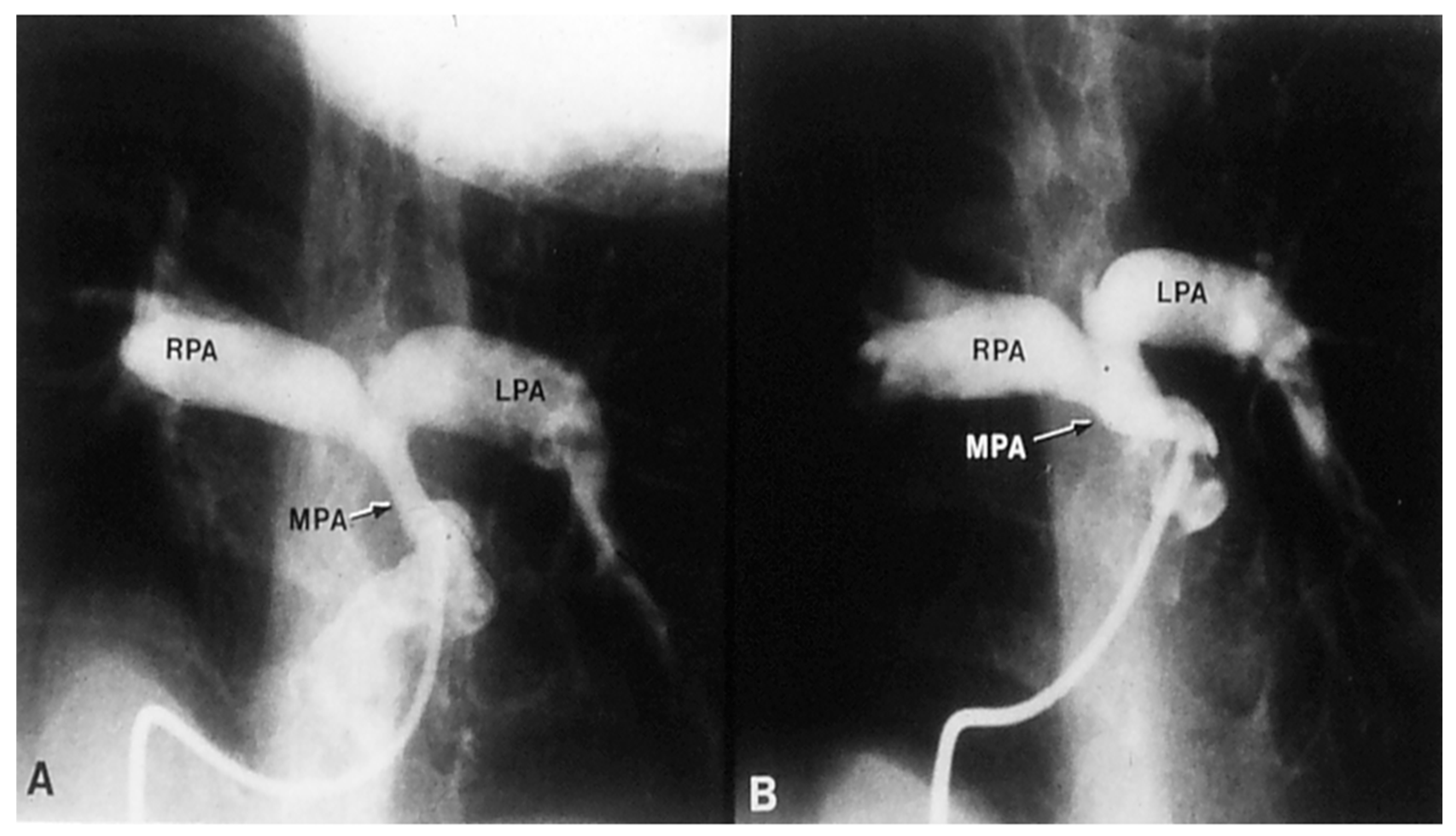
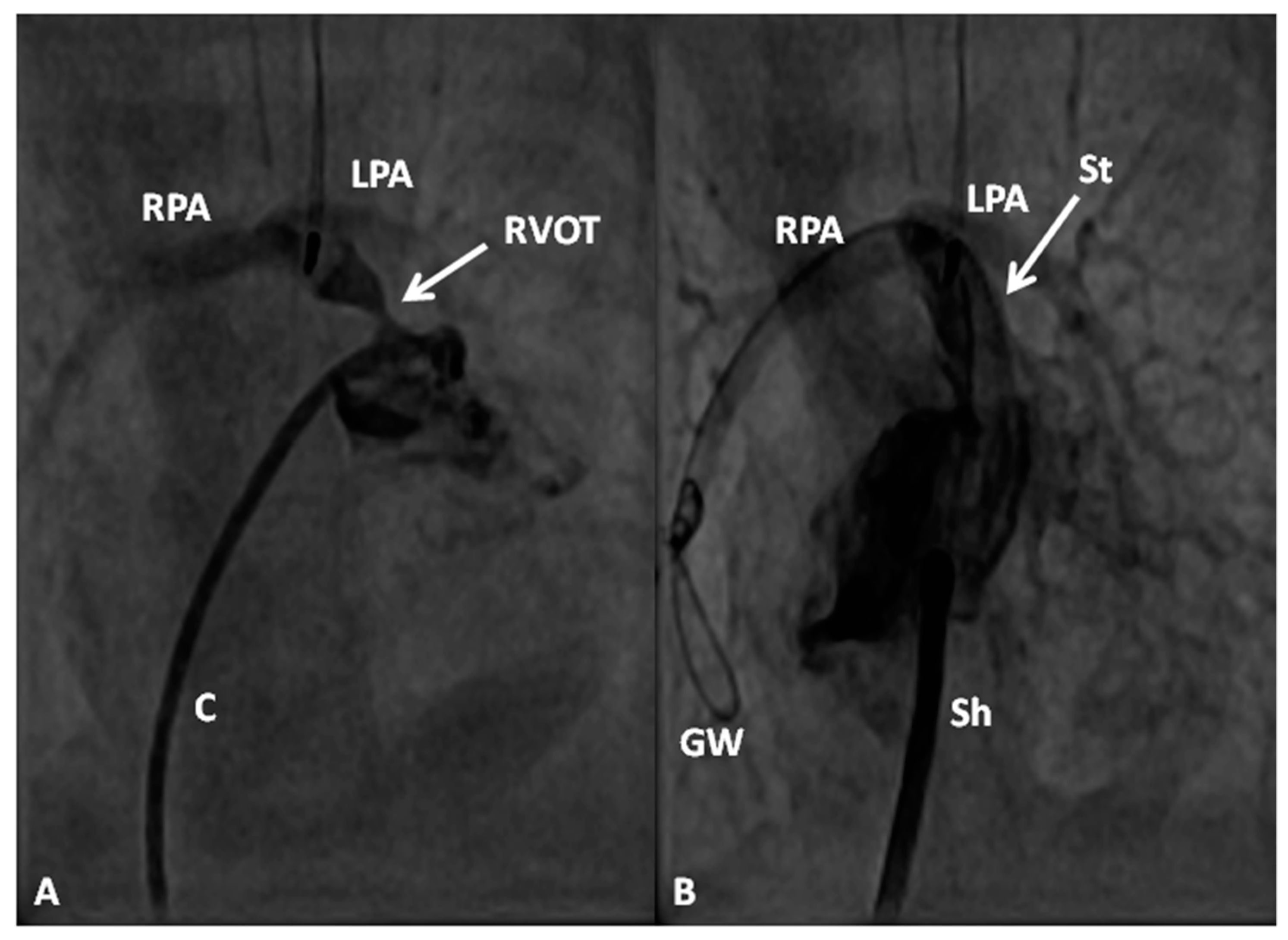
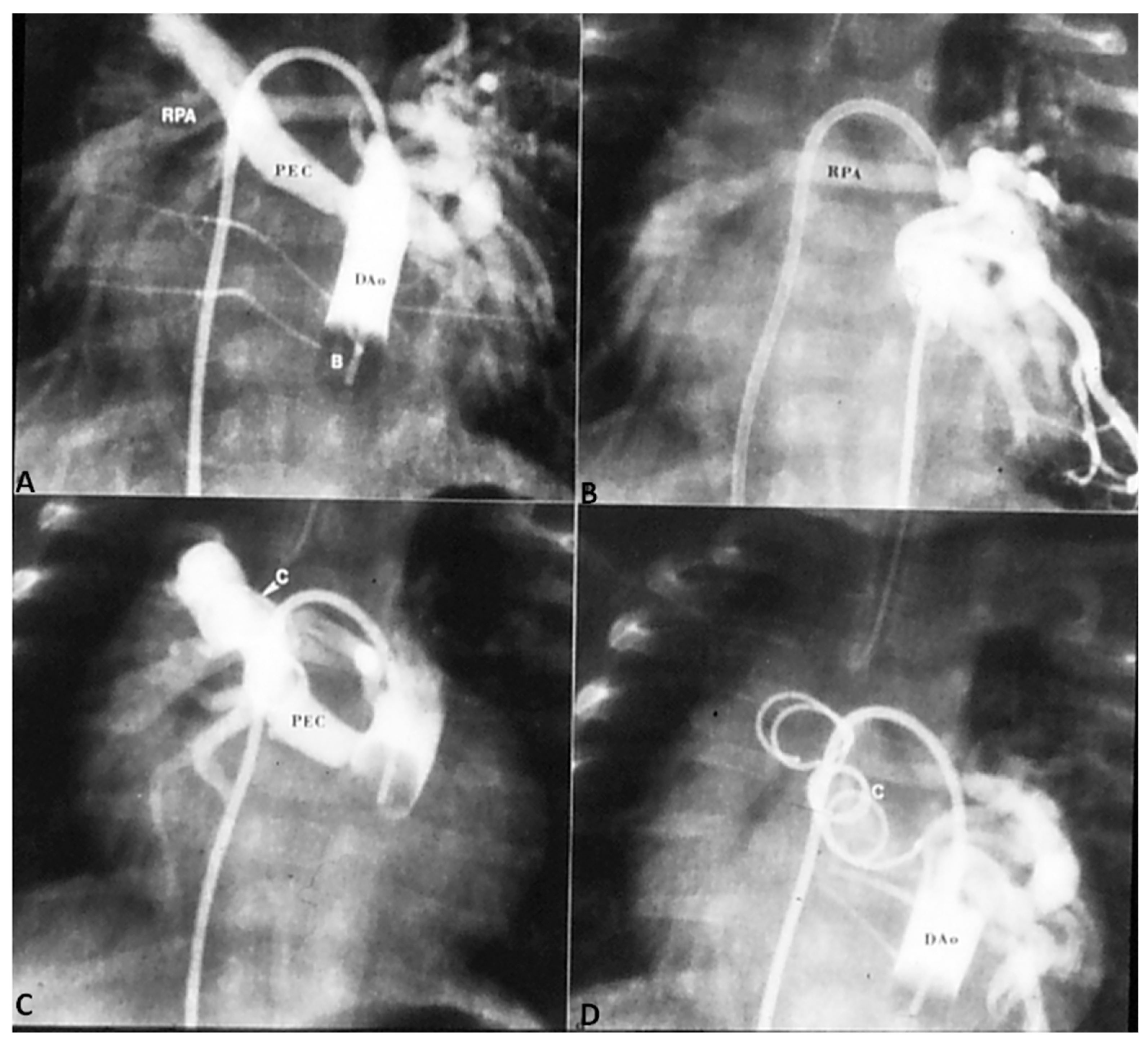
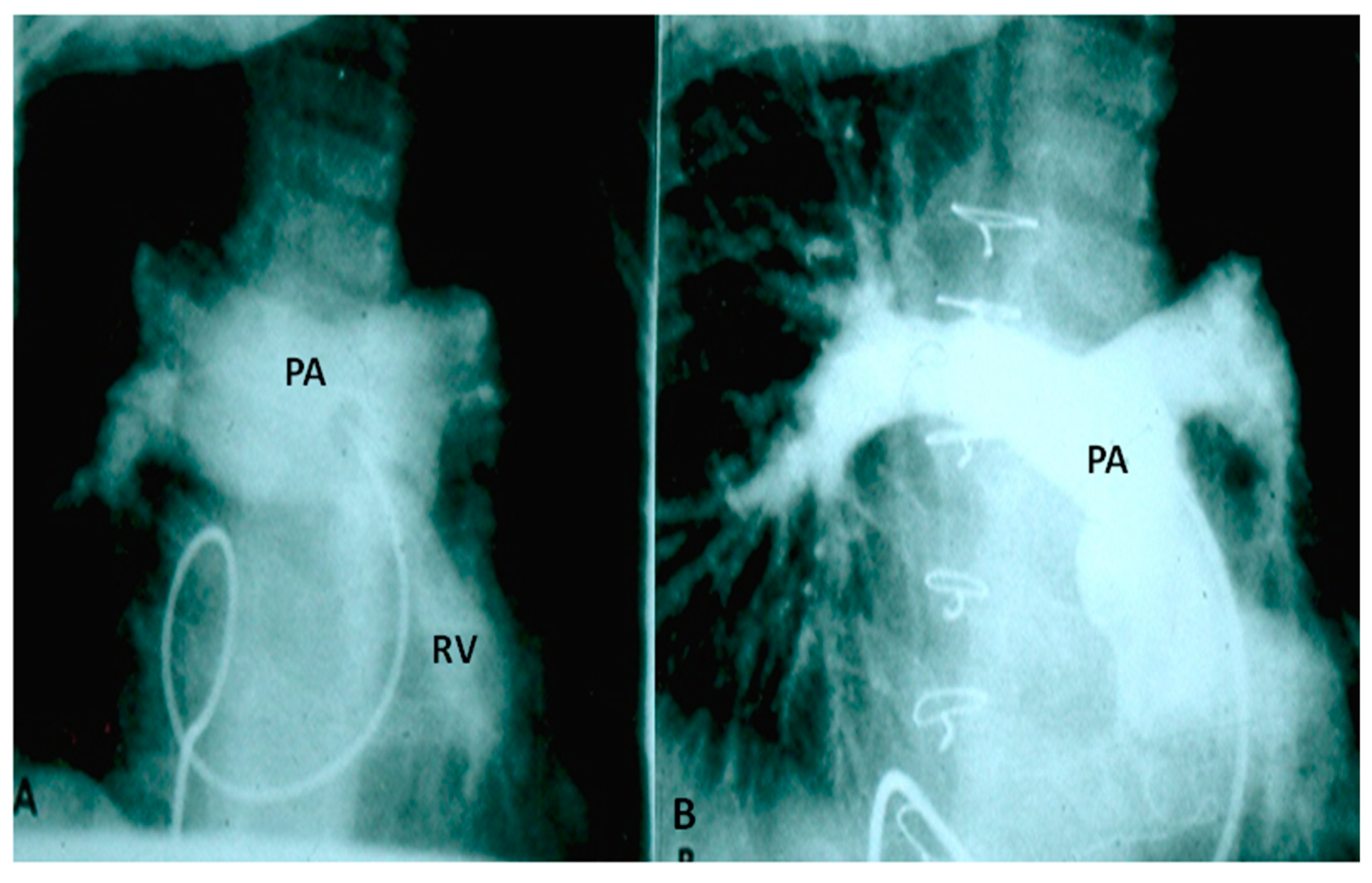
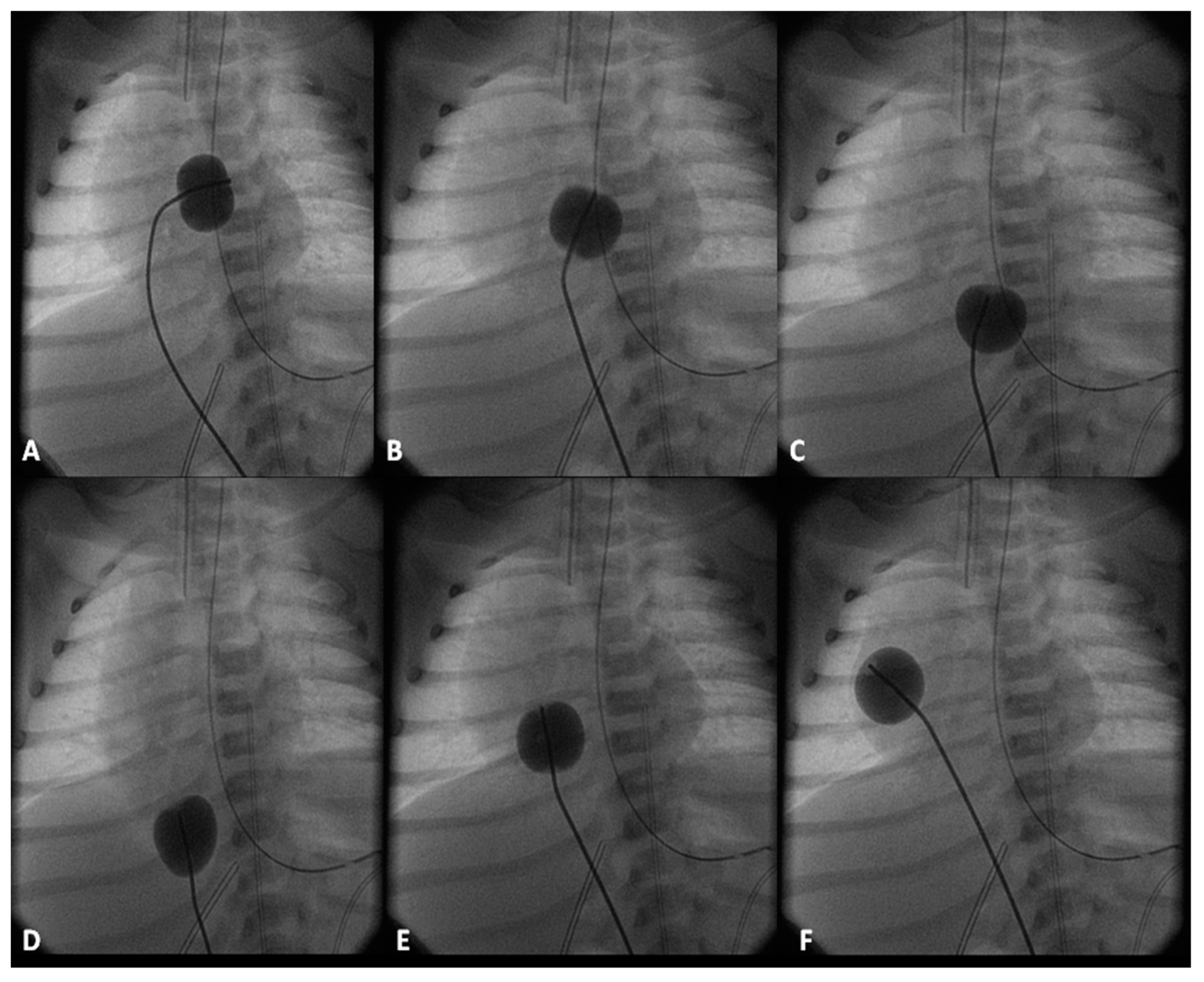
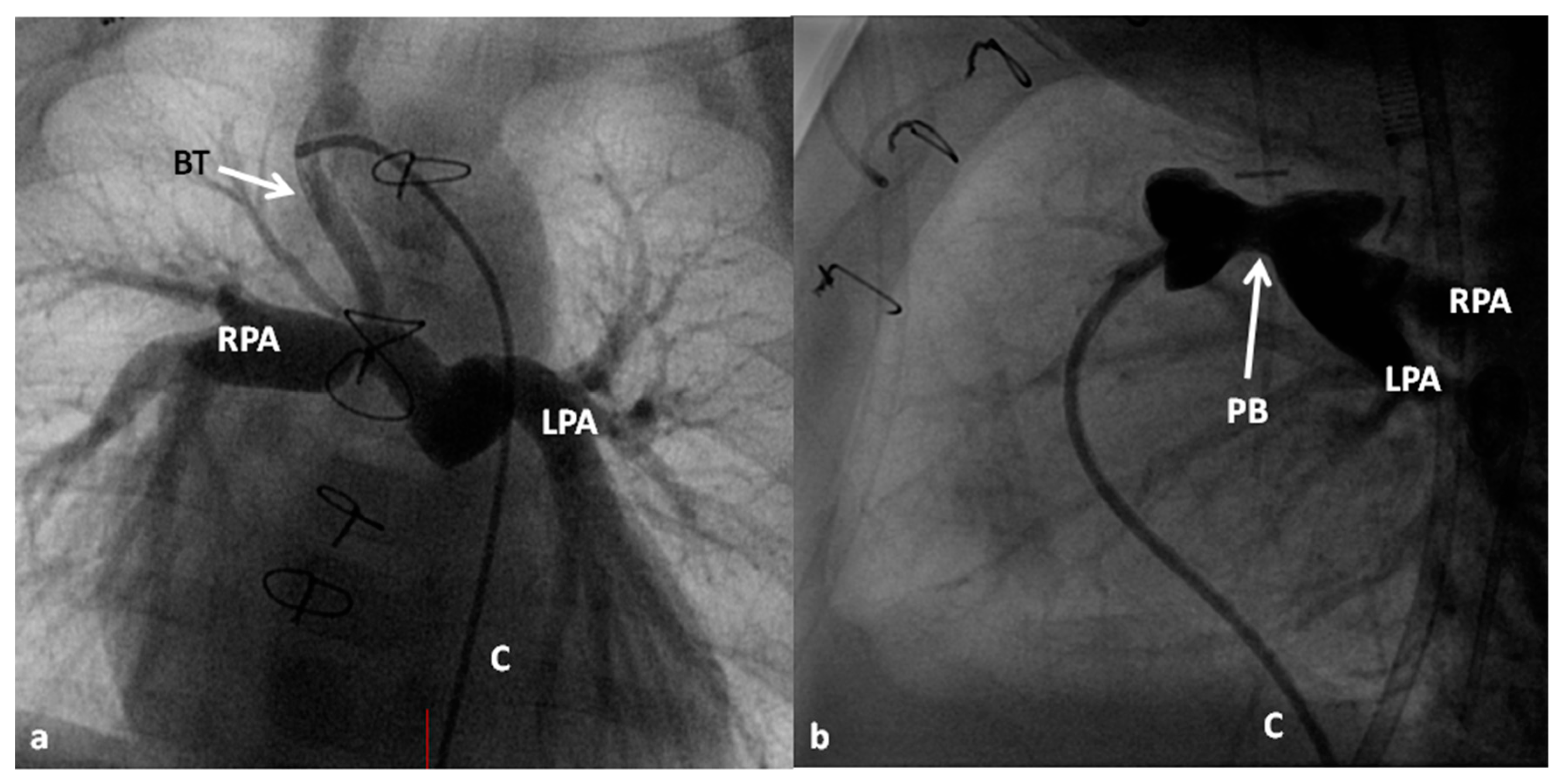
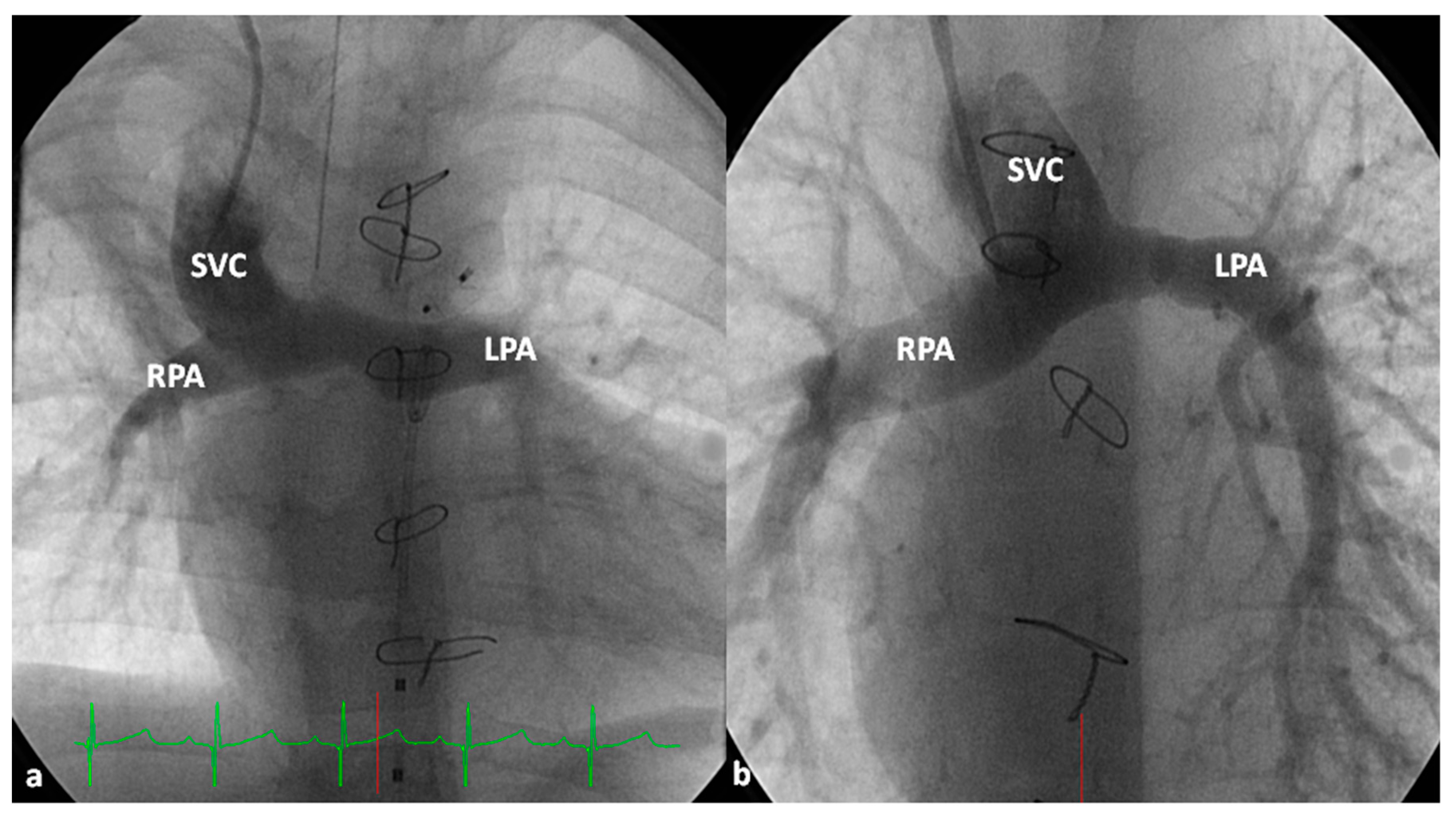
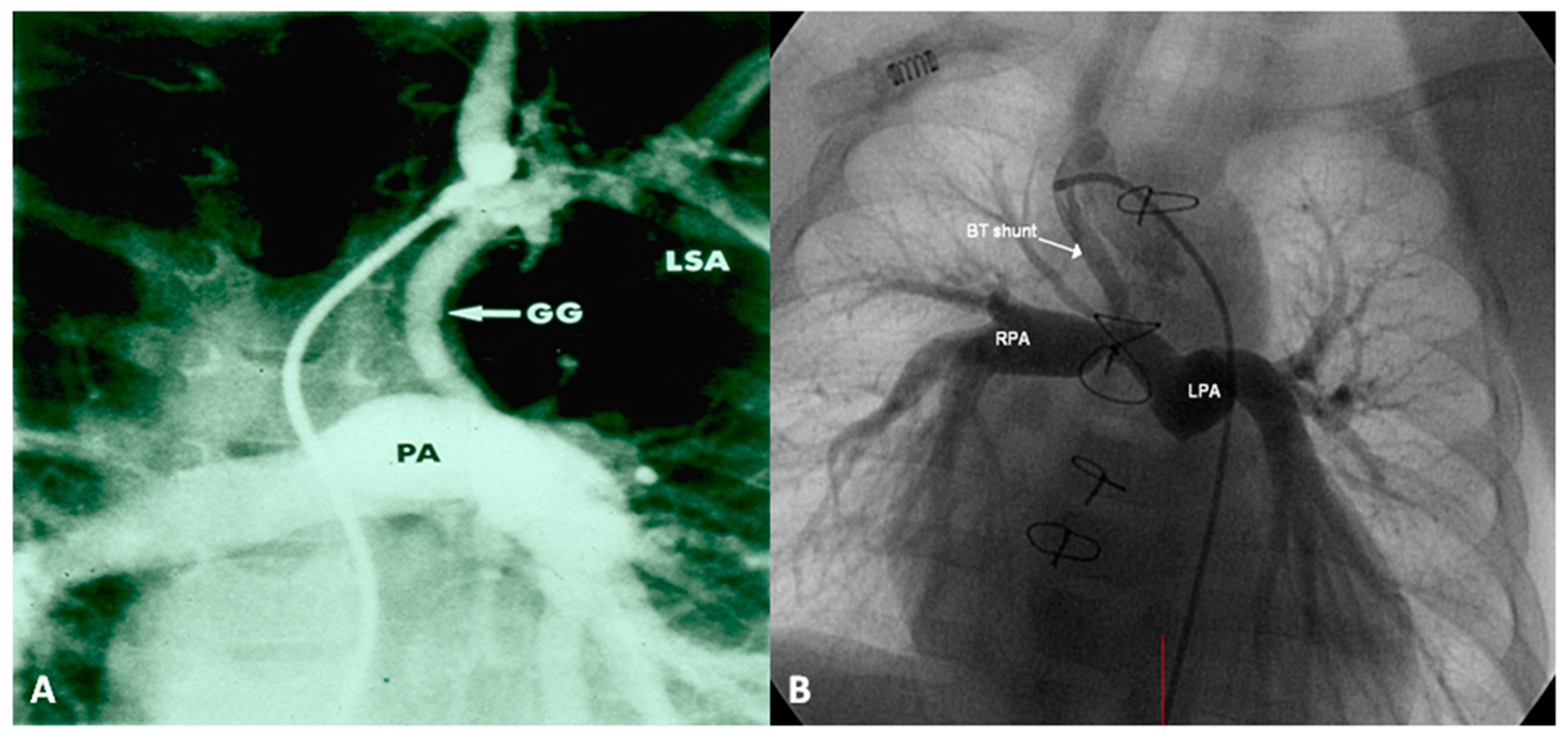
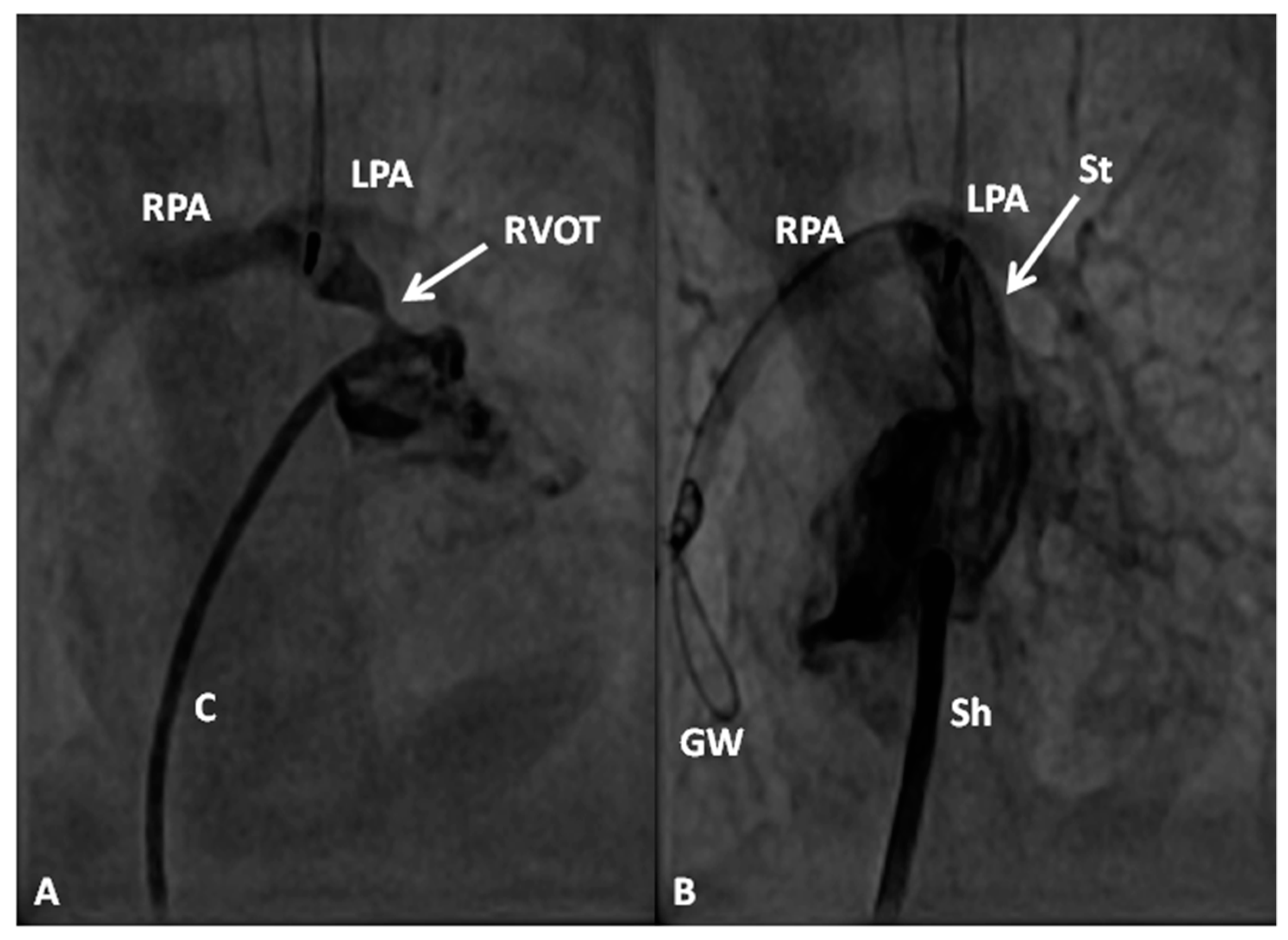
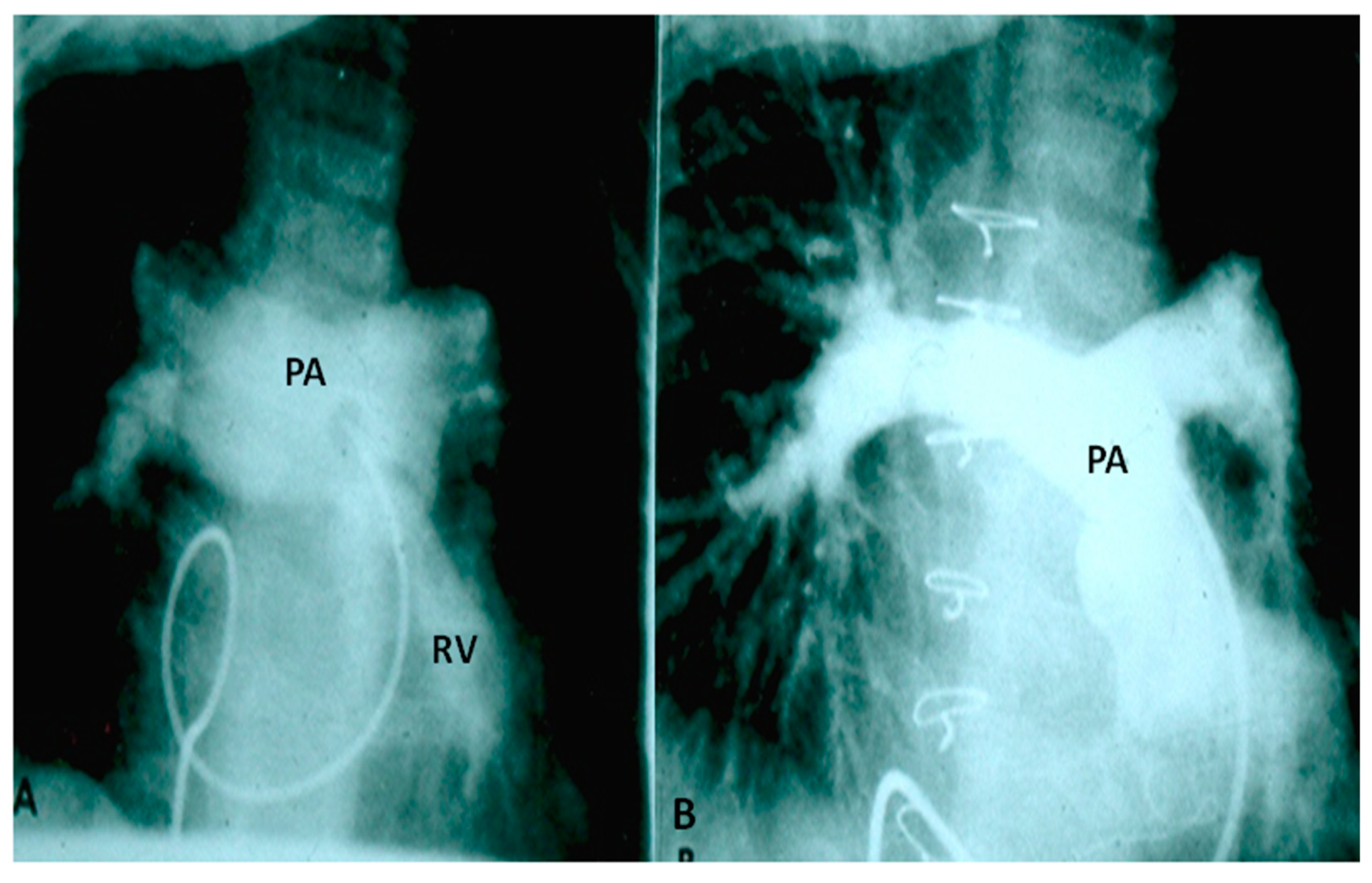
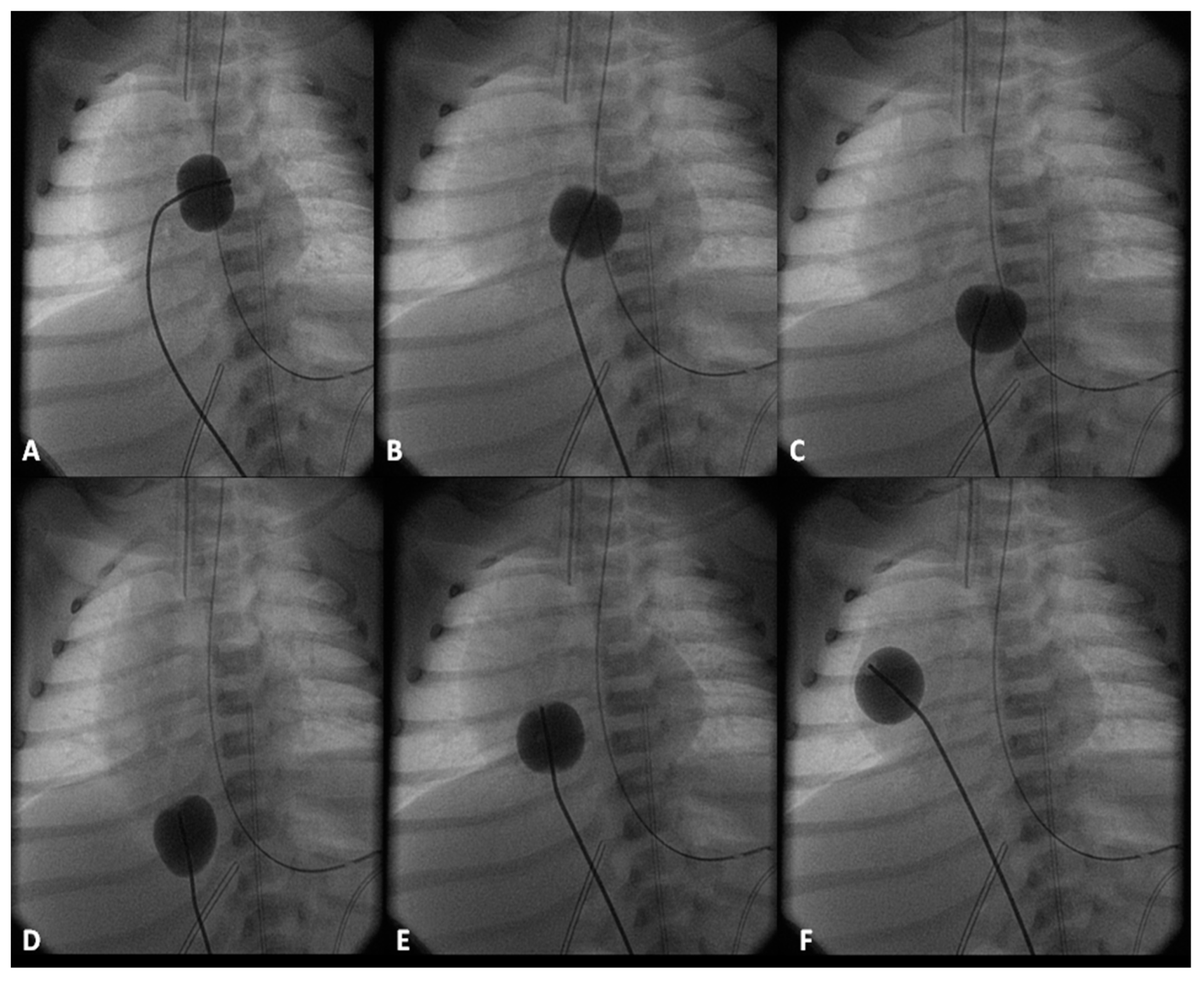
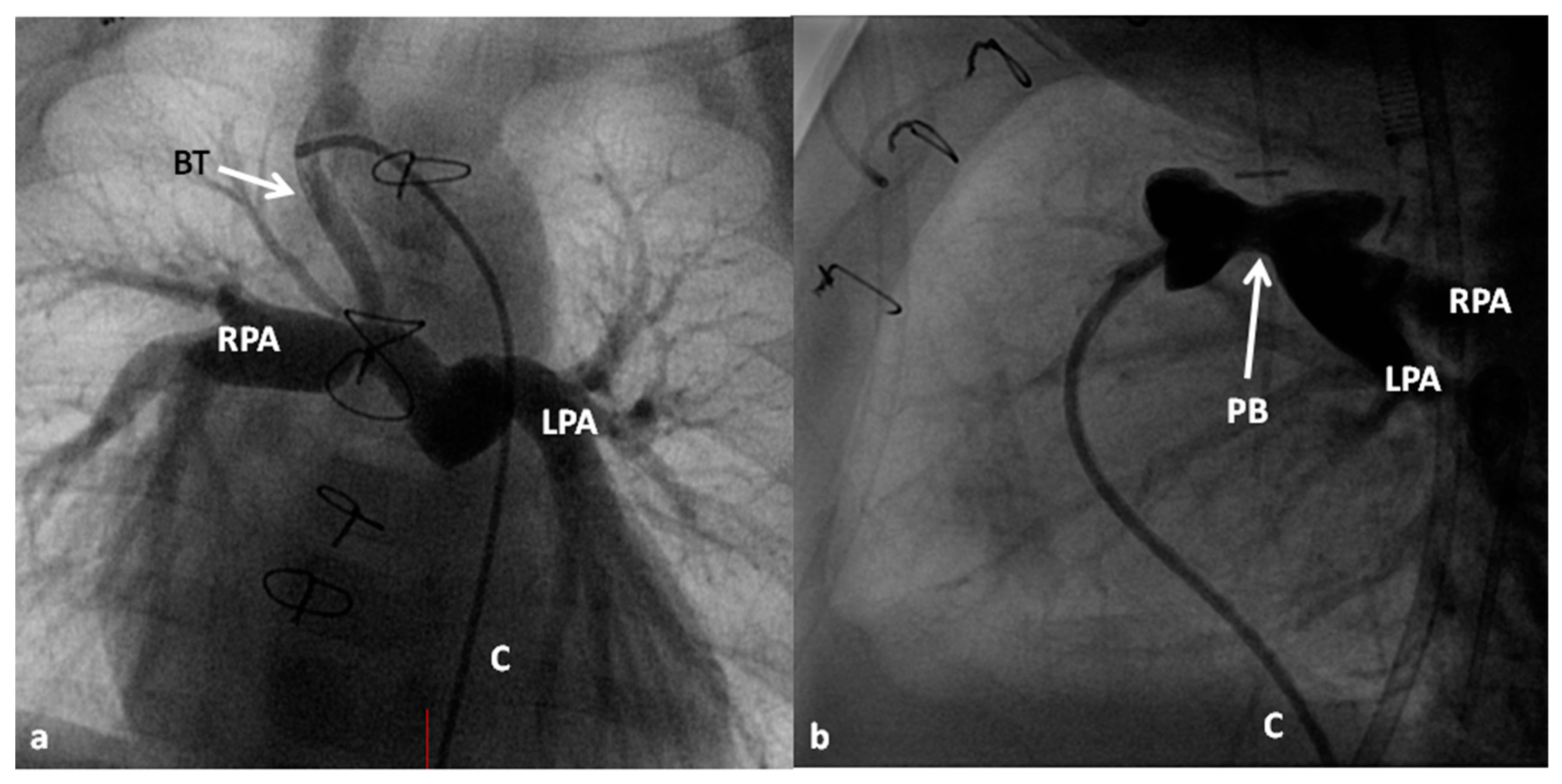
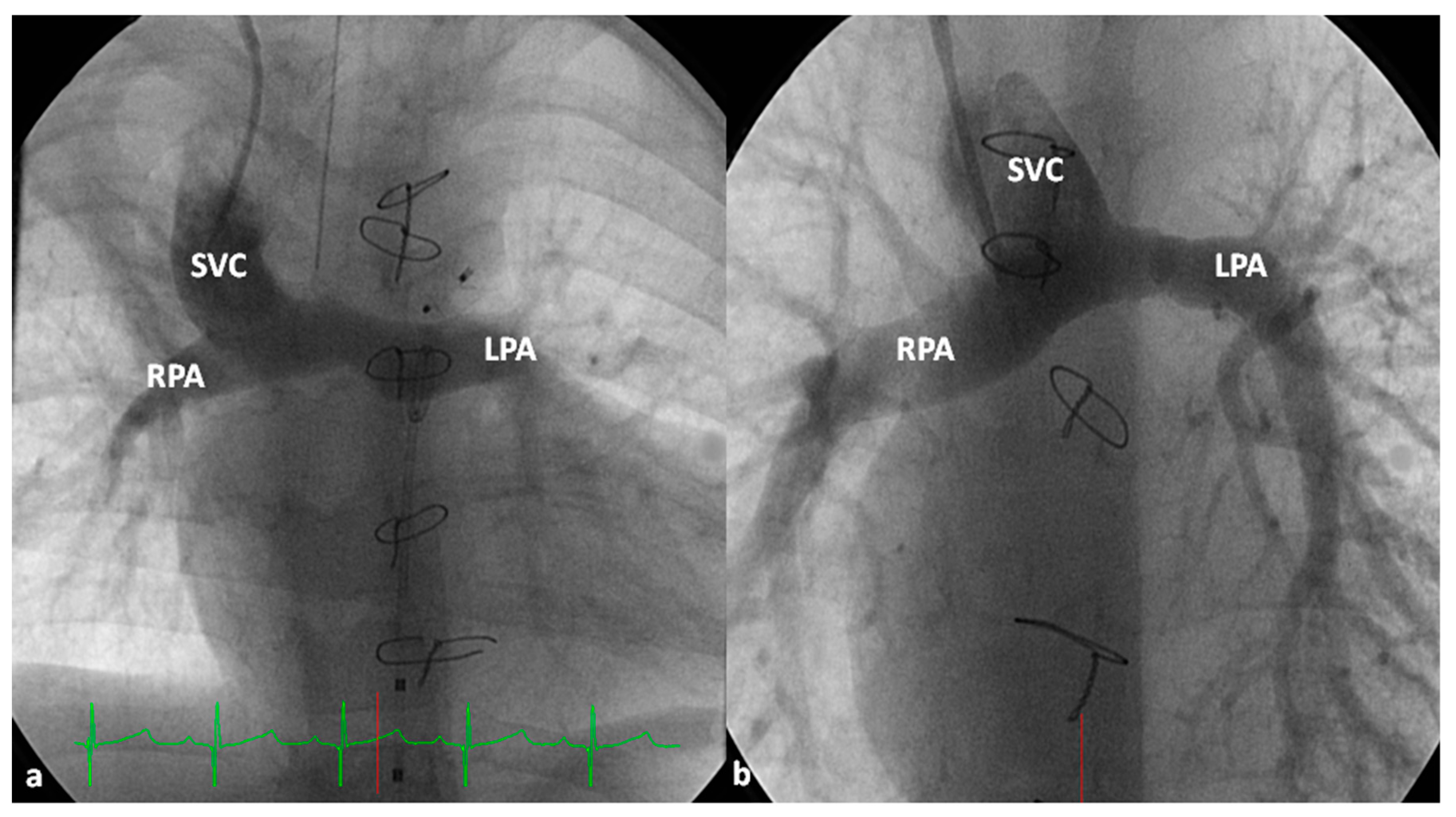
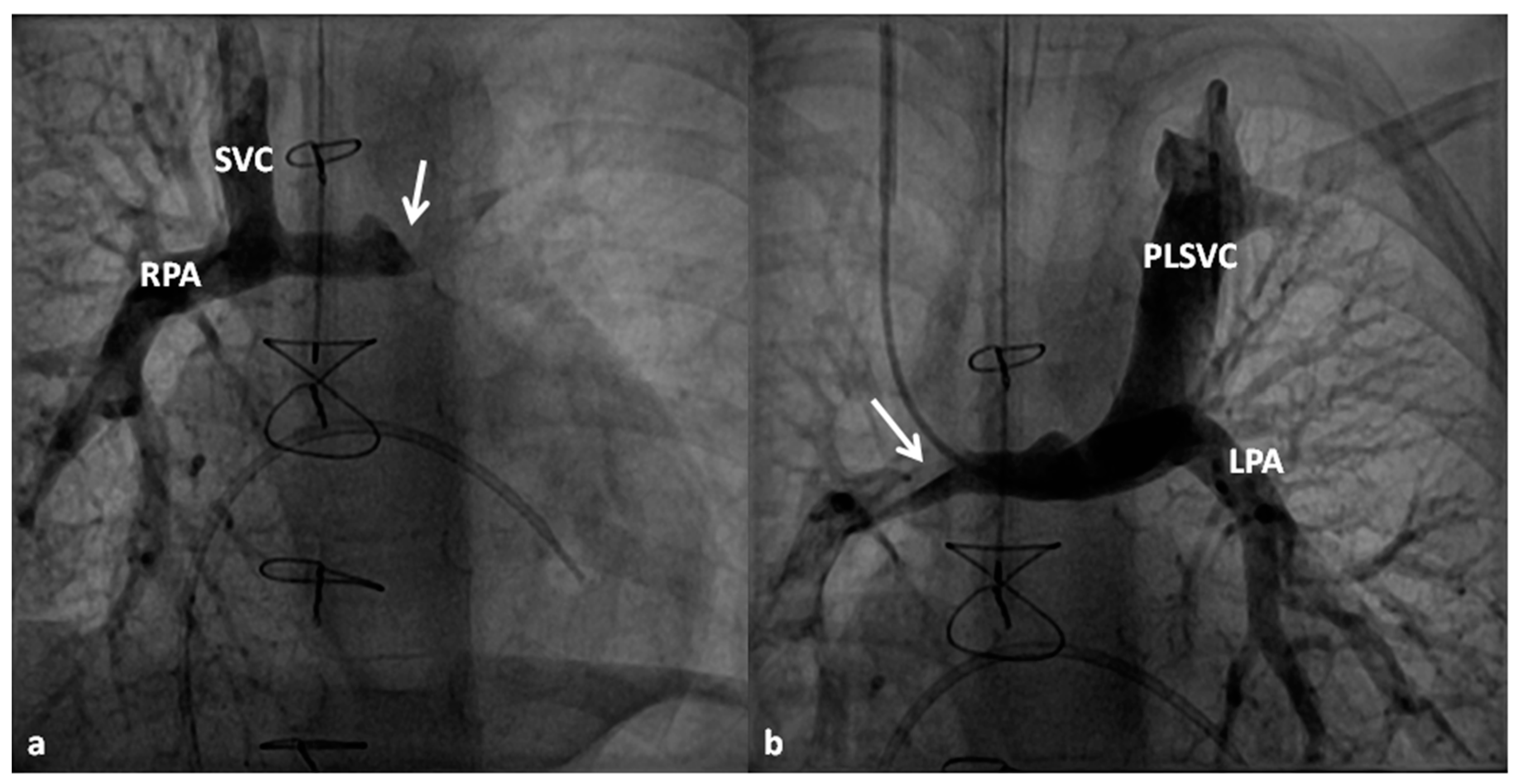
2.2.1. Palliative Procedures
2.2.2. Corrective Surgery
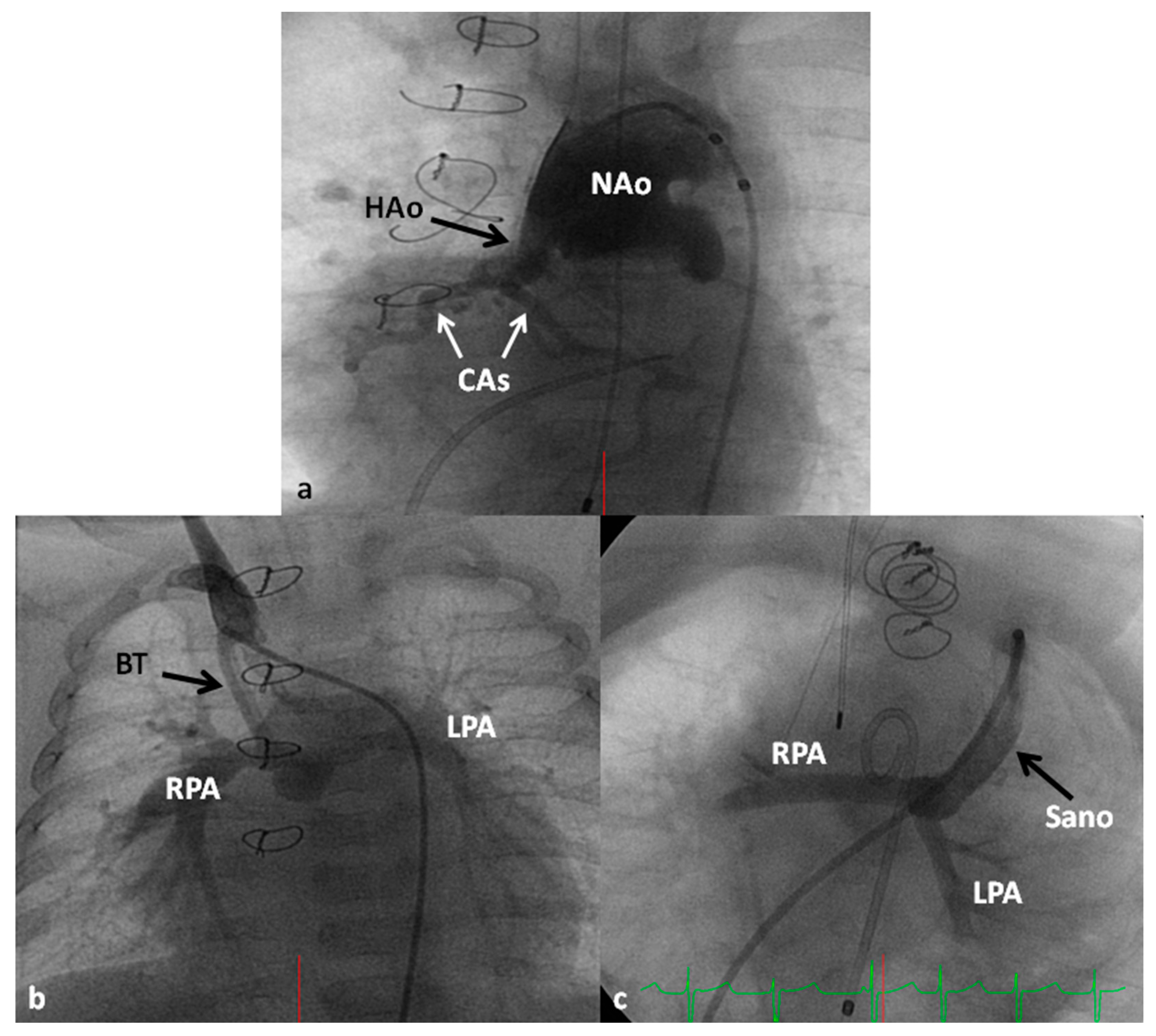
2.3. TOF with MAPCAs
2.4. TOF with Absent Pulmonary Valve Syndrome
3. Transposition of the Great Arteries
3.1. TGA with Intact Ventricular Septum
3.2. TGA with VSD
3.3. TGA with VSD and PS
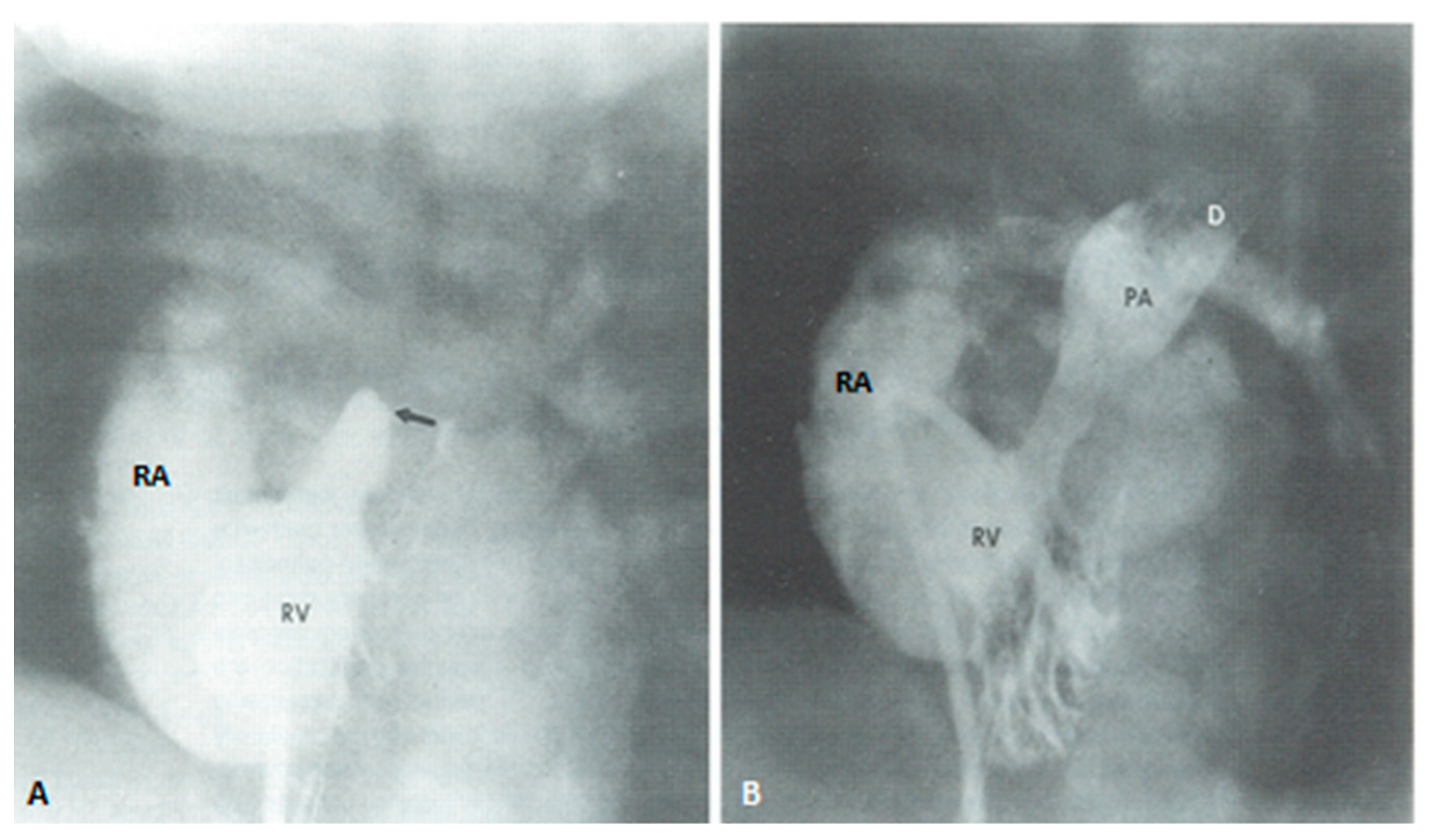
4. Tricuspid Atresia
4.1. Palliative Management
4.1.1. Decreased Pulmonary Blood Flow
4.1.2. Near Normal Pulmonary Blood Flow
4.1.3. Increased Pulmonary Blood Flow
4.1.4. Interatrial Obstruction
4.1.5. Interventricular Obstruction
4.2. “Corrective” Surgery
4.2.1. Stage I
4.2.2. Stage II
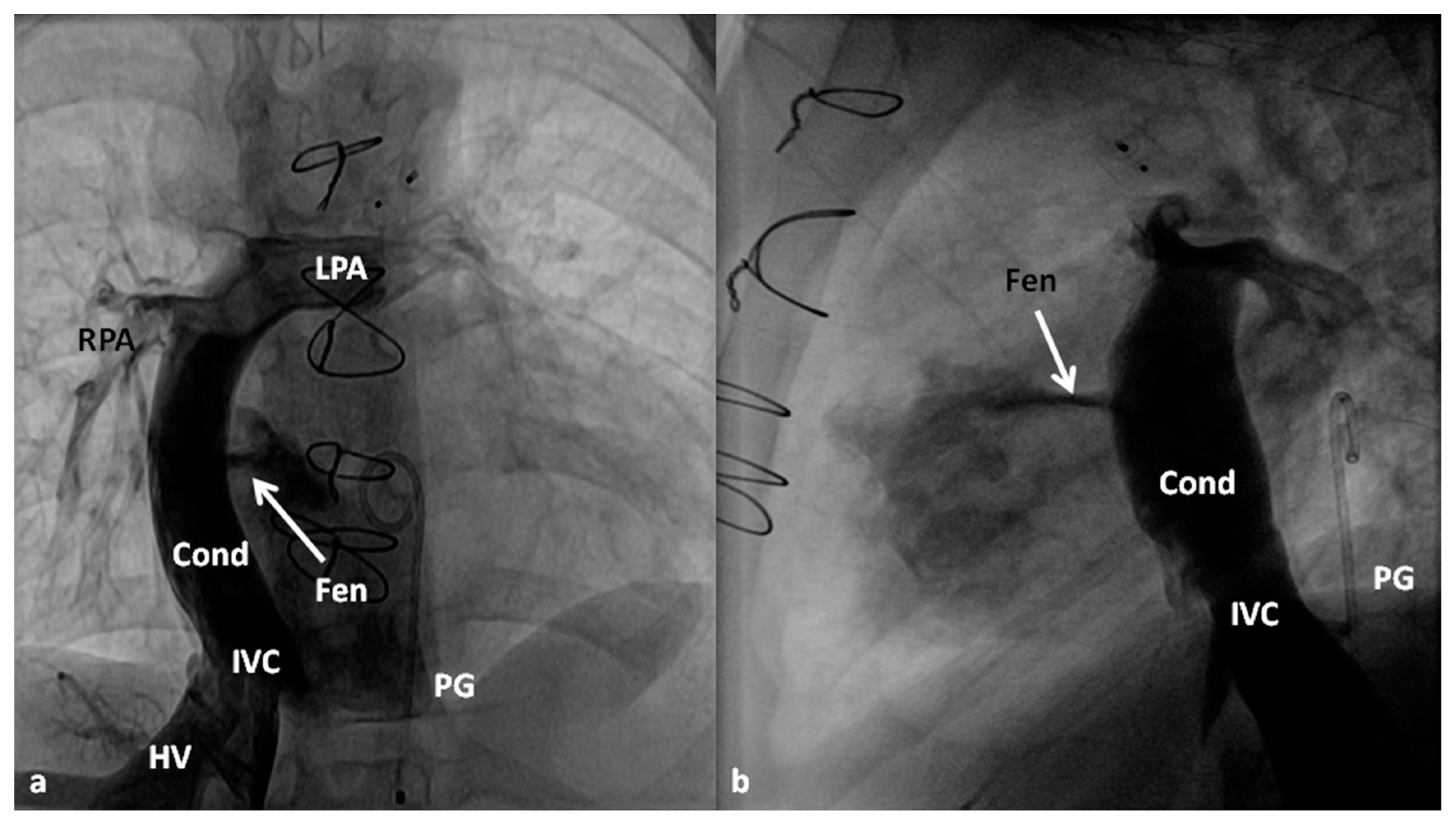
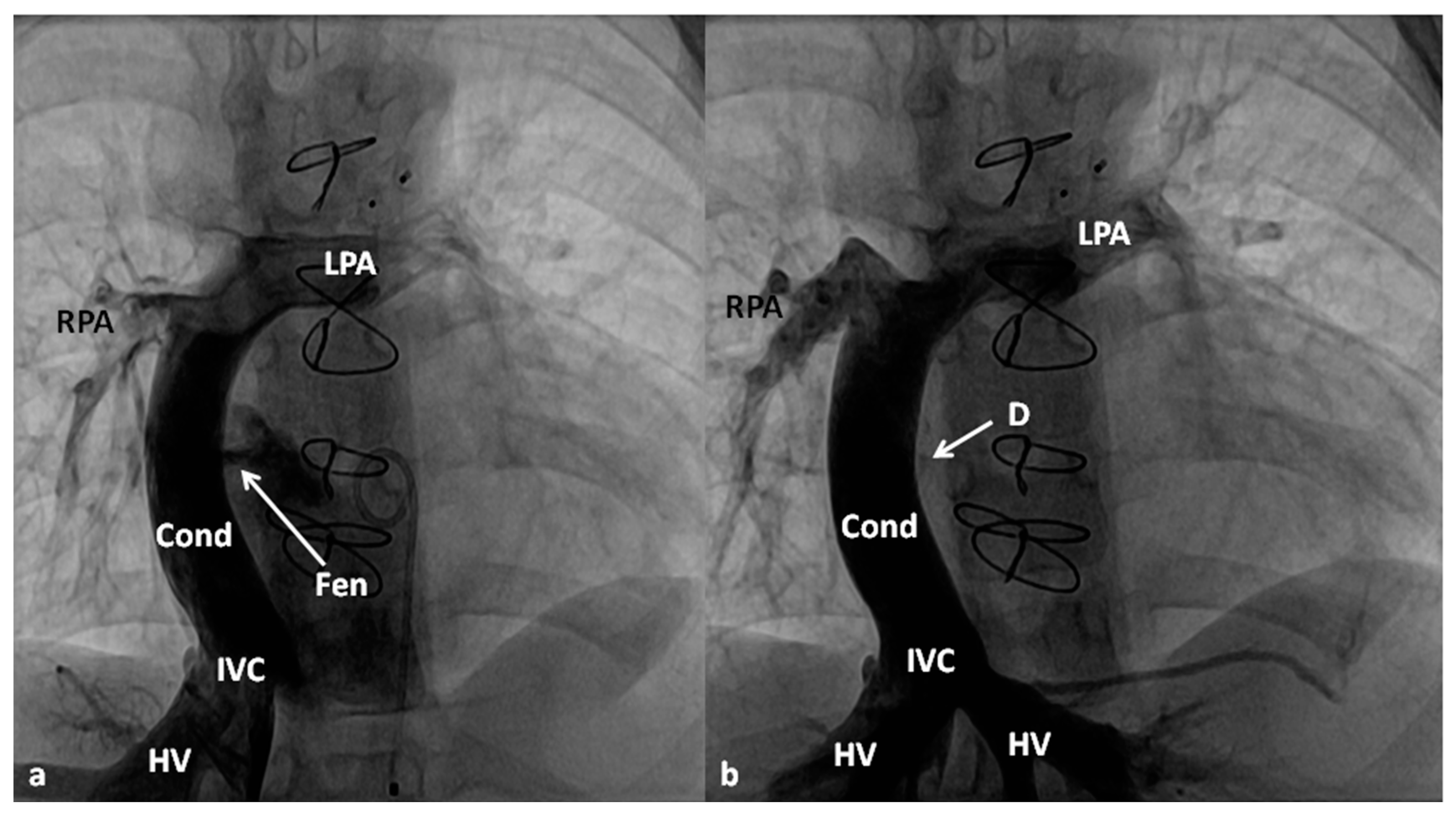
4.2.3. Stage IIIA
4.2.4. Stage IIIB
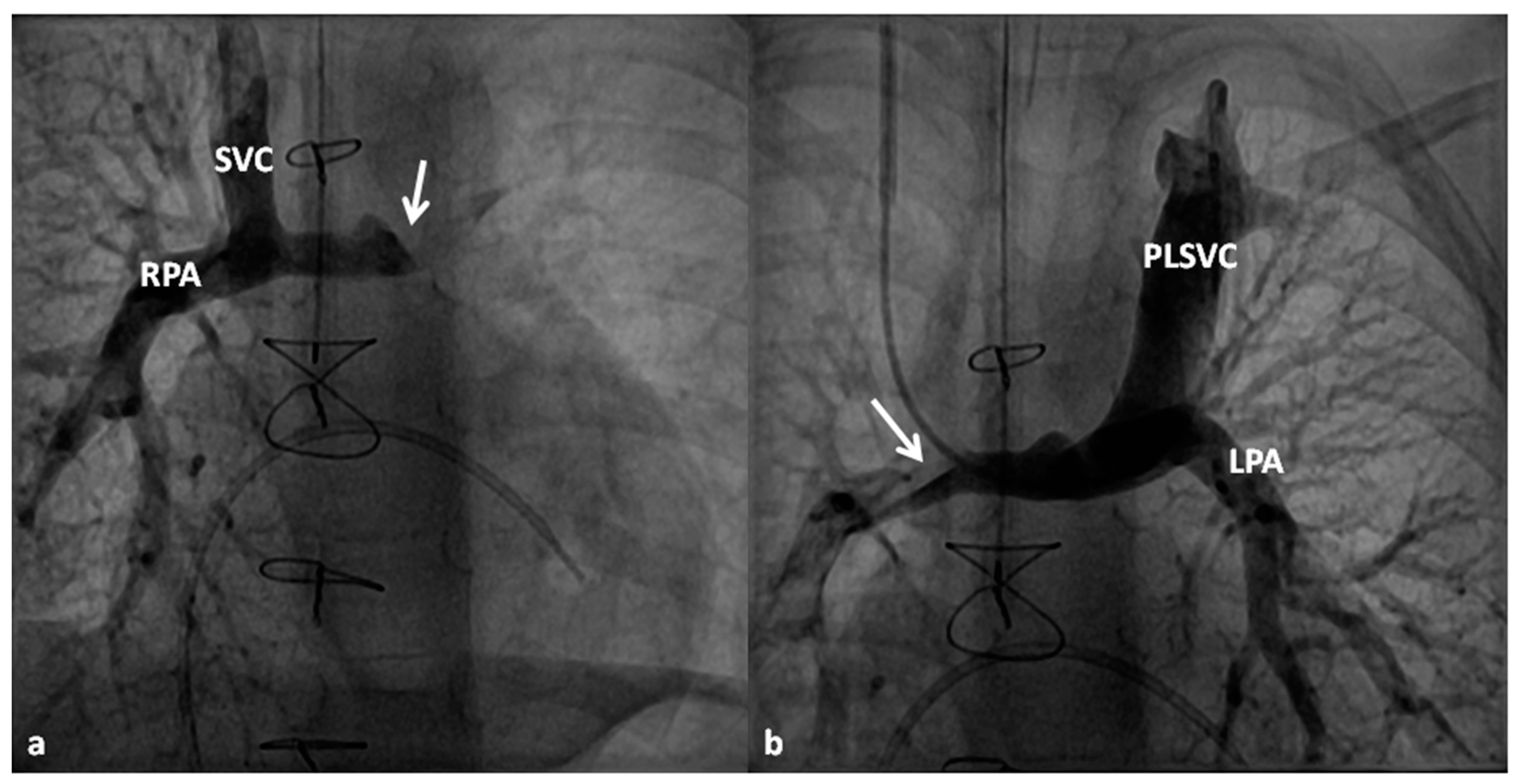
5. Total Anomalous Pulmonary Venous Connection
6. Truncus Arteriosus
7. Hypoplastic Left Heart Syndrome
7.1. Prenatal Identification
7.2. Postnatal Presentation
7.3. Management at Presentation
7.3.1. Maintain Systemic Flow
7.3.2. Limit Excessive Pulmonary Flow
7.3.3. Ensure Adequate Egress of the Left Atrial Blood
7.3.4. Other Measures
7.4. “Corrective” Surgery
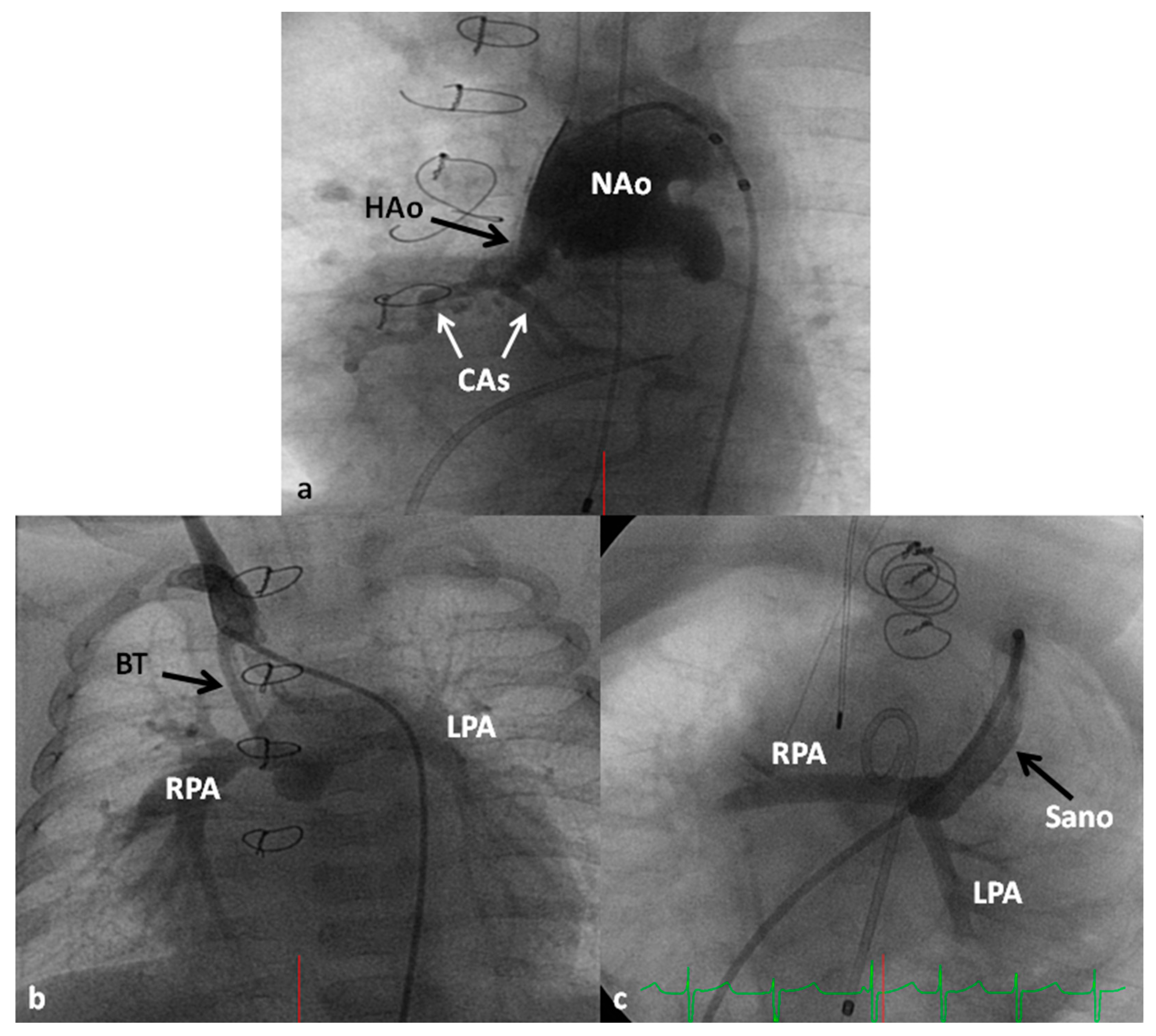
7.4.1. Stage I (Norwood Procedure)
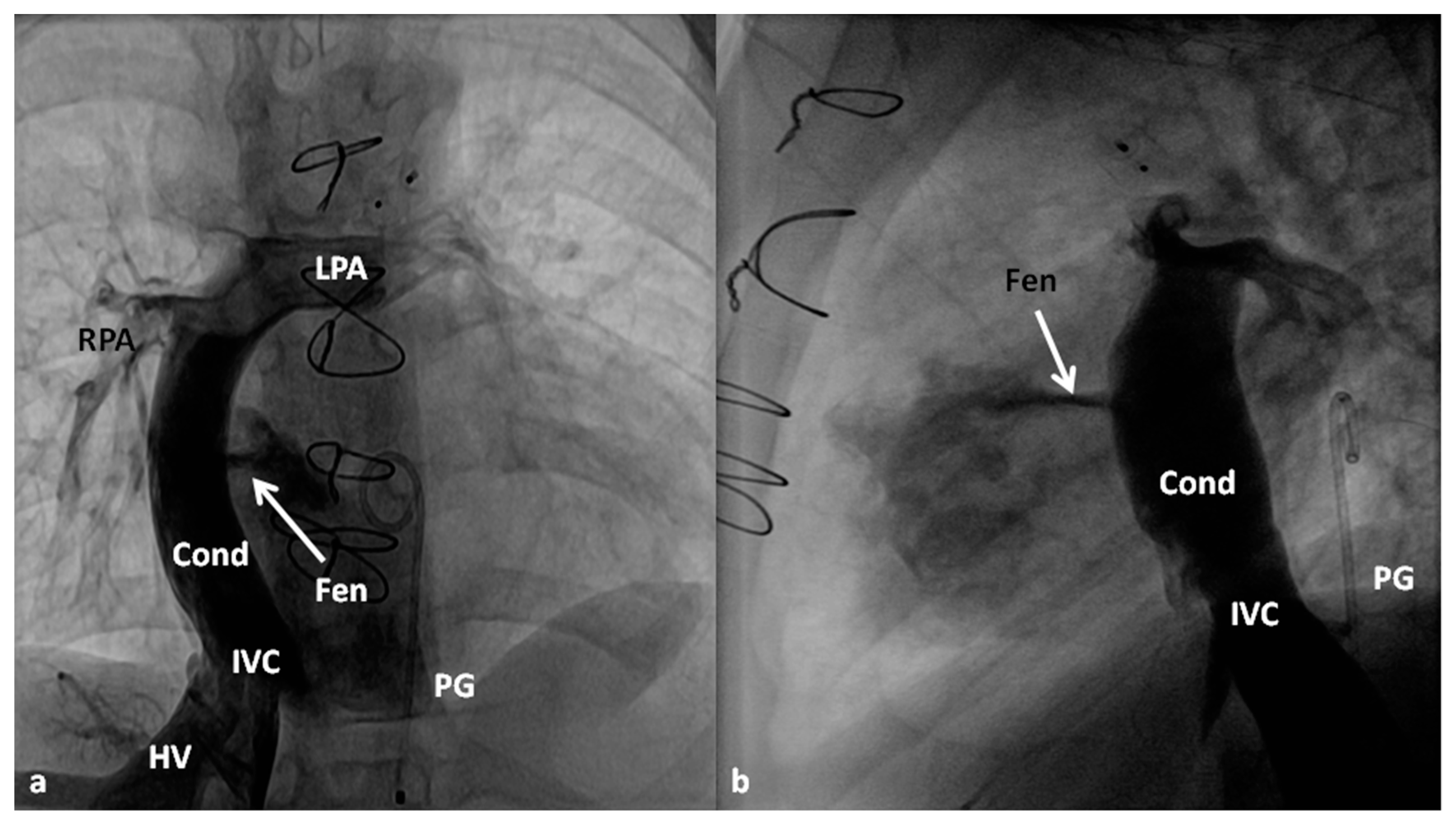
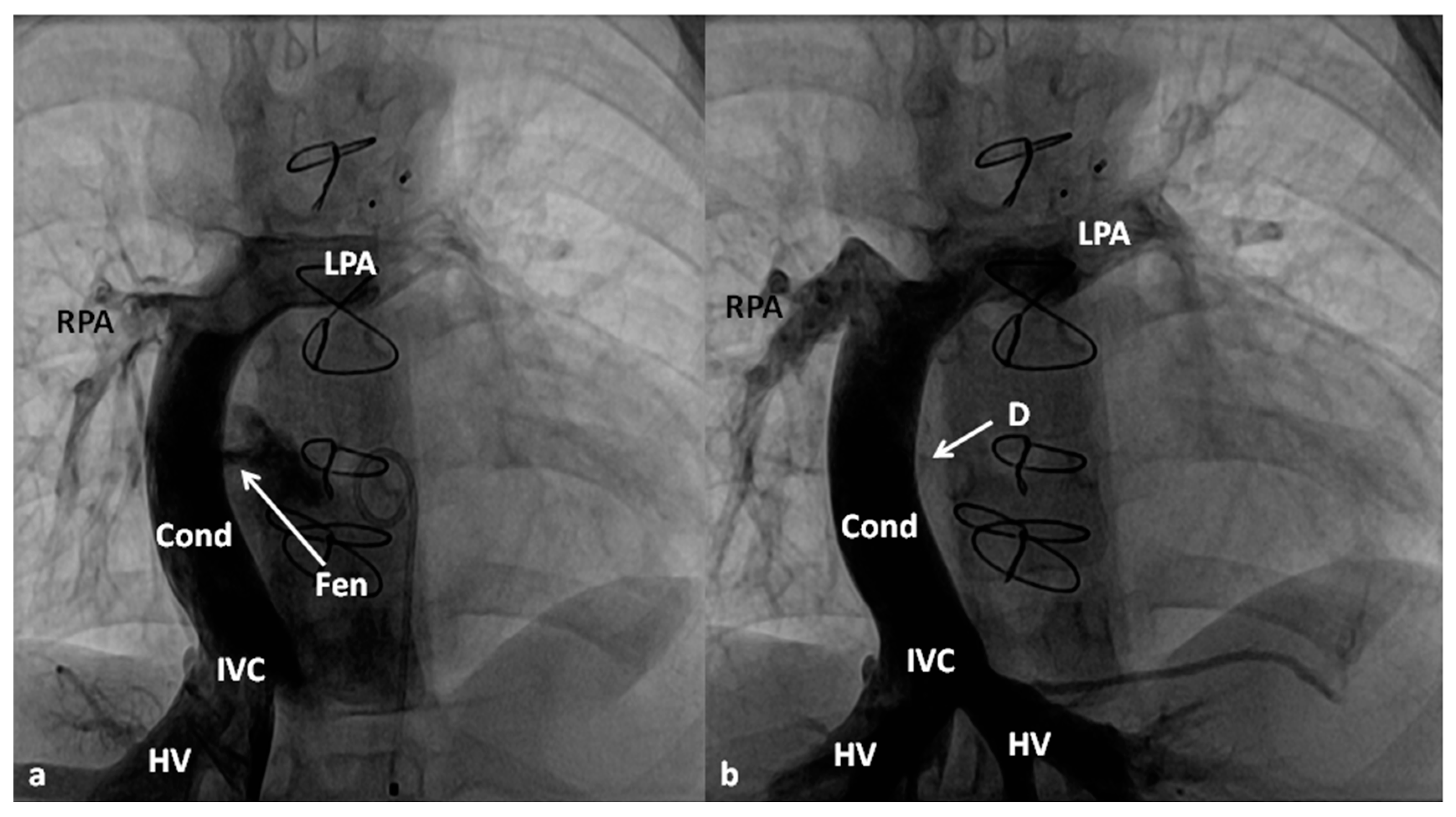
7.4.2. Stages II (Bidirectional Glenn), IIIA (Fontan Completion) and IIIB (Fenestration Closure)
7.5. Cardiac Transplantation
7.6. Alternative Approaches
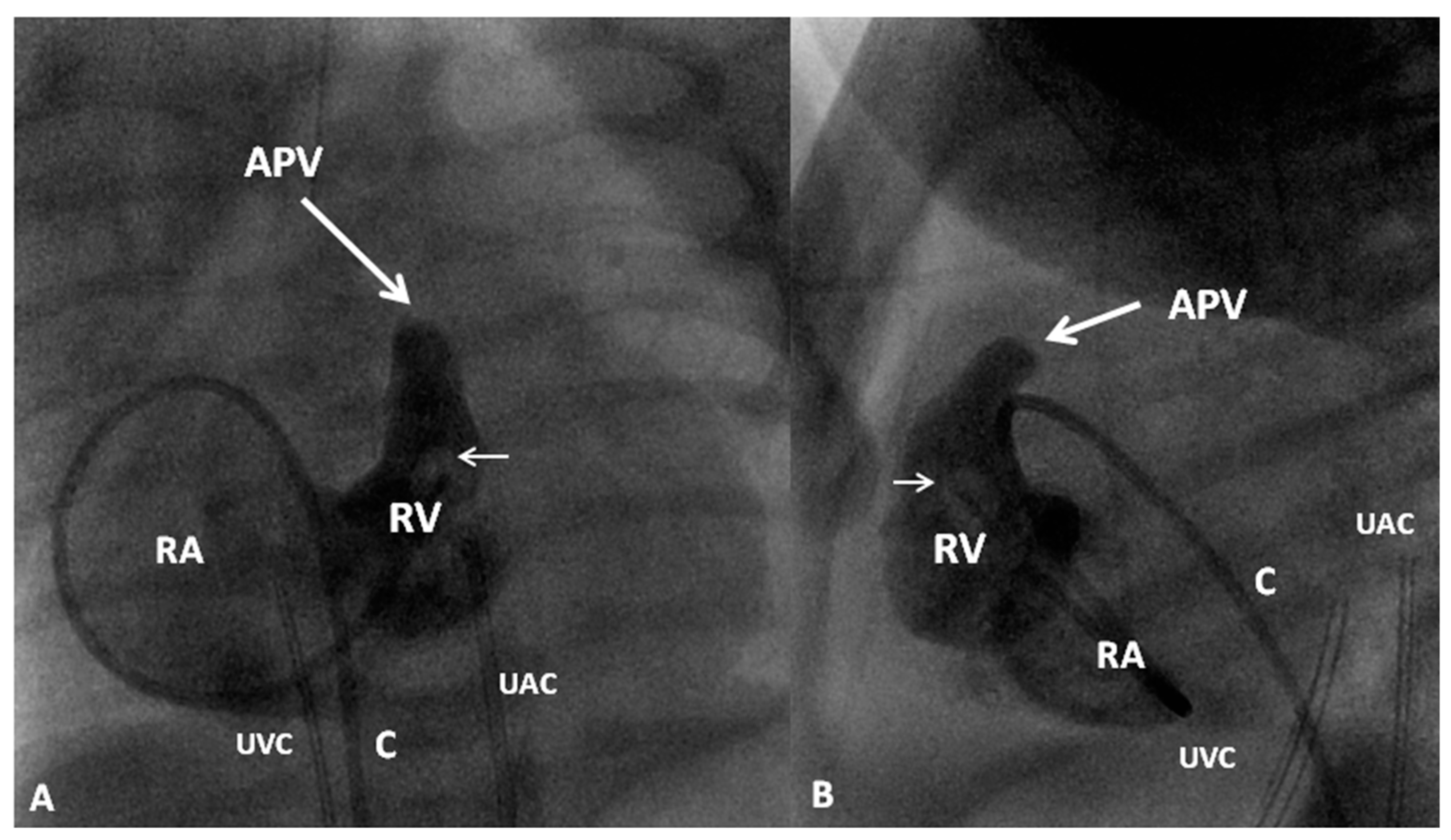
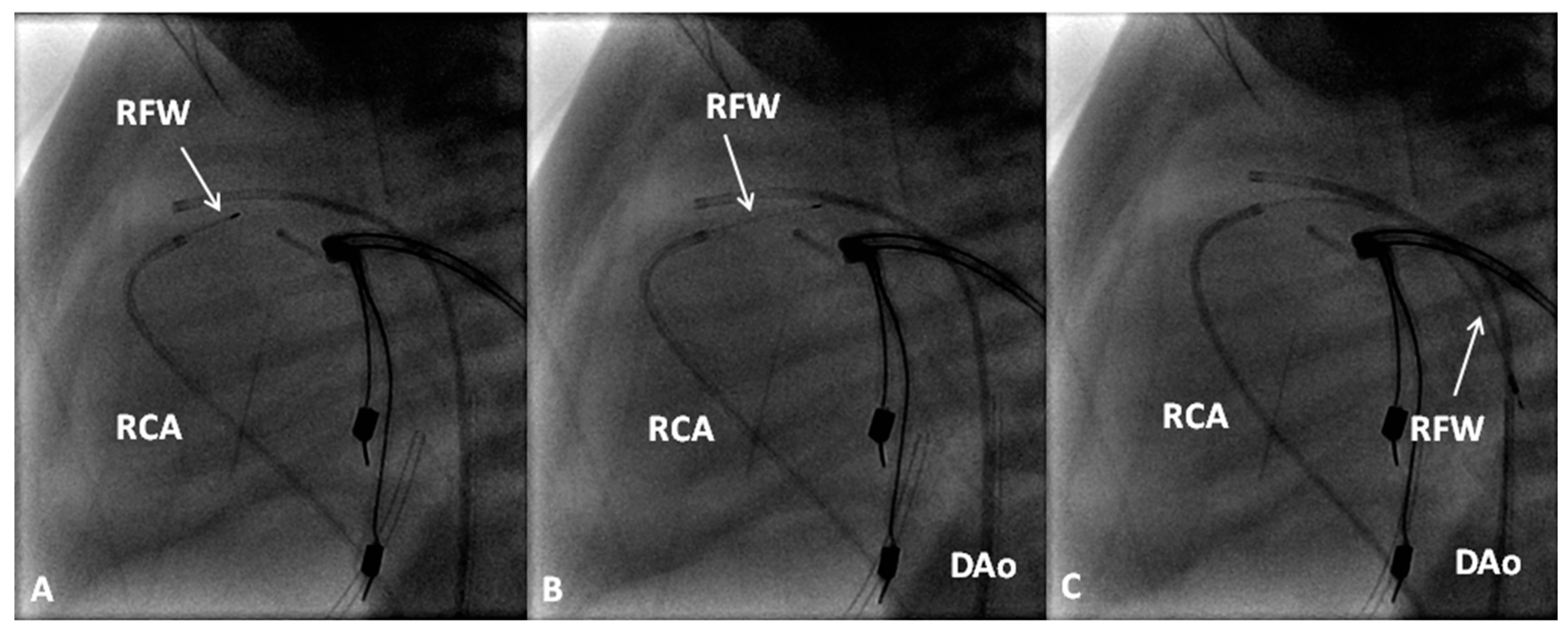
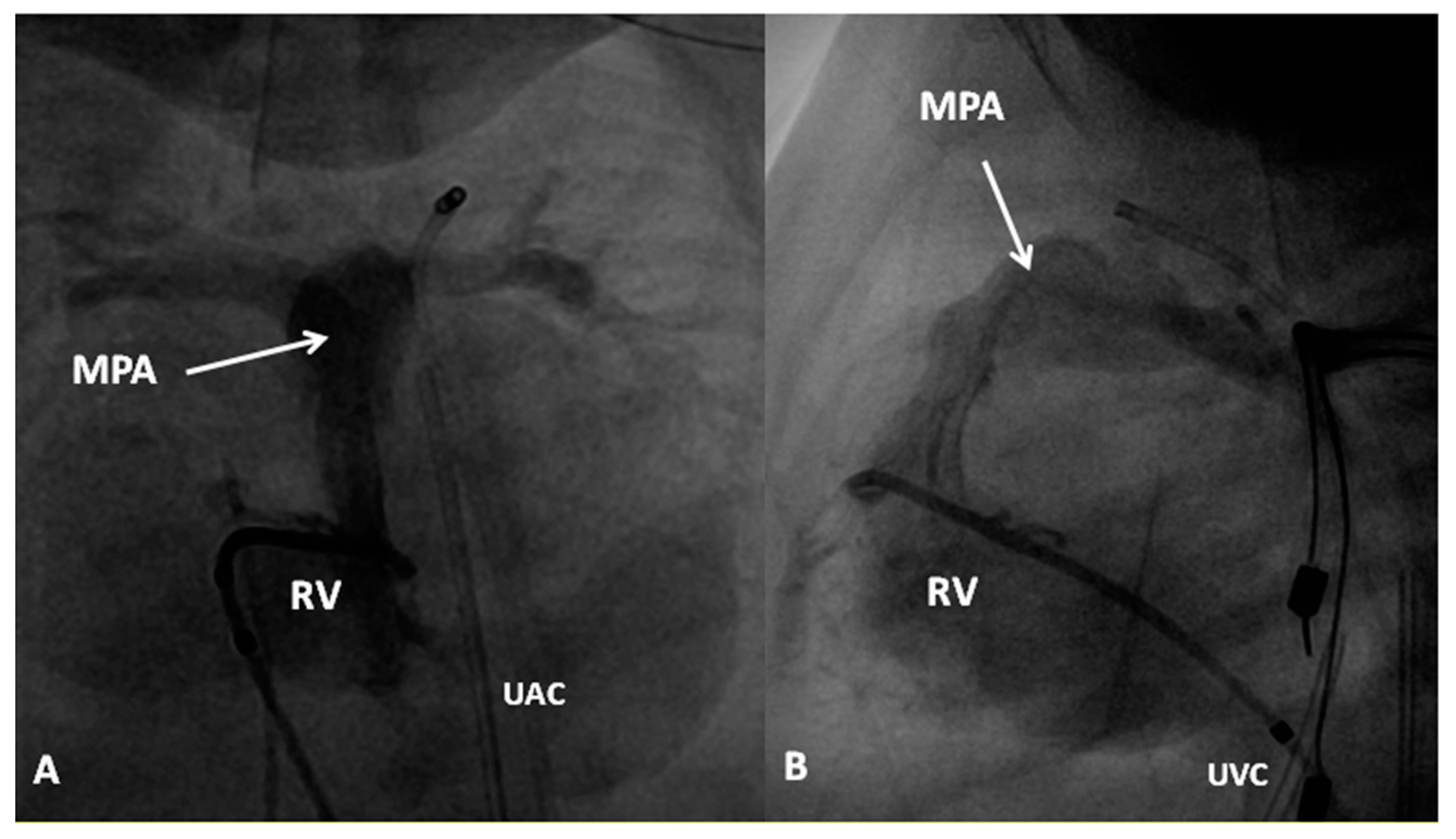
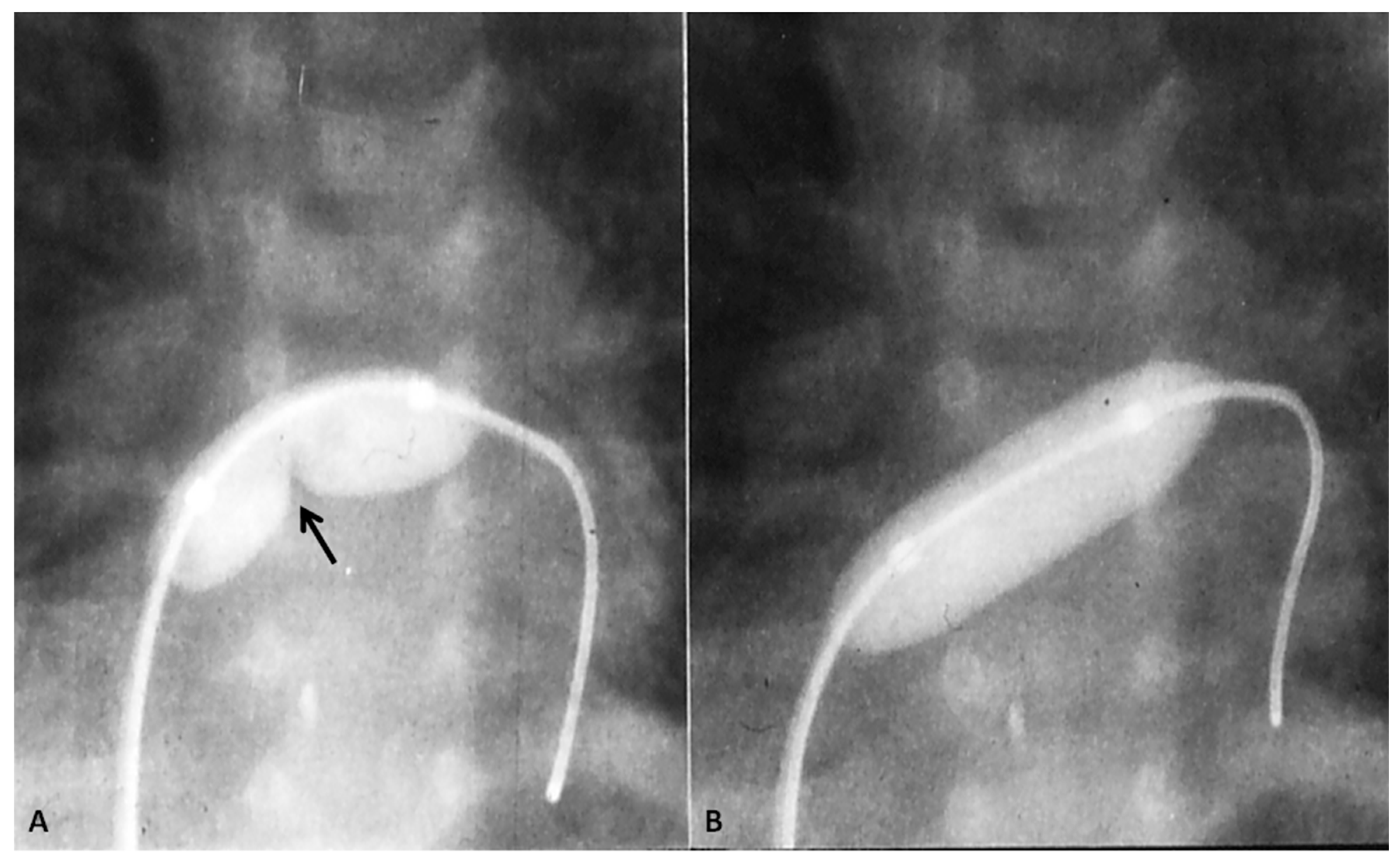
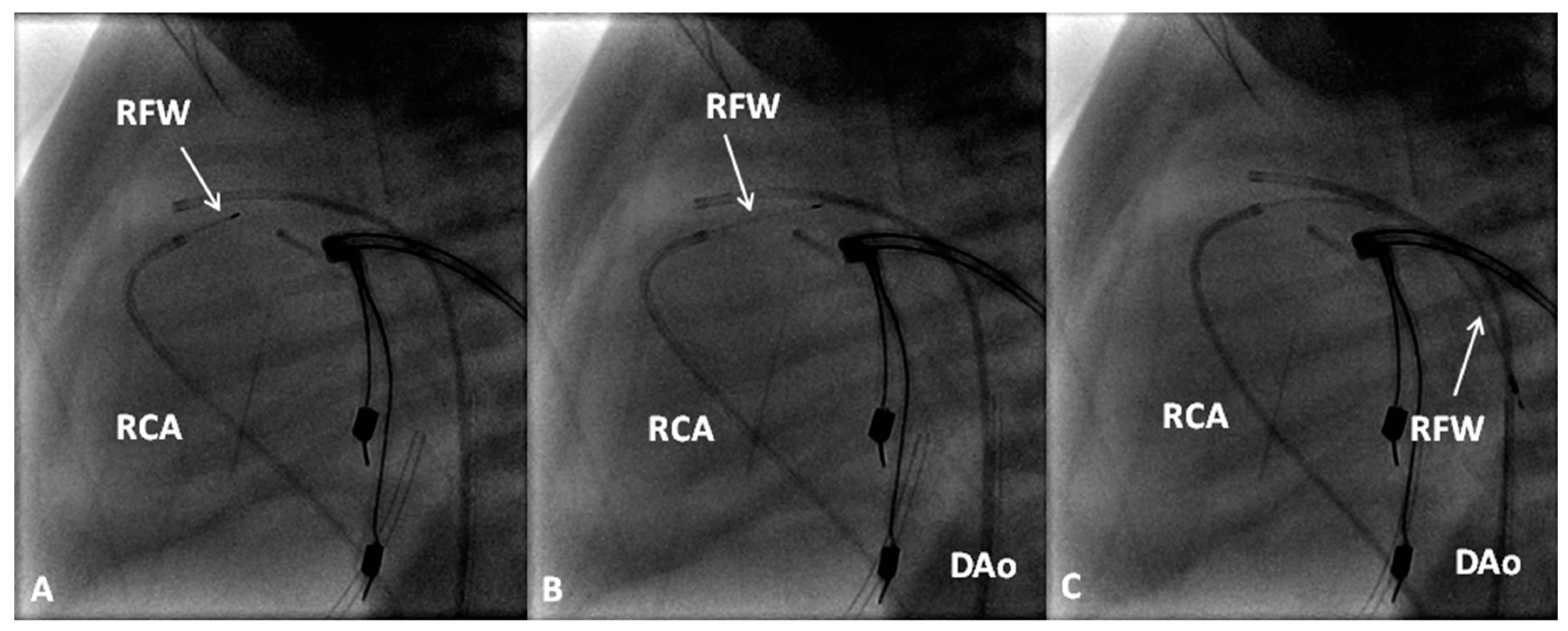
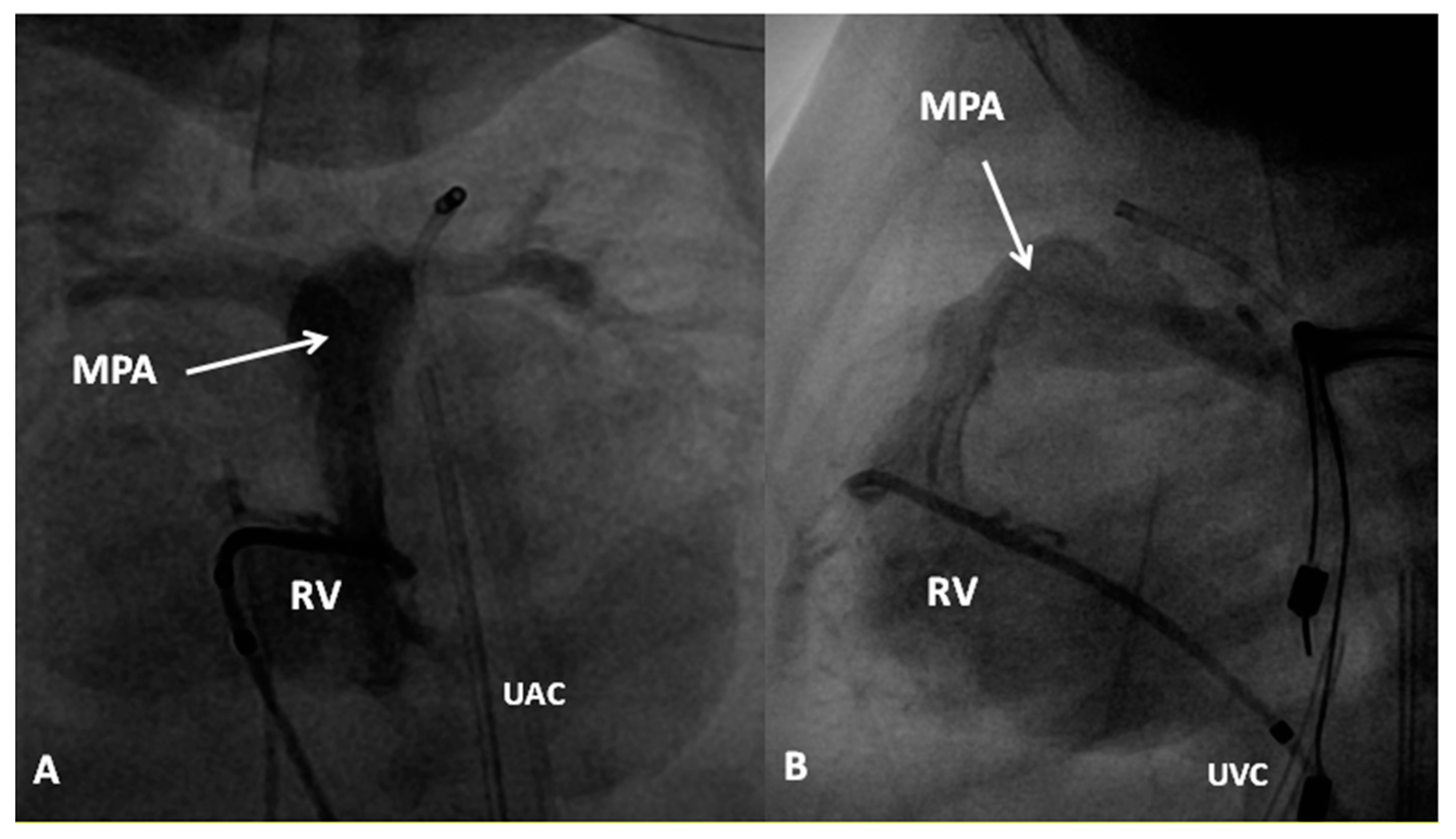
8. Pulmonary Atresia with Intact Ventricular Septum
9. Double-Outlet Right Ventricle (DORV)
10. Double-Inlet Left Ventricle and Univentricular Hearts
11. Other Defects
Conflicts of Interest
References
