Abstract
Neuroblastoma is a tumor with great clinical heterogeneity. Patients in North America are risk-stratified using a number of features including age at diagnosis, disease stage, tumor histology, MYCN status (amplified versus nonamplified), and tumor cell ploidy. In this paper, we review the evidence for utilizing these features in the risk classification of neuroblastic tumors. Additionally, we review the clinical and biologic criteria used by various cooperative groups to define low, intermediate, and high-risk disease populations in clinical trials, highlighting the differences in risk classification internationally. Finally, we discuss the development of the International Neuroblastoma Risk Group classification system, designed to begin worldwide standardization of neuroblastoma pretreatment risk classification and allow comparison of clinical trials conducted through different cooperative groups.
1. Introduction
One of the hallmarks of neuroblastoma is its clinical heterogeneity. While children with low-risk disease may be observed or undergo surgery and those with intermediate-risk disease may receive chemotherapy and undergo surgical resection, those with high-risk disease receive intensive, multimodality therapy that includes chemotherapy, surgery, myeloablative chemotherapy with autologous stem cell rescue, radiation, and immunotherapy with an anti-GD2 antibody [1,2,3]. Thus, it is critical to appropriately risk-stratify patients to ensure that they receive the optimal treatment regimen. Over the last several decades, numerous clinical and biologic factors have been incorporated into the risk classification system for neuroblastoma to categorize patients as having low, intermediate, or high-risk disease. Figure 1 illustrates major events contributing to modern risk classification and will be discussed in this review. Efforts have been made to uniformly define risk groups across cooperative groups through the International Neuroblastoma Risk Group (INRG) classification system [4].
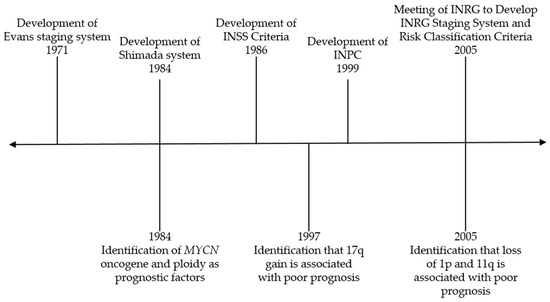
Figure 1.
Timeline of major events contributing to risk classification for neuroblastoma. INSS—International Neuroblastoma Staging System; INPC—International Neuroblastoma Pathology Classification; INRG—International Neuroblastoma Risk Group.
2. Risk Classification: Key Clinical and Biologic Factors
The Children’s Oncology Group (COG) has traditionally used the following factors to stratify patient risk. (1) Age at diagnosis, (2) stage to define extent of disease by International Neuroblastoma Staging System (INSS), (3) tumor histology using International Neuroblastoma Pathology Classification (INPC) criteria, (4) MYCN status, and (5) DNA index or tumor cell ploidy.
2.1. Age at Diagnosis
Older age has been prognostic of poor outcome in neuroblastoma since the 1970s. Earlier studies demonstrated that patients over 12 months of age at diagnosis had inferior outcomes [5]. Upon retrospective review of 3666 patients treated on Pediatric Oncology Group (POG) or Children’s Cancer Group (CCG) studies (POG and CCG being predecessors to the COG), London et al. [6] demonstrated that the prognostic significance of age on outcome is continuous in nature and suggested that an age cutoff between 12 and 18 months, rather than 365 days, could be utilized for clinical risk stratification. The CCG and POG groups demonstrated favorable prognosis in children 12–18 months of age with metastatic, MYCN nonamplified disease and children with metastatic, MYCN nonamplified, hyperdiploid disease, compared to older children thus supporting a prognostic age cut-off of 18 months and potentially less intensive therapy for those 12–18 months with other favorable prognostic markers [7,8]. Metastatic disease in children under 18 months of age is not associated with the poor outcomes that result in older patients.
During development of the INRG risk classification system (detailed later in this review), an analysis of non-COG patients in the INRG cohort supported an age cutoff between 15 and 19 months. While it was again acknowledged that the prognostic significance of age is continuous, an age cutoff of 18 months (547 days) was chosen by the INRG task force for clinical purposes. However, for a subgroup of patients with diploid, MYCN nonamplified tumors and distant metastatic disease, the INRG task force recommended an age cutoff of 12 months (365 days) for the INRG classification system [4]. Additional analyses showed that age retained prognostic significance in more modern cohorts treated with intensified therapy and supported an older age cutoff of greater than 18 months at diagnosis as a risk criterion [4,9].
