
Cardiac Hypertrophy: A Comprehensive Review from Prenatal Life to Young Adulthood

 , , , and
, , , and  on behalf of the Working Group on Congenital Heart Disease Cardiovascular Prevention in Paediatric Age of the Italian Society of Cardiology Sic
on behalf of the Working Group on Congenital Heart Disease Cardiovascular Prevention in Paediatric Age of the Italian Society of Cardiology Sic
Abstract
1. Introduction
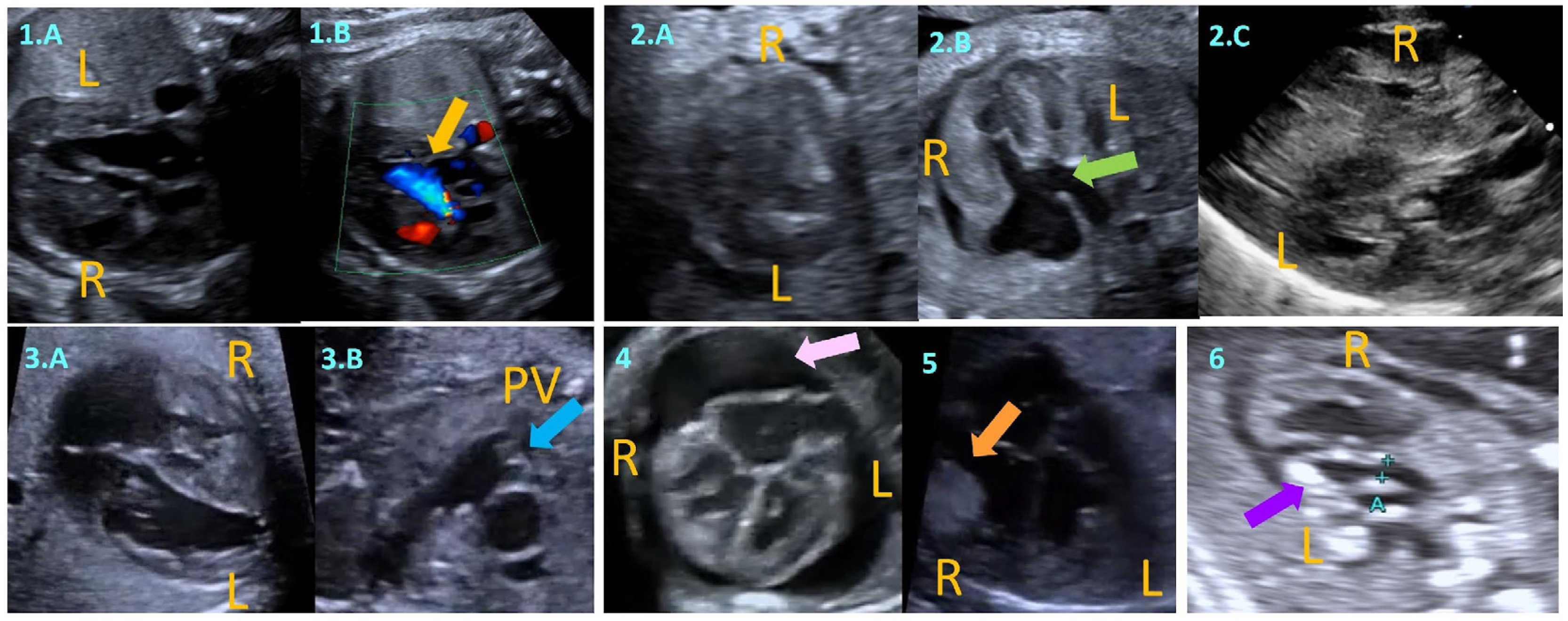
2. Fetal Myocardial Hypertrophy
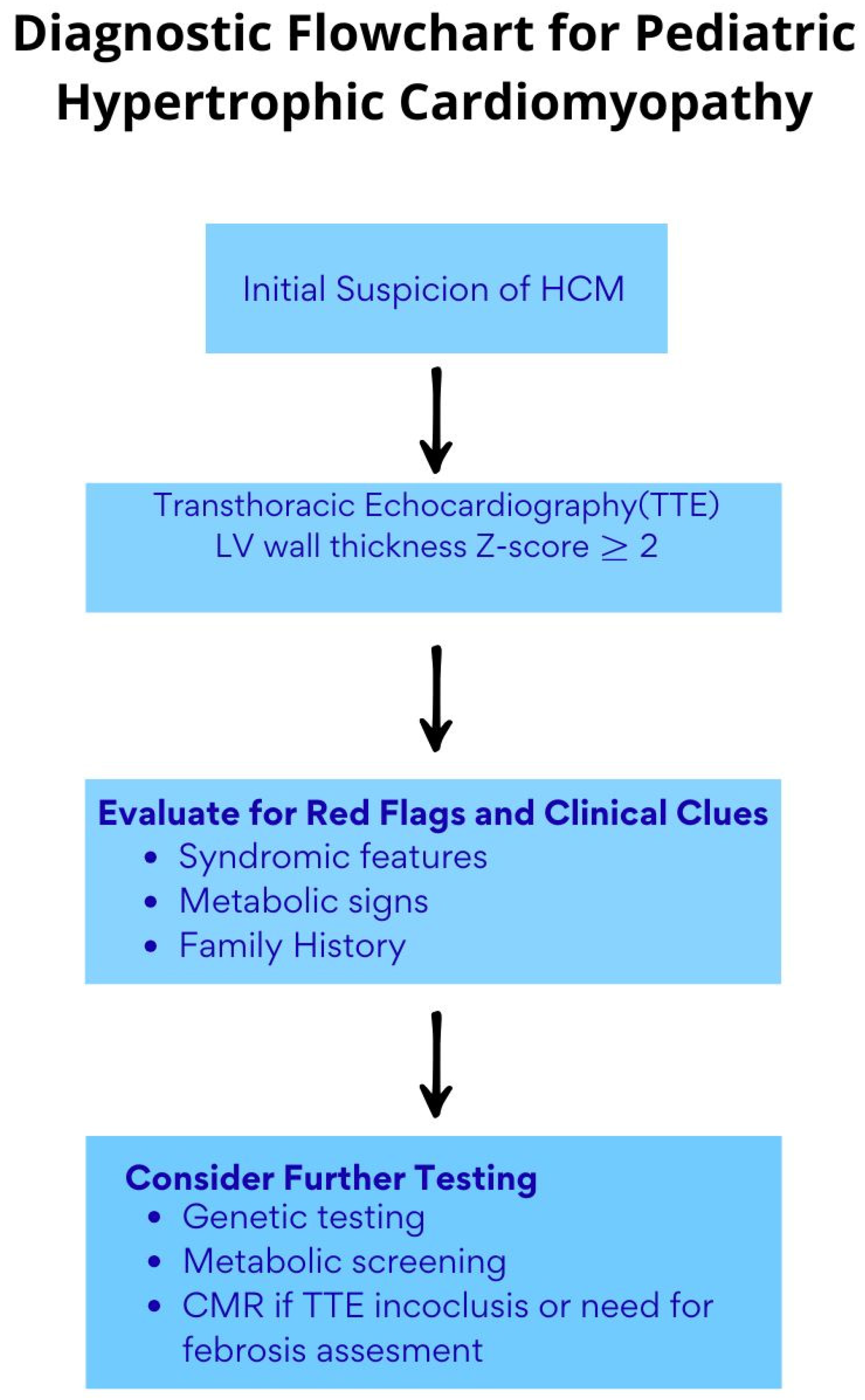