- Rao, P.S. Management of congenital heart disease: State of the art—Part I—Acyanotic heart defects. Children 2019, 6, 42. [Google Scholar] [CrossRef]
- Rao, P.S. Principles of management of the neonate with congenital heart disease. Neonatol. Today 2007, 2, 1–10. [Google Scholar]
- Rao, P.S. Principles of management of the neonate with congenital heart disease. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 13. [Google Scholar]
- Alapati, S.; Rao, P.S. Tetralogy of Fallot in the neonate. Neonatol. Today 2011, 6, 1–10. [Google Scholar]
- Alapati, S.; Rao, P.S. Tetralogy of Fallot. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 29. [Google Scholar]
- Fallot, E. Contribution a l’anatomie pathologique de la maladie bleu (cyanose cardiaque). Marseille Med. 1888, 25, 418–422. [Google Scholar]
- Rao, P.S. Consensus on timing of intervention for common congenital heart diseases: Part II—Cyanotic heart defects. Indian J. Pediatr. 2013, 80, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Blalock, A.; Taussig, H.B. The surgical treatment of malformations of the heart in which there is pulmonary stenosis or pulmonary atresia. J. Am. Med. Assoc. 1945, 128, 189–194. [Google Scholar] [CrossRef]
- Rao, P.S.; Covitz, W.; Moore, H.V. Principles of palliative management of patients with tricuspid atresia. In Tricuspid Atresia; Rao, P.S., Ed.; Futura Publishing Co.: Mount Kisco, NY, USA, 1982; pp. 233–253. [Google Scholar]
- Rao, P.S.; Covitz, W.; Chopra, P.S. Principles of palliative management of patients with tricuspid atresia. In Tricuspid Atresia, 2nd ed.; Rao, P.S., Ed.; Futura Publishing Co.: Mount Kisco, NY, USA, 1992; pp. 297–320. [Google Scholar]
- de Leval, M.; McKay, R.; Jones, M.; Stark, J.; Macartney, F.J. Modified Blalock-Taussig shunt: Use of subclavian orifice as a flow regulator in prosthetic systemic-pulmonary artery shunts. J. Thorac. Cardovasc. Surg. 1981, 81, 112–119. [Google Scholar]
- Rao, P.S.; Brais, M. Balloon pulmonary valvuloplasty for congenital cyanotic heart defects. Am. Heart J. 1988, 115, 1105–1110. [Google Scholar] [CrossRef]
- Rao, P.S.; Wilson, A.D.; Thapar, M.K.; Brais, M. Balloon pulmonary valvuloplasty in the management of cyanotic congenital heart defects. Catheter. Cardiovasc. Diagn. 1992, 25, 16–24. [Google Scholar] [CrossRef]
- Rao, P.S. Transcatheter management of cyanotic congenital heart defects: A review. Clin. Cardiol. 1992, 15, 483–496. [Google Scholar] [CrossRef]