2.2. Disease Stage
In 1971, Evans et al. [10] published the Evans Staging System based on extent of disease including a IV-S category, recognizing that there is a cohort of patients with metastatic disease limited to the skin, liver, and bone marrow, who have superior outcomes. Subsequently in 1986, an international group convened to develop a surgical staging system to aid in comparison of outcomes and therapies between countries, as various staging systems were being used worldwide [11]. The International Neuroblastoma Staging System (INSS) was developed taking into account degree of tumor resection, presence of ipsilateral or contralateral lymph nodes involvement, tumor infiltration across the midline of the body, and separation of patients with INSS stage 4S (infants with specific metastatic disease pattern including only liver, skin, and bone marrow) from other children with metastatic disease (INSS stage 4) to harmonize staging across groups, see Table 1 for definitions of stages [11]. Survival among patients with INSS stage 4 disease was significantly worse than those with stages 1, 2, 3, or 4S disease [12]. Staging was again addressed in 2005 during a meeting of the International Neuroblastoma Risk Group (INRG) task force and a system utilizing image-defined risk factors in place of degree of surgical resection was developed (INRG Staging System, or INRGSS) [4,13,14]. This system would allow for pretreatment staging rather than postsurgical staging. This is important in patients with localized disease who do not require surgical resection such as those with perinatally diagnosed disease. These patients could not be properly staged with INSS as surgical resection is required to define stage 1 or 2 disease. European groups had already adopted the image-defined risk factors to eliminate surgical style or skill from the evaluation of risk. In addition to the use of radiographic features in place of surgical resection, several other changes were included in the INRG system including elimination of lymph node assessment and midline nature of tumors and use of 18 months instead of 12 months to define MS disease. The INRGSS (Table 2) is now being incorporated into new protocols developed through the Children’s Oncology Group.

Table 1.
International Neuroblastoma Staging System (INSS).
Table 2.
International Neuroblastoma Risk Group Staging Sysem (INRGSS).
The methods used to perform the staging evaluation have changed over time as well. Bone scans were previously used to asses for metastatic disease to the bone. This has been replaced by the use of I-123-metaiodobenzylguanidine (I-123-MIBG) scans [2]. I-123-MIBG is a radiotracer that is taken up by the norepinephrine transporter. The majority of neuroblastoma cells take up this tracer, allowing for a more specific mechanism to detect metastatic disease. In patients with MIBG non-avid tumors, fluorodexoyglucose (FDG)-positron emission topography (PET) is used. CT and/or MRI is used to evaluate for soft tissue disease. Bilateral bone marrow aspirate and biopsy is used to evaluate for the presence of metastatic disease in the bone marrow.
2.3. Tumor Histology
Shimada et al. [15] developed the first histology grading system in 1984 to classify neuroblastic tumors by histologic features. Features including degree of stroma present, grade of differentiation, mitosis-karyorrhexis index (MKI) [16], presence of nodules, and age were utilized to define groups with either favorable or unfavorable prognosis. The International Neuroblastoma Pathology Classification (INPC) was then developed in 1999 to update these histologic factors impacting prognosis [17,18]. INPC incorporates multiple factors consisting of diagnostic category (accounting for quantity of Schwannian stromal development and grade of tumor differentiation), MKI, and age to ultimately define tumor histology as favorable versus unfavorable (Table 3). Diagnostic categories include ganglioneuroma (Schwannian stroma-dominant) with mature or maturing subtypes; ganglioneuroblastoma, intermixed (Schwannian stroma-rich); ganglioneuroblastoma, nodular (composite Schwannian stroma-rich/stroma-dominant and stroma-poor); and neuroblastoma (Schwannian stroma-poor) with undifferentiated, poorly differentiated, and differentiating subtypes. This provides greater subdivision than the Shimada classification that only defined stroma-poor and stroma-rich. Grade was further subdivided as well to include three categories (differentiating, poorly differentiated, and undifferentiated) instead of the two included in the Shimada system (differentiating and undifferentiated). MKI reflects the degree of cell replication seen in a high-power microscope field and categorized as low (<2% or <100/5000 mitotic and karyorrhectic cells), intermediate (<2%–4% or <100–200/5000 mitotic and karyorrhectic cells), and high (>4% or >200/5000 mitotic and karyorrhectic cells). Age is not incorporated into prognostic grouping for ganglioneuroblastoma intermixed and ganglioneuroma, which fall within the favorable histology group or ganglioneuroblastoma, nodular which is considered an unfavorable histology. Age and MKI, however, impact prognostic grouping within tumors categorized as neuroblastoma. Neuroblastoma tumors with favorable histology follow a framework of age-linked maturation and include poorly differentiated (age <1.5 years) to differentiating (age <5 years) neuroblastoma and should have low (age <5 years) or up to intermediate (age <1.5 years) MKI. In contrast, tumors with unfavorable histology demonstrate features that suggest aggressive growth and have immature histologies for the age of the patient. Within neuroblastoma tumors, the unfavorable histology group includes undifferentiated histology (in any age) or poorly differentiated subtype (age ≥1.5 years) or any subtype (age ≥5 years). Further, those with high MKI (in any age) or intermediate (age ≥1.5 years) qualify as having unfavorable histology [17]. Age is included in INPC and contributes to the prognostic significance of the histologic groups. Thus, within the risk classification system, age is weighted twice—both within INPC and independently.
Table 3.
International Neuroblastoma Pathology Classificaiton (INPC) histology definitions.