- Rao, P.S. Pulmonary valve in cyanotic heart defects with pulmonary oligemia. In Percutaneous Interventions in Congenital Heart Disease; Sievert, H., Qureshi, S.A., Wilson, N., Hijazi, Z., Eds.; Informa Health Care: Oxford, UK, 2007; pp. 197–200. [Google Scholar]
- Rao, P.S. Pulmonary valve disease: Pulmonary valve in cyanotic heart defects with pulmonary oligemia. In Interventions in Structural, Valvular and Congenital Heart Disease; Sievert, H., Qureshi, S.A., Wilson, N., Hijazi, Z., Eds.; CRC Press: Boca Raton, FL, USA, 2014; pp. 297–308, Print ISBN 978-1-4822-1563-2; eBook ISBN 978-1-4822-1564-9. [Google Scholar]
- Kohli, V.; Azad, S.; Sachdev, M.S.; Joshi, R.; Joshi, R.; Ebeid, M.R. Balloon dilation of the pulmonary valve in premature infants with tetralogy of Fallot. Pediatr. Cardiol. 2008, 29, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Laudito, A.; Bandisode, V.M.; Lucas, J.F.; Radtke, W.A.; Adamson, W.T.; Bradley, S.M. Right ventricular outflow tract stent as a bridge to surgery in a premature infant with tetralogy of Fallot. Ann. Thorac. Surg. 2006, 81, 744–746. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.S. Stents in the management of vascular obstructive lesions associated with congenital heart disease. In Cardiac Catheterization and Imaging (From Pediatrics to Geriatrics); Vijayalakshmi, I.B., Ed.; Jaypee Publications: New Delhi, India, 2015; pp. 573–598. [Google Scholar]
- Rao, P.S. Congenital heart disease. In Conn’s Current Therapy; Rakel, R.E., Ed.; W.B. Saunders: Philadelphia, PA, USA, 1989; pp. 2010–2013. ISBN 0-7216-2581-9. [Google Scholar]
- Rao, P.S. Diagnosis and management of cyanotic congenital heart disease: Part I. Indian J. Pediatr. 2009, 76, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.C.; Rao, P.S. Pediatric cardiac emergencies. Emerg. Med. 2013, 3, 164. [Google Scholar] [CrossRef]
- Gibbs, J.L.; Rothman, M.T.; Rees, M.; Parsons, J.M.; Blackburn, M.E.; Ruiz, C.E. Stenting of the arterial duct: a new approach to palliation for pulmonary atresia. Br. Heart J. 1992, 67, 240–245. [Google Scholar] [CrossRef]
- Siblini, G.; Rao, P.S.; Singh, G.K.; Tinker, K.; Balfour, I.C. Transcatheter management of neonates with pulmonary atresia and intact ventricular septum. Catheter. Cardiovasc. Diagn. 1997, 42, 395–402. [Google Scholar] [CrossRef]
- Alwi, M.; Choo, K.K.; Latiff, H.A.; Kandavello, G.; Samion, H.; Mulyadi, M.D. Initial results and medium-term follow-up of stent implantation of patent ductus arteriosus in duct-dependent pulmonary circulation. J. Am. Coll. Cardiol. 2004, 44, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.A.; Mulla, N.F.; Dyar, D.; Cephus, C.; Larsen, R.L. Valve perforation and balloon pulmonary valvuloplasty in an infant with tetralogy of Fallot and pulmonary atresia. Catheter. Cardiovasc. Diagn. 1997, 40, 403–406. [Google Scholar] [CrossRef]
- Rao, P.S. Transcatheter perforation of atretic pulmonary valve in pulmonary atresia with ventricular septal defect. Congenit. Cardiol. Today 2018, 16, 1–8. [Google Scholar]
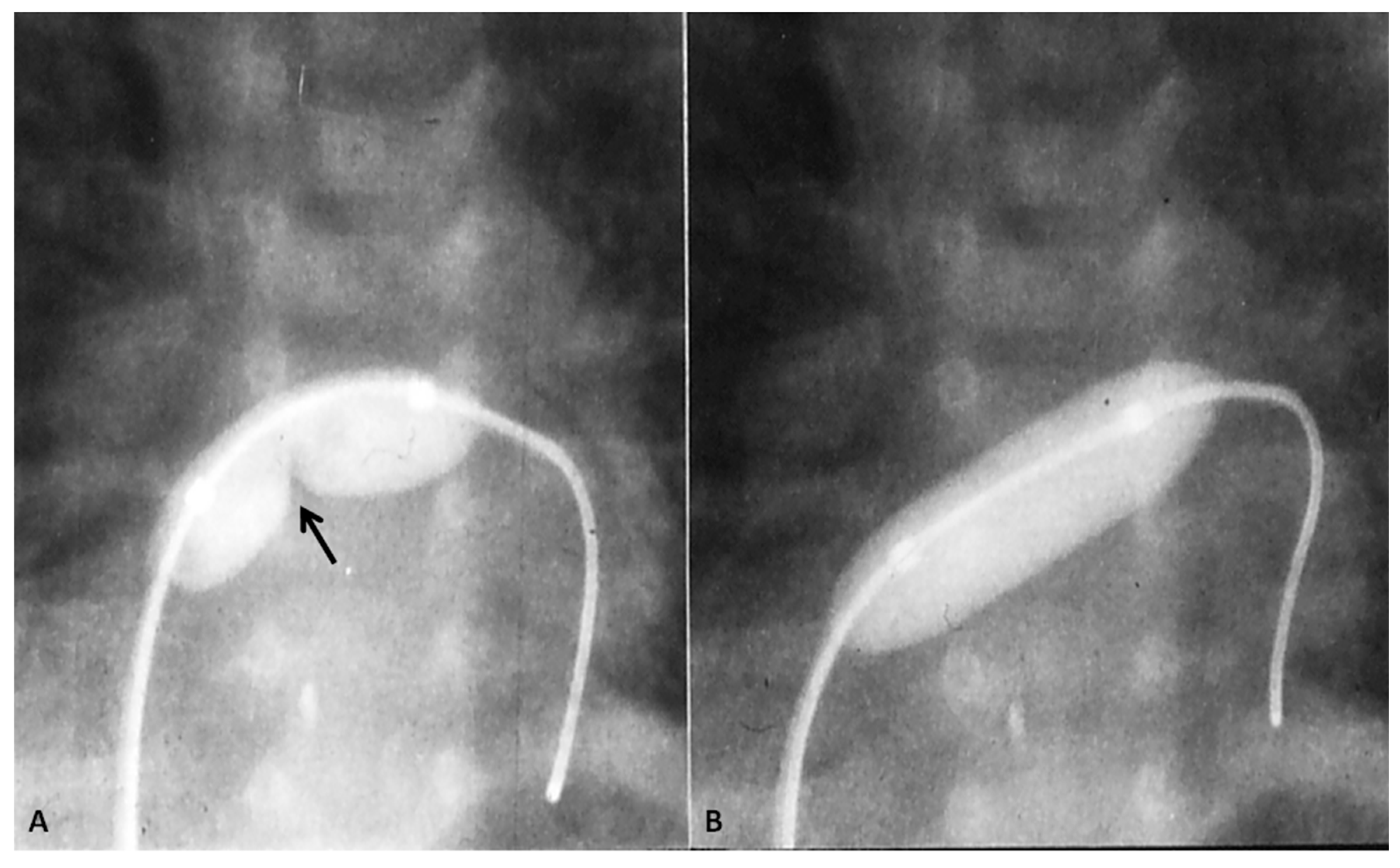
- Latson, L.A. Nonsurgical treatment of a neonate with pulmonary atresia and intact ventricular septum by transcatheter puncture and balloon dilatation of the atretic valve membrane. Am. J. Cardiol. 1991, 68, 277–279. [Google Scholar] [CrossRef]
- Rao, P.S. Pulmonary atresia with intact ventricular septum. Curr. Treat. Opt. Cardiovasc. Med. 2002, 4, 321–336. [Google Scholar] [CrossRef]
- Rao, P.S. Role of interventional cardiology in the treatment of neonates: Part III. Congenit. Cardiol. Today 2008, 6, 1–10. [Google Scholar]
- Liao, P.K.; Edwards, W.D.; Julsrud, P.R.; Puga, F.J.; Danielson, G.K.; Feldt, R.H. Pulmonary blood supply in patients with pulmonary atresia and ventricular septal defect. J. Am. Coll. Cardiol. 1985, 6, 1343–1350. [Google Scholar] [CrossRef]
- Rao, P.S. Transcatheter embolization of unwanted blood vessels in children. In Catheter Based Devices for Treatment of Noncoronary Cardiovascular Disease in Adults and Children; Rao, P.S., Kern, M.J., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2003; pp. 457–473. [Google Scholar]
- Nakata, S.; Imai, Y.; Takanashi, Y.; Kurosawa, H.; Tezuka, K.; Nakazawa, M.; Ando, M.; Takao, A. A new method for the quantitative standardization of cross-sectional areas of the pulmonary arteries in congenital heart diseases with decreased pulmonary blood flow. J. Thorac. Cardiovasc. Surg. 1984, 88, 610–619. [Google Scholar] [PubMed]
- Puga, F.J.; Leoni, F.E.; Julsrud, P.R.; Mair, D.D. Complete repair of pulmonary atresia, ventricular septal defect, and severe peripheral arborization abnormalities of the central pulmonary arteries: Experience with preliminary unifocalization procedures in 38 patients. J. Thorac. Cardiovasc. Surg. 1989, 98, 1018–1028. [Google Scholar] [PubMed]
- Arensman, F.W.; Francis, P.D.; Helmsworth, J.A.; Benzing, G.; Schreiber, J.T.; Schwartz, D.C.; Kaplan, S. Early medical and surgical intervention for tetralogy of Fallot with absence of pulmonic valve. J. Thorac. Cardiovasc. Surg. 1982, 84, 430–436. [Google Scholar]