2.4. MYCN Status
Several genetic factors predictive of outcome in neuroblastoma have been identified, beginning in the 1980s with MYCN (or N-myc) status and DNA index, or tumor cell ploidy. MYCN is an oncogene located on the short arm of chromosome 2. Amplification of MYCN was identified in neuroblastoma cell lines and then in untreated tumors. Brodeur et al. [19] demonstrated an association between amplification of MYCN and higher stage tumors in early analyses. Seeger et al. [20] identified that MYCN amplification was associated with shorter progression free survival in all stages of disease. MYCN was the first clinically relevant genetic biomarker in cancer [21]. Approximately 20% of primary neuroblastoma tumors demonstrate MYCN amplification. Biologically, MYCN is involved in many processes leading to aggressive disease including migration/metastases, cell survival, apoptosis, proliferation, pluripotency, self-renewal, angiogenesis, and blocking cell cycle arrest, immune surveillance, and differentiation [22]. MYCN amplification remains one of the strongest predictors of high-risk disease [23]. Currently, COG uses fluorescence in situ hybridization to determine MYCN status [24]. Samples with a 4-fold increase in signal compared to a centromeric reference are considered amplified. Those with 2–3-fold increase have MYCN gain. Other methods can be used to determine amplification as well including polymerase chain reaction, array-based comparative genomic hybridization, and multiplex ligation-dependent probe amplification [25].
2.5. Tumor Cell Ploidy
Tumor ploidy was identified as a prognostic factor around the time the role of MYCN amplification in neuroblastoma was identified. Tumors with DNA index less than or equal to 1 have poorer outcomes than hyperdiploid tumors [26,27]. In 1984, Look et al. [27] showed that higher DNA index was associated with better response to therapy in infants with unresectable tumors. Kaneko et al. then showed the association between near triploid tumors and favorable disease. Diploid and near-tetraploid tumors were associated with more aggressive disease [28]. Patients between 12 and 18 months with MYCN nonamplified, hyperdiploid tumors were found to have superior outcomes than the same group with diploid tumors [8].
2.6. Chromosomal Aberrations
Although MYCN and tumor cell ploidy are the two biologic markers that have been classically utilized by the COG to assign risk groups, chromosomal aberrations have been identified and used by different groups. Certain segmental chromosome aberrations were found to be recurrent and associated individually with inferior outcomes. Loss of heterozygosity at 1p and 11q and 17q gain have been found to be predictive of poorer survival in patients with neuroblastoma [29,30]. COG utilized 1p and 11q status for treatment assignment on the intermediate risk study ANBL0531 (NCT NCT00499616), such that loss of heterozygosity (LOH) at 1p36 and/or 11q23 precluded patients from receiving therapy reduction [31]. Because 1p loss and 17q gain are associated with MYCN amplification—but 11q loss is not—11q aberration was incorporated into the INRG risk classification as a predictor of poor outcome that is independent of MYCN status [4,29,30].
The presence of whole versus partial chromosomal aberrations has also been found to correlate with clinical behavior of the tumor. Schleiermacher and colleagues evaluated types of chromosomal changes and their impact on outcomes in patients with MYCN nonamplified tumors [32]. They identified that patients with whole chromosome gains and losses had significantly better outcomes than patients with partial or segmental chromosome aberrations. The presence of segmental chromosome aberrations has been incorporated as an unfavorable genomic feature in COG ANBL1232 (NCT02176967), an ongoing study that is using response and biologic criteria to decrease therapy in a select cohort with localized non-high-risk disease. A favorable genomic profile, including absence of any segmental chromosome aberration (1p, 3p, 4p, or 11q loss or 1q, 2p, or 17q gain) and one or more whole chromosome gains, and favorable histology allow for observation, rather than initiating chemotherapy, for patients with L2 tumors. The ability to observe patients with large tumors with favorable biology remains a study question.
2.7. Other Lab Findings
Other labs including ferritin and lactic dehydrogenase (LDH) have been found to prognostic as well. Elevated ferritin has been found to be correlated with worse outcome in patients with Evans stage III and IV disease [33]. It is also elevated more often in patients with Evans stage IV disease in comparison to those with stage IV-S disease [34]. Elevated LDH has similarly been correlated with poorer prognosis [35]. These markers were some of the earliest indicators of poor prognosis. Currently, these markers are not routinely used in evaluation of patients with neuroblastoma as they are not specific, but they may be useful in combination with other more specific prognostic features.
3. Variation in Risk Classification Among Cooperative Groups and the Development of the INRG Risk Classification System
Cooperative groups have used different sets of prognostic factors to define risk groups, making it difficult to compare treatments between studies. Several studies have been completed around the world in patients with low, intermediate, and high-risk disease using various inclusion criteria (Table 4). Between 1998 and 2004, the COG in North America, New Zealand, and Australia completed a study in low risk patients (COG P9641) using the INSS stage, age, MYCN status, INPC, and tumor cell ploidy to determine eligibility with a goal of showing that surgery alone was sufficient in low risk patients with stage 2 disease [36]. During the same time, the International Society of Pediatric Oncology Europe Neuroblastoma (SIOPEN) group completed a low risk study, SIOPEN LNESG1, that required patients to have surgically resectable localized tumors that were MYCN nonamplified [37]. This study also wanted to show that all patients with localized resectable disease, except those with stage 2 MYCN amplified tumors, could be successfully treated with surgery alone. The German Society of Pediatric Oncology and Hematology (GPOH) also conducted trials (GPOH NB95-S and NB97) during this period evaluating only infants less than 12 months of age with localized MYCN nonamplified tumors and looking at the ability to treat without cytotoxic chemotherapy [38]. In these low risk studies all completed between 1995 and 2004, only COG utilized histology and tumor cell ploidy. Additionally, only COG included patients with INSS stage 4S disease with this group of low risk patients. COG did not exclude patients with MYCN amplified tumors in the case of stage 1 tumors or infants under 12 months with stage 2 tumors, or older patients with stage 2 tumors with favorable histology by INPC. In the low risk patients, all groups looked at eliminating chemotherapy unless a patient was either symptomatic or had high risk features such as MYCN amplification.
Table 4.
Studies performed and the factors used for risk stratification.
Several intermediate risk studies were completed between 1997 and 2006 as well. COG A3961, a study conducted through COG utilized certain factors similar to the low risk study including age, MYCN status, INSS stage, INPC, and tumor cell ploidy to evaluate the ability to maintain excellent outcomes in patients with intermediate risk disease with reduced intensity chemotherapy [39]. Infants under 365 days with MYCN nonamplified, INSS stage 3 and 4 disease or 4s disease with either unfavorable histology per INPC or diploid tumors were included. Older patients were included with MYCN nonamplified INSS stage 3 disease with favorable histology by INPC. In Europe, SIOPEN completed several studies to evaluate these intermediate risk patients. SIOPEN 99.1 enrolled infants less than 12 months with localized, unresectable MYCN nonamplified tumors and the ability to utilize low dose chemotherapy and surgery alone [40]. SIOPEN 99.2 enrolled infants with MYCN nonamplified, metastatic disease that did not have metastases to the bone, central nervous system, lung, or pleura to evaluate the ability to only give chemotherapy for symptomatic patients [41]. In parallel, SIOPEN 99.3 enrolled infants with MYCN non-amplified disease who were not eligible for 99.2. This study evaluated the ability to give low dose chemotherapy in this cohort. SIOPEN EUNS evaluated patients older than 12 months with MYCN nonamplified localized, unresectable tumors and the ability to give low dose chemotherapy in these patients [42]. Similar to the low risk studies, only COG included tumor cell ploidy and histology. While there is likely overlap between the SIOPEN and COG intermediate risk cohorts, it is unclear if the populations are truly comparable due to the differences in criteria used to determine risk. Each of these intermediate risk studies evaluated the ability to give low dose chemotherapy without sacrificing the good outcomes expected in this group.
Among high-risk trials, inclusion criteria also varied among cooperative groups. In the United Kingdom, the Children’s Cancer and Leukemia Group completed CCLG-NB-1990-11 enrolling all patients over 12 months with metastatic disease to evaluate if a shortened interval between chemotherapy cycles will lead to improved survival [43]. SIOPEN enrolled patients with Evans stage 3 or 4 disease responsive to induction chemotherapy on ENSG1 to evaluate the role of high dose chemotherapy in this population [44]. The CCG completed CCG 3891 to evaluate the role of autologous stem cell transplantation versus chemotherapy for consolidation followed by 13-cis-retionoic acid for the treatment of high-risk neuroblastoma. This study utilized age, INSS stage, MYCN status, INPC histology, and ferritin in risk classification and study inclusion criteria [45]. They included patients over 12 months with INSS stage 4 disease, INSS stage 3 disease with either MYCN amplification, elevated ferritin, or unfavorable histology, INSS stage 2 disease with MYCN amplification, or INSS stage 1 or 2 disease that recurs after initial resection. GPOH completed a study (GPOH NB97) looking at patients over 12 months with INSS stage 4 disease and patients with MYCN amplified tumors of other stages comparing outcomes with autologous stem cell transplant versus maintenance chemotherapy [46]. After the CCG study, COG completed COG A3973 using age, MYCN status, INSS stage, and INPC including patients under 12 months with MYCN amplified INSS stage 3, 4, or 4s disease, patients over 12 months with INSS stage 4 or stage 3 disease with MYCN amplification or unfavorable histology or stage 2 disease with unfavorable histology and MYCN amplification, as well as patients who originally had low stage disease that returned with metastatic disease to determine whether purging neuroblastoma cells from autologous stem cells improves outcomes [47]. Finally, SIOPEN completed a SIOPEN HR-NBL1 evaluating patients over 12 months with INSS stage 4 disease and patients with stage 2, 3, or 4 disease with MYCN amplification to evaluate the need for IL-2 in addition to dinutuximab during maintenance therapy [48]. In general, CCG and then COG included additional features beyond what the various European groups utilized. All groups incorporated stage, age, and MYCN status, while CCG and COG also utilized ferritin and histology via INPC as in the lower risk groups.