- Heinemann, M.K.; Hanley, F.L. Preoperative management of neonatal tetralogy of Fallot with absent pulmonary valve syndrome. Ann. Thorac. Surg. 1993, 55, 172–174. [Google Scholar] [CrossRef]
- Rao, P.S.; Lawrie, G.M. Absent pulmonary valve syndrome: Surgical correction with pulmonary arterioplasty. Br. Heart J. 1983, 50, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Ilbawi, M.N.; Idriss, F.S.; Muster, A.J.; Wessel, H.U.; Paul, M.H.; DeLeon, S.Y. Tetralogy of Fallot with absent pulmonary valve: Should valve insertion be a part of the intracardiac repair? J. Thorac. Cardiovasc. Surg. 1981, 81, 906–915. [Google Scholar] [PubMed]
- Rao, P.S. Transposition of the great arteries in the neonate. Neonatol. Today 2010, 5, 1–8. [Google Scholar]
- Rao, P.S. Transposition of the great arteries. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 28. [Google Scholar]
- Rashkind, W.J.; Miller, W.W. Creation of an atrial septal defect without thoracotomy. J. Am. Med. Assoc. 1966, 196, 991–992. [Google Scholar] [CrossRef]
- Rao, P.S. Role of interventional cardiology in neonates: Part I. Non-surgical atrial septostomy. Neonatol. Today 2007, 2, 9–14. [Google Scholar]
- Rao, P.S. Transcatheter blade atrial septostomy. Catheter. Cardiovasc. Diagn. 1984, 10, 335–342. [Google Scholar] [CrossRef]
- Senning, A. Surgical correction of transposition of the great vessels. Surgery 1959, 45, 966–980. [Google Scholar] [PubMed]
- Mustard, W.T.; Keith, J.D.; Trusler, G.A.; Fowler, R.; Kidd, L. The surgical management of transposition of the great vessels. J. Thorac. Cardiovasc. Surg. 1964, 48, 953–958. [Google Scholar] [PubMed]
- Jatene, A.D.; Fontes, V.F.; Paulista, P.P.; Souza, L.C.; Neger, F.; Galantier, M.; Sousa, J.E. Anatomic correction of transposition of the great vessels. J. Thorac. Cardiovasc. Surg. 1976, 72, 364–370. [Google Scholar]
- LeCompte, Y.; Zannini, L.; Hazan, E.; Jarreau, M.M.; Bex, J.P.; Tu, T.V.; Neveux, J.Y. Anatomic correction of transposition of the great arteries. J. Thorac. Cardiovasc. Surg. 1981, 82, 629–631. [Google Scholar]
- Yacoub, M.H.; Radley-Smith, R.; Hilton, C.J. Anatomical correction of complete transposition of the great arteries and ventricular septal defect in infancy. Br. Med. J. 1976, 1, 1112–1114. [Google Scholar] [CrossRef]