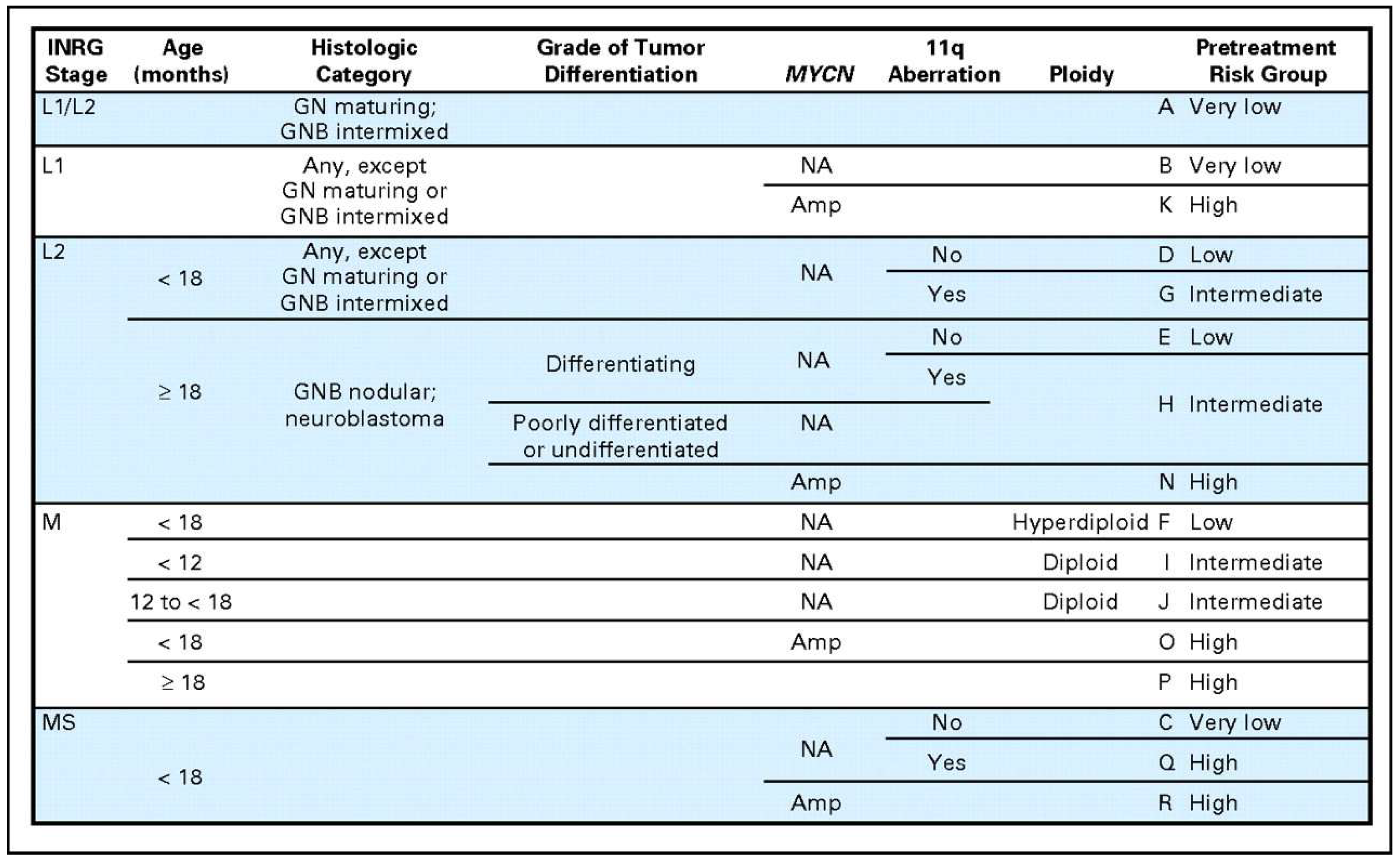
In order to address this problem of non-uniformly defined risk groups, a taskforce was assembled to develop the International Neuroblastoma Risk Group (INRG) classification system [4]. First, a new staging system was established that was based on image-defined risk factors of the original tumor rather than degree of surgical resection and allows for pretreatment risk stratification [13,49]. With the new staging system defined, a new risk stratification system was created as well. This was done by analyzing data collected on 8800 patients diagnosed between 1990 and 2002 in North America, Australia, Europe, and Japan. From this cohort, the most predictive factors were identified including INRG stage, age, histologic category, grade of differentiation, MYCN status, 11q aberration, and tumor cell ploidy. These features were used to define 17 cohorts that were categorized as very low, low, intermediate, or high risk. Patients categorized as very low risk had 5-year EFS of >85%, low from >75% to ≤85%, intermediate from >50 to <75%, and high <50% [4]. These groups were put into a risk stratification table that could be adopted to standardize the risk stratification of neuroblastoma patients (Figure 2).
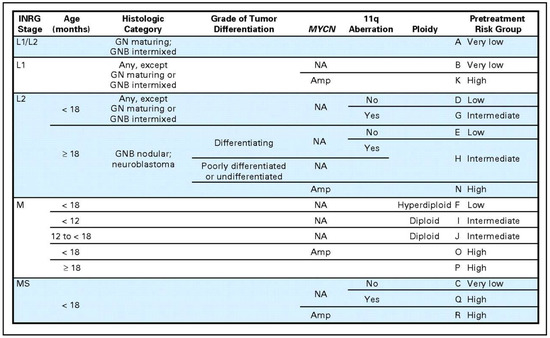
Figure 2.
International Neuroblastoma Risk Group (INRG) classification system [4]. Reprinted with permission from American Society of Clinical Oncology © 2009, The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report; published by American Society of Clinical Oncology, 2009.
As more data becomes available, the INRG risk classification system will continue to evolve. With this, it will be possible to better identify and define very low-risk or very high risk, or “ultra-high-risk” subgroups and therapy can be tailored accordingly. Morgenstern et al. [50] was able to identify a very high-risk subset of SIOPEN patients from the SIOPEN HR-NBL1 study using age, LDH, and number of metastatic sites. Saarinen-Pihkala et al. [51] identified an ultra-high-risk cohort as well using MYCN amplification and presence of bone metastases at diagnosis. Using the INRG database, a larger group of patients could be used to further investigate the definition of very or ultra-high-risk. Also with ongoing research into better ways to refine risk-stratificaiton, there will be novel factors added to the risk stratification schema. For instance, TERT rearrangements and ATRX mutations lead to alterations in telomere maintenance and are associated with more aggressive disease [52,53]. Circulating tumor DNA is another area of active research being done to identify less invasive way to characterize and monitor tumors [54]. These or other similar findings can be added to the risk stratification schema to better subdivide patients and assign the most appropriate treatment regimens.
4. Conclusions
This INRG risk stratification system has not yet been adopted by all of the cooperative groups, but it represents the first step in being able to perform cross-cooperative group studies in order to optimize therapy. The INRG has already served to increase collaboration and data sharing between cooperative groups. Additionally, as technology and our understanding of the underlying biology of neuroblastoma improve, genomic features beyond MYCN amplification, ploidy, and segmental chromosome aberrations will likely be incorporated in risk classification. Additional refinement of risk classification will allow patients to receive the ideal therapy for his/her tumor in order to optimize outcomes but limit exposure to treatment-related toxicities.
Author Contributions
Conceptualization, E.S. and A.V.D.; Writing—Original Draft Preparation, E.S. and A.V.D.; Writing—Review and Editing, E.S. and A.V.D.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, M.M.; London, W.B.; Ambros, F.P.; Nakagawara, A.; Berhold, F.; Schleirmacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackal, C.L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Prim. 2016, 2, 16078. [Google Scholar] [CrossRef] [PubMed]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.L.; Chon, S.L.; Hogarty, M.; COG Neuroblastoma Committee. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr. Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Moniclar, T.; Amros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iheara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Breslow, N.; McCann, B. Statistical estimation of prognosis for children with neuroblastoma. Cancer Res. 1971, 31, 2098–2103. [Google Scholar] [PubMed]
- London, W.B.; Castleberry, R.P.; Matthay, K.K.; Look, A.T.; Seeger, R.C.; Shimada, S.; Thorner, P.; Brodeur, G.; Maris, J.M.; Reznolds, C.P.; et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J. Clin. Oncol. 2005, 23, 6459–6465. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.L.; Lal, A.; Seeger, R.C.; Maris, J.M.; Shimada, H.; O’Leary, M.; Gerbing, R.B.; Matthay, K.K. Favorable prognosis for patients 12 to 18 months of age with stage 4 nonamplified MYCN neuroblastoma: A Children’s Cancer Group Study. J. Clin. Oncol. 2005, 23, 6474–6480. [Google Scholar] [CrossRef] [PubMed]
- George, R.E.; London, W.B.; Cohn, S.L.; Maris, J.M.; Kretschmar, C.; Diller, L.; Brodeur, G.M.; Castleberry, R.P.; Look, A.T.; et al. Hyperdiploidy plus nonamplified MYCN confers a favorable prognosis in children 12 to 18 months old with disseminated neuroblastoma: A Pediatric Oncology Group study. J. Clin. Oncol. 2005, 23, 6466–6473. [Google Scholar] [CrossRef]
- Moroz, V.; Machin, D.; Faldum, A.; Hero, B.; Iehara, T.; Mosseri, V.; Ladenstein, R.; De Bernardi, B.; Rubie, H.; Berthold, F.; et al. Changes over three decades in outcome and the prognostic influence of age-at-diagnosis in young patients with neuroblastoma: A report from the International Neuroblastoma Risk Group Project. Eur. J. Cancer 2011, 47, 561–571. [Google Scholar] [CrossRef]
- Evans, A.E.; D’Angio, G.J.; Randolph, J. A proposed staging for children with neuroblastoma. Children’s cancer study group A. Cancer 1971, 27, 374–378. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Barrett, A. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J. Clin. Oncol. 1988, 6, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Iehara, T.; Tsuchida, Y.; Kaneko, M.; Hata, J.; Naito, H.; Iwafuchi, M.; Ohnuma, N.; Mugishima, H.; Toyoda, Y.; et al. Experience with International Neuroblastoma Staging System and Pathology Classification. Br. J. Cancer 2002, 86, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Monclair, T.; Mosseri, V.; Cecchetto, G.; De Bernardi, B.; Michon, J.; Holmes, K. Influence of image-defined risk factors on the outcome of patients with localised neuroblastoma. A report from the LNESG1 study of the European International Society of Paediatric Oncology Neuroblastoma Group. Pediatr. Blood Cancer 2015, 62, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Chatten, J.; Newton, W.A.; Sachs, N.; Hamoudi, A.B.; Chiba, T.; Marsden, H.B.; Misugi, K. Histopathologic prognostic factors in neuroblastic tumors: Definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J. Natl. Cancer Inst. 1984, 73, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Teshiba, R.; Kawano, S.; Wang, L.L.; He, L.; Naranjo, A.; London, W.B.; Seeger, R.C.; Gastier-Foster, J.M.; Look, A.T.; Hogarty, M.D.; et al. Age-dependent prognostic effect by Mitosis-Karyorrhexis Index in neuroblastoma: A report from the Children’s Oncology Group. Pediatr. Dev. Pathol. 2014, 17, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Ambros, I.M.; Dehner, L.P.; Hata, J.; Joshi, V.V.; Roald, B. Terminology and morphologic criteria of neuroblastic tumors: Recommendations by the International Neuroblastoma Pathology Committee. Cancer 1999, 86, 349–363. [Google Scholar] [CrossRef]