- Jonas, R.A.; Giglia, T.M.; Sanders, S.P.; Wernovsky, G.; Nadal-Ginard, B.; Mayer, J.E.; Castaneda, A.R. Rapid, two-stage arterial switch for transposition of the great arteries and intact ventricular septum beyond the neonatal period. Circulation 1989, 80, I203–I208. [Google Scholar]
- Rastelli, G.C.; McGoon, D.C.; Wallace, R.B. Anatomic correction of transposition of the great arteries with ventricular septal defect and subpulmonary stenosis. J. Thorac. Cardiovasc. Surg. 1969, 58, 545–552. [Google Scholar]
- Kawashima, Y.; Fujita, T.; Miyamoto, T.; Manabe, H. Intraventricular rerouting of blood for the correction of Taussig-Bing malformation. J. Thorac. Cardiovasc. Surg. 1971, 62, 825–829. [Google Scholar]
- Nikaidoh, H. Aortic translocation and biventricular outflow tract reconstruction. A new surgical repair for transposition of the great arteries associated with ventricular septal defect and pulmonary stenosis. J. Thorac. Cardiovasc. Surg. 1984, 88, 365–372. [Google Scholar] [PubMed]
- Rao, P.S. Tricuspid Atresia; Futura Publishing Co.: Mount Kisco, NY, USA, 1982. [Google Scholar]
- Rao, P.S. Tricuspid atresia. In Fetal and Neonatal Cardiology; Long, W.A., Ed.; W.B. Saunders: Philadelphia, PA, USA, 1990; pp. 525–540. [Google Scholar]
- Rao, P.S. Tricuspid Atresia, 2nd ed.; Futura Publishing Co.: Mount Kisco, NY, USA, 1992. [Google Scholar]
- Rao, P.S. Tricuspid atresia. In Pediatric Cardiovascular Medicine, 2nd ed.; Moller, J.H., Hoffman, J.I.E., Eds.; Wiley-Blackwell: Hoboken, NJ, USA; John Wiley & Sons Ltd.: Oxford, UK, 2012; pp. 487–508. [Google Scholar]
- Rao, P.S. Tricuspid atresia. In A Comprehensive Approach to Management of Congenital Heart Diseases; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publications: New Delhi, India, 2013; pp. 397–413. [Google Scholar]
- Rao, P.S. A unified classification for tricuspid atresia. Am. Heart J. 1980, 99, 799–804. [Google Scholar] [CrossRef]
- Fontan, F.; Baudet, E. Surgical repair of tricuspid atresia. Thorax 1971, 26, 240–248. [Google Scholar] [CrossRef]
- Kreutzer, G.; Bono, H.; Galindez, E. Una operacion para la correccion de la atresia tricuspidea. In Proceedings of the Ninth Argentinean Congress of Cardiology, Buenos Aires, Argentina, 31 October–6 November 1971. [Google Scholar]
- Annecchino, F.P.; Fontan, F.; Chauve, A.; Quaegebeur, J. Palliative reconstruction of the right ventricular outflow tract in tricuspid atresia: A report of 5 patients. Ann. Thorac. Surg. 1980, 29, 317–321. [Google Scholar] [CrossRef]
- Rao, P.S.; Sissman, N.J. Spontaneous closure of physiologically advantageous ventricular septal defects. Circulation 1971, 43, 83–90. [Google Scholar] [CrossRef]
- Rao, P.S. Natural history of the ventricular septal defect in tricuspid atresia and its surgical implications. Br. Heart J. 1977, 39, 276–288. [Google Scholar] [CrossRef]
- Rao, P.S. Further observations on the spontaneous closure of physiologically advantageous ventricular septal defects in tricuspid atresia: Surgical implications. Ann. Thorac. Surg. 1983, 35, 121–131. [Google Scholar] [CrossRef]
- Rao, P.S. Natural history of ventricular septal defects in tricuspid atresia. In Tricuspid Atresia, 2nd ed.; Rao, P.S., Ed.; Futura Publishing Co.: Mount Kisco, NY, USA, 1992; pp. 261–293. [Google Scholar]
- Bonnet, D.; Sidi, D.; Vouhé, P.R. Absorbable pulmonary artery banding in tricuspid atresia. Ann. Thorac. Surg. 2001, 71, 360–361. [Google Scholar] [CrossRef]
- Rao, P.S. Absorbable pulmonary artery band in tricuspid atresia (Editorial). Ann. Thorac. Surg. 2001, 71, 361–362. [Google Scholar] [CrossRef]
- Dick, M.; Fyler, D.C.; Nadas, A.S. Tricuspid atresia: Clinical course in 101 patients. Am. J. Cardiol. 1975, 36, 327–337. [Google Scholar] [CrossRef]
- Rao, P.S. Cardiac catheterization in tricuspid atresia. In Tricuspid Atresia; Rao, P.S., Ed.; Futura Publishing Co.: Mount Kisco, NY, USA, 1982; pp. 153–178. [Google Scholar]
- Park, S.C.; Neches, W.H.; Zuberbuhler, J.R.; Lenox, C.C.; Mathews, R.A.; Fricker, F.J.; Zoltun, R.A. Clinical use of blade atrial septostomy. Circulation 1978, 58, 600–606. [Google Scholar] [CrossRef]
- Salim, M.; Muster, A.J.; Paul, M.H.; Benson, D.W., Jr. Relation between preoperative left ventricular muscle mass and outcome of the Fontan procedure in patients with tricuspid atresia. J. Am. Coll. Cardiol. 1989, 14, 750–755. [Google Scholar] [CrossRef]
- Chopra, P.S.; Rao, P.S. Corrective surgery for tricuspid atresia: Which modifications of Fontan-Kreutzer procedure should be used? A review. Am. Heart J. 1992, 123, 758–767. [Google Scholar] [CrossRef]
- Rao, P.S.; Chopra, P.S. Modification of Fontan-Kreutzer procedures for tricuspid atresia: Can a choice be made? In Tricuspid Atresia, 2nd ed.; Rao, P.S., Ed.; Futura Publishing Co.: Mount Kisco, NY, USA, 1992; pp. 361–375. [Google Scholar]
- Rao, P.S. Pediatric Tricuspid Atresia. Medscape Drugs & Diseases. Updated 5 January 2016. Available online: http://emedicine.medscape.com/article/900832-overview (accessed on 4 April 2019).