- Shimada, H.; Ambros, I.M.; Dehner, L.P.; Joshi, V.V.; Roald, B.; Stram, D.O.; Gerbing, R.B.; Lukens, J.N.; Matthay, K.K.; Castleberry, R.P.; et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 1999, 86, 364–372. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Seeger, R.C.; Brodeur, G.M.; Sather, H. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Cao, Y.; Jin, Y.; Yu, J.; Wang, J.; Yan, J.; Zhao, Q. Research progress of neuroblastoma related gene variations. Oncotarget 2017, 8, 18444–18455. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; Cohn, S.L. Genetic discoveries and treatment advances in neuroblastoma. Curr. Opin. Pediatr. 2016, 28, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Gastier-Foster, J.M.; Mann, M. Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children’s Oncology Group. Cancer 2017, 123, 4224–4235. [Google Scholar] [CrossRef] [PubMed]
- Ambros, P.F.; Ambros, I.M.; Brodeur, G.M.; Khan, J.; Haber, M.; Pearson, A.D.J.; Choln, S.L.; London, W.B.; Maris, J.M.; Spitz, R.; et al. International consensus for neuroblastoma molecular diagnostics: Report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br. J. Cancer 2009, 100, 1471–1482. [Google Scholar] [CrossRef] [PubMed]
- Oppedal, B.R.; Storm-Mathisen, I.; Lie, S.O.; Brandtzaeg, P. Prognostic factors in neuroblastoma. Clinical, histopathologic, and immunohistochemical features and DNA ploidy in relation to prognosis. Cancer 1988, 62, 772–780. [Google Scholar] [CrossRef]
- Look, A.T.; Hayes, F.A.; Nitschke, R.; McWilliams, N.B.; Green, A.A. Cellular DNA content as a predictor of response to chemotherapy in infants with unresectable neuroblastoma. N. Engl. J. Med. 1984, 311, 231–235. [Google Scholar] [CrossRef]
- Kaneko, Y.; Kanda, N.; Maseki, N.; Sakurai, M.; Takeda, T.; Tsuchida, Y.; Okabe, I. Different karyotypic patterns in early and advanced stage neuroblastomas. Cancer Res. 1987, 47, 311–318. [Google Scholar]
- Lastowska, M.; Cotterill, S.; Pearson, A.D.; Roberts, P.; McGuckin, A.; Lewis, I.; Bown, N. Gain of chromosome arm 17q predicts unfavourable outcome in neuroblastoma patients. U.K. Children’s Cancer Study Group and the U.K. Cancer Cytogenetics Group. Eur. J. Cancer 1997, 33, 1627–1633. [Google Scholar] [CrossRef]
- Attiyeh, E.F.; London, W.B.; Mosse, Y.P.; Wang, Q.; Winter, C.; Khazi, D.; Seeger, R.C.; Look, A.T.; Shimada, H.; Brouder, G.M.; et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N. Engl. J. Med. 2005, 353, 2243–2253. [Google Scholar] [CrossRef]
- Twist, C.; London, W.B.; Naranjo, A.; Smidt, L.M. Maintaining outstanding outcomes using response- and biology-based therapy for intermediate-risk neuroblastoma: A report from the Children’s Oncology Group study ANBL0531. J. Clin. Oncol. 2014, 32 (Suppl. 15), 10006. [Google Scholar] [CrossRef]
- Schleiermacher, G.; Michon, J.; Huon, I.; Plantaz, D.; Riberio, A.; Mosseri, V.; Munzer, C.; Thomas, C.; Rubie, H.; Countrier, J.; et al. Chromosomal CGH identifies patients with a higher risk of relapse in neuroblastoma without MYCN amplification. Br. J. Cancer 2007, 97, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Hann, H.W.; Evans, A.E.; Siegel, S.E.; Wong, K.Y.; Dalton, A.; Salther, H.; Hammond, D.; Seeger, R.C. Prognostic importance of serum ferritin in patients with Stages III and IV neuroblastoma: The Childrens Cancer Study Group experience. Cancer Res. 1985, 45, 2843–2848. [Google Scholar] [PubMed]
- Hann, H.W.; Evans, A.E.; Cohen, I.J.; Leitmeyer, J.E. Biologic differences between neuroblastoma stages IV-S. and IV. Measurement of serum ferritin and E-rosette inhibition in 30 children. N. Engl. J. Med. 1981, 305, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.J.; Altman, A.J.; Frantz, C.N. Serum lactic dehydrogenase, an indicator of tumor activity in neuroblastoma. J. Pediatr. 1980, 97, 89–91. [Google Scholar] [CrossRef]
- Strother, D.R.; London, W.B.; Schmidt, M.L.; Brouder, G.M.; Shimada, H.; Torner, P.; Collins, M.H.; Tagge, E.; Murray, K.; Cholin, L.S.; et al. Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: Results of Children’s Oncology Group study P9641. J. Clin. Oncol. 2012, 30, 1842–1848. [Google Scholar] [CrossRef] [PubMed]