- de Leval, M.R.; Kilner, P.; Gewilling, M.; Bull, C. Total cavopulmonary connection: A logical alternative to atriopulmonary connection for complex Fontan operation. J. Thorac. Cardiovasc. Surg. 1988, 96, 682–695. [Google Scholar] [PubMed]
- Marcelletti, C.; Corno, A.; Giannico, S.; Marino, B. Inferior vena cavapulmonary artery extra-cardiac conduit. A new form of right heart bypass. J. Thorac. Cardiovasc. Surg. 1990, 100, 228–232. [Google Scholar]
- Bridges, N.D.; Lock, J.E.; Castaneda, A.R. Baffle fenestration with subsequent transcatheter closure: Modification of the Fontan operation for patients with increased risk. Circulation 1990, 82, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Laks, H.; Pearl, J.M.; Haas, G.S.; Drinkwater, D.C.; Milgalter, E.; Jarmakani, J.M.; Isabel-Jones, J.; George, B.L.; Williams, R.G. Partial Fontan advantages of an adjustable interatrial communication. Ann. Thorac. Surg. 1991, 52, 1084–1094. [Google Scholar] [CrossRef]
- Rao, P.S. Fontan operation: Indications, short and long term outcomes. Indian J. Pediatr. 2015, 82, 1147–1156. [Google Scholar] [CrossRef]
- Azzolina, G.; Eufrate, S.; Pensa, P. Tricuspid atresia: Experience in surgical management with a modified cavopulmonary anastomosis. Thorax 1972, 27, 111–115. [Google Scholar] [CrossRef]
- Kumar, S.P.; Rubinstein, C.S.; Simsic, J.M.; Taylor, A.B.; Saul, J.P.; Bradley, S.M. Lateral tunnel versus extracardiac conduit Fontan procedure: A concurrent comparison. Ann. Thorac. Surg. 2003, 76, 1389–1396. [Google Scholar] [CrossRef]
- Whitfield, C.K.; Rao, P.S. Total anomalous pulmonary venous connection in the neonate. Neonatol. Today 2013, 8, 1–7. [Google Scholar]
- Rao, P.S. Total anomalous pulmonary venous connection. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 31. [Google Scholar]
- Rao, P.S.; Silbert, D.R. Superior vena caval obstruction in total anomalous pulmonary venous return. Br. Heart J. 1974, 36, 228–232. [Google Scholar] [CrossRef]
- Bando, K.; Turrentine, M.W.; Ensing, G.J.; Sun, K.; Sharp, T.G.; Sekine, Y.; Girod, D.A.; Brown, J.W. Surgical management of total anomalous pulmonary venous connection. Circulation 1996, 94, 12–16. [Google Scholar]
- Rao, P.S.; Balaguru, D. Truncus arteriosus in the neonate. Neonatol. Today 2013, 8, 1–6. [Google Scholar]
- Balaguru, D.; Rao, P.S. Truncus arteriosus. In A Comprehensive Approach to Management of Congenital Heart Diseases; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publications: New Delhi, India, 2013; pp. 600–613. [Google Scholar]
- Balaguru, D.; Rao, P.S. Truncus arteriosus. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 32. [Google Scholar]
- Collett, R.W.; Edwards, J.E. Persistent truncus arteriosus: A classification according to anatomic types. Surg. Clin. N. Am. 1949, 29, 1245–1270. [Google Scholar] [CrossRef]
- Ebert, P.A.; Turley, K.; Stanger, P.; Hoffman, J.I.; Heymann, M.A.; Rudolph, A.M. Surgical treatment of truncus arteriosus in the first 6 months of life. Ann. Surg. 1984, 200, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Noonan, J.A.; Nadas, A.S. The hypoplastic left heart syndrome. Pediatr. Clin. N. Am. 1958, 5, 1029–1056. [Google Scholar] [CrossRef]
- Rao, P.S.; Striepe, V.; Merrill, W.H. Hypoplastic left heart syndrome. In Cardiac Anesthesia for Infants and Children; Kambam, J., Ed.; Mosby-Year Book: St. Louis, MO, USA, 1994; pp. 296–309. [Google Scholar]
- Rao, P.S.; Turner, D.R.; Forbes, T.J. Hypoplastic left heart syndrome. eMedicine from WebMD. Updated 22 September 2009. Available online: http://emedicine.medscape.com/article/890196-overview (accessed on 4 April 2019).
- Alapati, S.; Rao, P.S. Hypoplastic left heart syndrome in the neonate. Neonatol. Today 2011, 6, 1–9. [Google Scholar]
- Rao, P.S.; Alapati, S. Hypoplastic left heart syndrome. In A Comprehensive Approach to Management of Congenital Heart Diseases; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publications: New Delhi, India, 2013; pp. 662–678. [Google Scholar]
- Rao, P.S.; Alapati, S. Hypoplastic left heart syndrome. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 33. [Google Scholar]
- Norwood, W.I.; Lang, P.; Hansen, D.D. Physiologic repair of aortic atresia-hypoplastic left heart syndrome. N. Engl. J. Med. 1983, 308, 23–26. [Google Scholar] [CrossRef]
- Norwood, W.I.; Lang, P.; Castaneda, A.R.; Campbell, D.N. Experience with operation for hypoplastic left heart syndrome. J. Thorac. Cardiovasc. Surg. 1981, 82, 511–519. [Google Scholar]
- Norwood, W.I., Jr. Hypoplastic left heart syndrome. Ann. Thorac. Surg. 1991, 52, 688–695. [Google Scholar] [CrossRef]
- Bailey, L.; Concepcion, W.; Shattuk, H.; Huang, L. Method of heart transplantation for treatment of hypoplastic left heart syndrome. J. Thorac. Cardiovasc. Surg. 1986, 92, 1–9. [Google Scholar]
- Day, R.W.; Barton, A.J.; Pysher, T.J.; Shaddy, R.E. Pulmonary vascular resistance of children treated with nitrogen during early infancy. Ann. Thorac. Surg. 1998, 65, 1400–1404. [Google Scholar] [CrossRef]
- Shrivatsava, S.; Radhakrishnan, S.; Dev, V.; Singh, L.S.; Rajani, M. Balloon dilatation of atrial septum in complete transposition of great arteries—A new technique. Indian Heart J. 1987, 39, 298–300. [Google Scholar]
- Rao, P.S. Static balloon dilatation of the atrial septum. Am. Heart J. 1993, 125, 1826–1828. [Google Scholar] [CrossRef]
- Gewillig, D.; Boshoff, L.; Mertens, L. Creation with a stent of an unrestrictive lasting atrial communication. Cardiol. Young 2002, 12, 404–407. [Google Scholar] [CrossRef]
- Sano, S.; Ishino, K.; Kawada, M.; Arai, S.; Kasahara, S.; Asai, T.; Masuda, Z.-I.; Takeuchi, M.; Ohtsuki, S.-I. Right ventricle-pulmonary artery shunt in first-stage palliation of hypoplastic left heart syndrome. J. Thorac. Cardiovasc. Surg. 2003, 126, 504–509. [Google Scholar] [CrossRef]
- Sano, S.; Ishino, K.; Kado, H.; Shiokawa, Y.; Sakamoto, K.; Yokota, M.; Kawada, M. Outcome of right ventricle-to-pulmonary artery shunt in first-stage palliation of hypoplastic left heart syndrome: A multi-institutional study. Ann. Thorac. Surg. 2004, 78, 1951–1957; discussion 1957–1958. [Google Scholar] [CrossRef]
- Ghanayem, N.S.; Jaquiss, R.D.B.; Cava, J.R.; Frommelt, P.C.; Mussatto, K.A.; Hoffman, G.M.; Tweddell, J.S. Right ventricle-to-pulmonary artery conduit versus Blalock-Taussig shunt: a hemodynamic comparison. Ann. Thorac. Surg. 2006, 82, 1603–1609. [Google Scholar] [CrossRef]
- Ohye, R.G.; Sleeper, L.A.; Mahony, L.; Newburger, J.W.; Pearson, G.D.; Lu, M.; Goldberg, C.S.; Tabbutt, S.; Frommelt, P.C.; Ghanayem, N.S.; et al. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N. Engl. J. Med. 2010, 362, 1980–1992. [Google Scholar] [CrossRef] [PubMed]
- Tweddell, J.S.; Hoffman, G.M.; Mussatto, K.A.; Fedderly, R.T.; Berger, S.; Jaquiss, R.D.B.; Ghanayem, N.S.; Frisbee, S.J.; Litwin, S.B. Improved survival of patients undergoing palliation of hypoplastic left heart syndrome: Lessons learned from 115 consecutive patients. Circulation 2002, 106, 82–89. [Google Scholar] [CrossRef]
- Bartram, U.; Grunenfelder, J.; Van Praagh, R. Causes of death after the modified Norwood procedure: A study of 122 postmortem cases. Ann. Thorac. Surg. 1997, 64, 1795–1802. [Google Scholar] [CrossRef]
- Galantowicz, M.; Cheatham, J.P. Lessons learned from the development of a new hybrid strategy for the management of hypoplastic left heart syndrome. Pediatr. Cardiol. 2005, 26, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Photiadis, J.; Sinzobahamvya, N.; Hraška, V.; Asfour, B. Does bilateral pulmonary banding in comparison to Norwood procedure improve outcome in neonates with hypoplastic left heart syndrome beyond second-stage palliation? A review of the current literature. J. Thorac. Cardiovasc. Surg. 2012, 60, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Hsia, T.Y.; Cosentino, D.; Corsini, C.; Pennati, G.; Dubini, G.; Migliavacca, F. Modeling of Congenital Hearts Alliance (MOCHA) Investigators. Use of mathematical modeling to compare and predict hemodynamic effects between hybrid and surgical Norwood palliations for hypoplastic left heart syndrome. Circulation 2011, 124, S204–S210. [Google Scholar] [CrossRef]
- Rao, P.S. Comprehensive management of pulmonary atresia with intact ventricular septum. Ann. Thorac. Surg. 1985, 40, 409–413. [Google Scholar] [CrossRef]
- Balaguru, D.; Rao, P.S. Pulmonary atresia with intact ventricular septum. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 34. [Google Scholar]
- Rao, P.S. Neonatal Catheter Interventions. In Cardiac Catheterization and Imaging (From Pediatrics to Geriatrics); Vijayalakshmi, I.B., Ed.; Jaypee Publications: New Delhi, India, 2015; pp. 388–432. [Google Scholar]
- Nathan, M.; Verma, R.; Balaguru, D.; Starr, J. A brief review of hybrid procedures for congenital heart disease. Heart Views 2009, 10, 156–161. [Google Scholar]
- Neufeld, H.N.; Lucas, R.V., Jr.; Lester, R.G.; Adams, P.; Anderson, R.C.; Edwards, J.E. Origin of both great vessels from the right ventricle without pulmonary stenosis. Br. Heart J. 1962, 24, 393–408. [Google Scholar] [CrossRef]
- Buck, S.H.; Rao, P.S.; Merrill, W.; Kambam, J. Double-outlet right ventricle. In Cardiac Anesthesia for Infants and Children; Kambam, J., Ed.; Mosby: St. Louis, MO, USA, 1993; pp. 312–339. [Google Scholar]
- Rao, P.S. Diagnosis and management of cyanotic congenital heart disease: Part II. Indian J. Pediatr. 2009, 76, 297–308. [Google Scholar] [CrossRef]
- Rao, P.S. Other cyanotic heart defects in the neonate. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 37. [Google Scholar]
- Rao, P.S. Subaortic obstruction after pulmonary artery banding in patients with tricuspid atresia and double-inlet left ventricle and ventriculoarterial discordance. J. Am. Coll. Cardiol. 1991, 18, 1585–1586. [Google Scholar] [CrossRef]
- Jureidini, S.B.; Marino, C.J.; Rao, P.S. Congenital coronary artery anomalies. Indian J. Pediatr. 1998, 65, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Kambam, J.; Rao, P.S. Anomalous origin of the left coronary artery (Bland-White-Garland Syndrome). In Cardiac Anesthesia for Infants and Children; Kambam, J., Ed.; Mosby-Year Book: St. Louis, MO, USA, 1994; pp. 361–367. [Google Scholar]
- Sharma, S.K.; Rao, P.S. Interrupted aortic arch. In The Encyclopedic Reference of Molecular Mechanisms of Disease; Lang, F., Ed.; Springer: Berlin/Heidelberg, Germany; New York, NY, USA; Tokyo, Japan, 2008. [Google Scholar]
- Rao, P.S. Tricuspid valve abnormalities other than tricuspid atresia. In Fetal and Neonatal Cardiology; Long, W.A., Ed.; W.B. Saunders: Philadelphia, PA, USA, 1990; pp. 541–550. [Google Scholar]
- Rao, P.S.; Kambam, J. Ebstein’s malformation of the tricuspid valve. In Cardiac Anesthesia for Infants and Children; Kambam, J., Ed.; Mosby-Year Book: St. Louis, MO, USA, 1994; pp. 320–332. [Google Scholar]
- Balaguru, D.; Rao, P.S. Diseases of the tricuspid valve (Ebstein’s Anomaly, Tricuspid Stenosis and Regurgitation). In A Comprehensive Approach to Management of Congenital Heart Diseasess; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publication: New Delhi, India, 2013; pp. 414–433. [Google Scholar]
- Balaguru, D.; Rao, P.S. Ebstein’s anomaly of the tricuspid valve. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 35. [Google Scholar]
- Balaguru, D.; Rao, P.S. Mitral atresia. In A Comprehensive Approach to Management of Congenital Heart Diseases; Vijayalakshmi, I.B., Rao, P.S., Chugh, R., Eds.; Jaypee Publications: New Delhi, India, 2013; pp. 458–467. [Google Scholar]
- Rao, P.S.; Leonard, T. Polysplenia syndrome. Cardiol. Dig. 1976, 11, 14–22. [Google Scholar]
- Rao, P.S. Dextrocardia: Systematic approach to differential diagnosis. Am. Heart J. 1981, 102, 389–403. [Google Scholar] [CrossRef]
- Rao, P.S. Cardiac malpositions including heterotaxy syndromes. In Perinatal Cardiology: A Multidisciplinary Approach; Rao, P.S., Vidyasagar, D., Eds.; Cardiotext Publishing: Minneapolis, MN, USA, 2015; Chapter 36. [Google Scholar]
- Rao, P.S. Cardiac malposition. In PG Textbook of Pediatrics; Gupta, P., Menon, P.S.N., Ramji, S., Lodha, R., Eds.; Jaypee Brothers Medical Publishers (P) Ltd.: New Delhi, India, 2015; pp. 1807–1816. [Google Scholar]
- Rao, P.S. Cardiac malposition. In PG Textbook of Pediatrics, 2nd ed.; Gupta, P., Menon, P.S.N., Ramji, S., Lodha, R., Eds.; Jaypee Brothers Medical Publishers (P) Ltd.: New Delhi, India, 2018; pp. 1807–1816. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5 Ts | Other Defects |
|---|---|
| Tetralogy of Fallot | Hypoplastic left heart syndrome |
| Transposition of the great arteries | Pulmonary atresia with intact ventricular septum |
| Tricuspid atresia | Double-outlet right ventricle |
| Total anomalous pulmonary venous connection | Double-inlet left ventricle and univentricular hearts |
| Truncus arteriosus |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, P.S. Management of Congenital Heart Disease: State of the Art—Part II—Cyanotic Heart Defects. Children 2019, 6, 54. https://doi.org/10.3390/children6040054
Rao PS. Management of Congenital Heart Disease: State of the Art—Part II—Cyanotic Heart Defects. Children. 2019; 6(4):54. https://doi.org/10.3390/children6040054
Chicago/Turabian StyleRao, P. Syamasundar. 2019. "Management of Congenital Heart Disease: State of the Art—Part II—Cyanotic Heart Defects" Children 6, no. 4: 54. https://doi.org/10.3390/children6040054
APA StyleRao, P. S. (2019). Management of Congenital Heart Disease: State of the Art—Part II—Cyanotic Heart Defects. Children, 6(4), 54. https://doi.org/10.3390/children6040054