- De Bernardi, B.; Mosseri, V.; Rubie, H.; Castel, V.; Conte, M.; Bruzzi, P.; Michon, J.; Laureyes, G.; Pearson, A.D.J.; Ambros, P.F.; et al. Treatment of localised resectable neuroblastoma. Results of the LNESG1 study by the SIOP Europe Neuroblastoma Group. Br. J. Cancer 2008, 99, 1027–1033. [Google Scholar] [CrossRef]
- Hero, B.; Simon, T.; Spitz, R. Localized infant neuroblastomas often show spontaneous regression: Results of the prospective trials NB95-S. and NB97. J. Clin. Oncol. 2008, 26, 1504–1510. [Google Scholar] [CrossRef]
- Baker, D.L.; Schmidt, M.L.; Cohn, S.L.; London, W.B.; Maris, J.M.; Stram, D.; Seeger, R.C.; Buxton, A.; Stram, D.; Sandler, A.; et al. Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N. Engl. J. Med. 2010, 363, 1313–1323. [Google Scholar] [CrossRef]
- Rubie, H.; De Bernardi, B.; Gerrard, M.; Canete, A.; Ladenistein, R.; Couturier, J.; Ambros, P.; Munzer, C.; Pearson, A.D.; Garaventa, A.; et al. Excellent outcome with reduced treatment in infants with nonmetastatic and unresectable neuroblastoma without MYCN amplification: Results of the prospective INES 99.1. J. Clin. Oncol. 2011, 29, 449–455. [Google Scholar] [CrossRef]
- De Bernardi, B.; Gerrard, M.; Boni, L. Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J. Clin. Oncol. 2009, 27, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Kohler, J.A.; Rubie, H.; Castel, V.; Beiske, K.; Holmes, K.; Gambini, C.; Casale, F.; Munzer, C.; Ermino, G.; Parodi, S.; et al. Treatment of children over the age of one year with unresectable localised neuroblastoma without MYCN amplification: Results of the SIOPEN study. Eur. J. Cancer 2013, 49, 3671–3679. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.D.; Pinkerton, C.R.; Lewis, I.J.; Imeson, J.; Ellershaw, C.; Machin, D. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: A randomised trial. Lancet Oncol. 2008, 9, 247–256. [Google Scholar] [CrossRef]
- Pritchard, J.; Cotterill, S.J.; Germond, S.M.; Imeson, J.; de Kraker, J.; Jones, D.R. High dose melphalan in the treatment of advanced neuroblastoma: Results of a randomised trial (ENSG-1) by the European Neuroblastoma Study Group. Pediatr. Blood Cancer 2005, 44, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Reynolds, C.P.; Seeger, R.C.; Shimada, H.; Adkis, E.S.; Haas-Kogan, D.; Gerbing, R.D.; London, W.B.; Vilablanca, J.G. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: A children’s oncology group study. J. Clin. Oncol. 2009, 27, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Berthold, F.; Boos, J.; Burdach, S.; Errtman, R.; Henze, G.; Klingebel, K.; Kremens, B.; Simon, T.; Hero, B.; Klingebiel, T.; et al. Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: A randomised controlled trial. Lancet Oncol. 2005, 6, 649–658. [Google Scholar] [CrossRef]
- Kreissman, S.G.; Seeger, R.C.; Matthay, K.K. Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 999–1008. [Google Scholar] [CrossRef]
- Ladenstein, R.L.; Poetschger, U.; Luksch, R. Busulphan-melphalan as a myeloablative therapy (MAT) for high-risk neuroblastoma: Results from the HR-NBL1/SIOPEN trial. J. Clin. Oncol. 2011, 29 (Suppl. 18), 2. [Google Scholar] [CrossRef]
- Avanzini, S.; Pio, L.; Erminio, G.; Granata, C.; Holmes, K.; Gambat, M.; Buffa, P.; Castel, V.; Pistorio, A.; Mattioli, G.; Sarnacki, S.; et al. Image-defined risk factors in unresectable neuroblastoma: SIOPEN study on incidence, chemotherapy-induced variation, and impact on surgical outcomes. Pediatr. Blood Cancer. 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, D.A.; Potschger, U.; Moreno, L. Risk stratification of high-risk metastatic neuroblastoma: A report from the HR-NBL-1/SIOPEN study. Pediatr. Blood Cancer 2018, 65, e27363. [Google Scholar] [CrossRef] [PubMed]
- Saarinen-Pihkala, U.M.; Jahnukainen, K.; Wikstrom, S.; Kouvusalo, A.; Karikoski, R.; Sariola, H.; Hovi, L. Ultrahigh-risk group within the high-risk neuroblastoma category. J. Pediatr. Hematol. Oncol. 2013, 35, e254–e259. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Hertwig, F.; Roels, F.; Menor, R.; Tomas, R.K.; Herrmann, C.; Eggert, A.; Peng, Z.; Zhao, C.; Bell, E.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.F.; Zhao, Q. TERT-mediated and ATRX-mediated Telomere Maintenance and Neuroblastoma. J. Pediatr. Hematol. Oncol. 2018, 40, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chicard, M.; Boyault, S.; Colmet Daage, L.; Bellini, A.; Clement, N.; Carrere, M.; Gambart, M.; Bernard, V.; Iacono, I.; Michon, J.; et al. Genomic Copy Number Profiling Using Circulating Free Tumor DNA Highlights Heterogeneity in Neuroblastoma. Clin. Cancer Res. 2016, 22, 5564–5573. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).