3. Etiology, Diagnosis, and Risk Stratification of Ventricular Hypertrophy in Children and Adolescents
- Etiological Spectrum Across Age Groups
- Differential Diagnosis of Childhood LVH
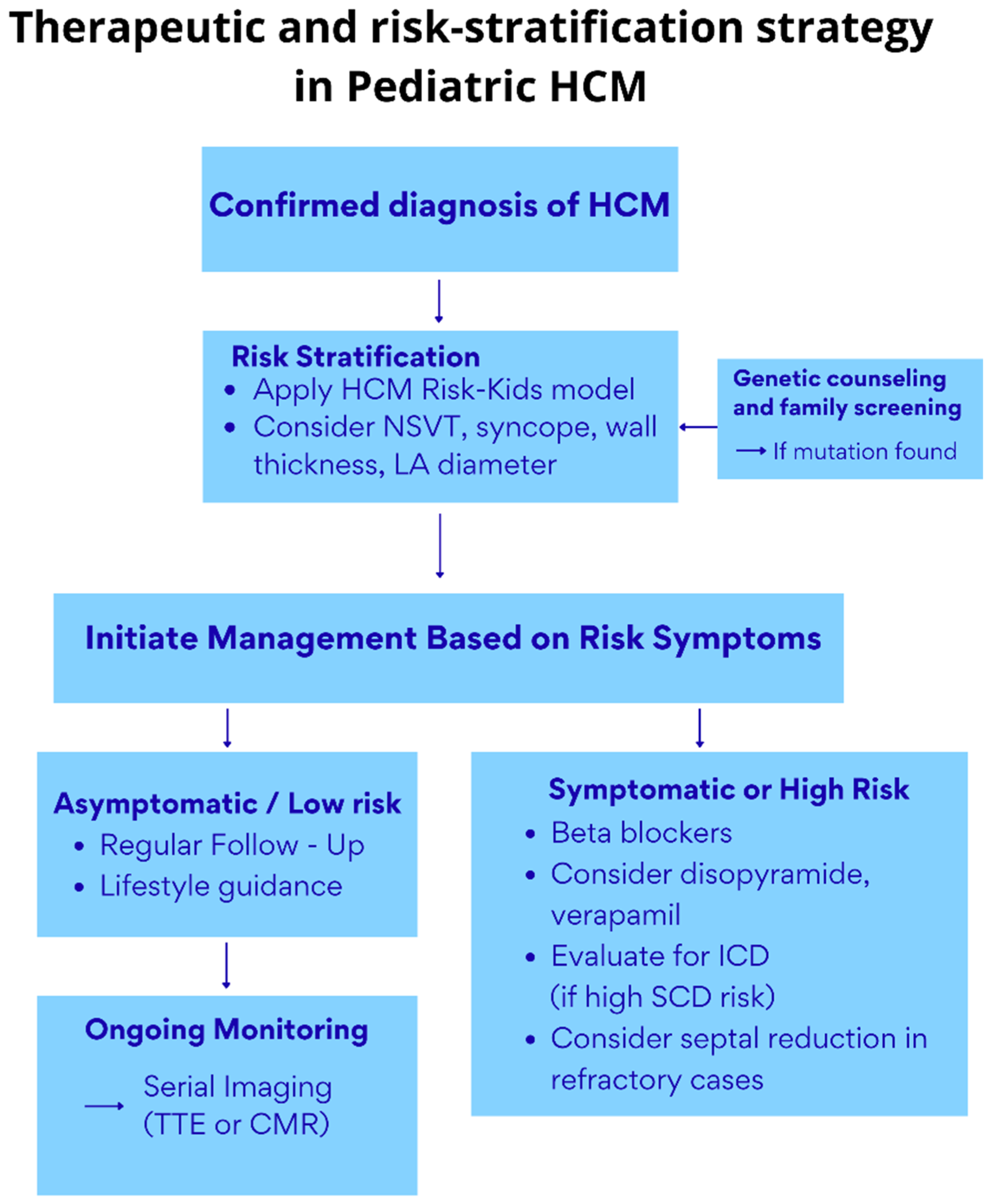
- Clinical Presentation and Risk Stratification of LVH in Childhood HCM
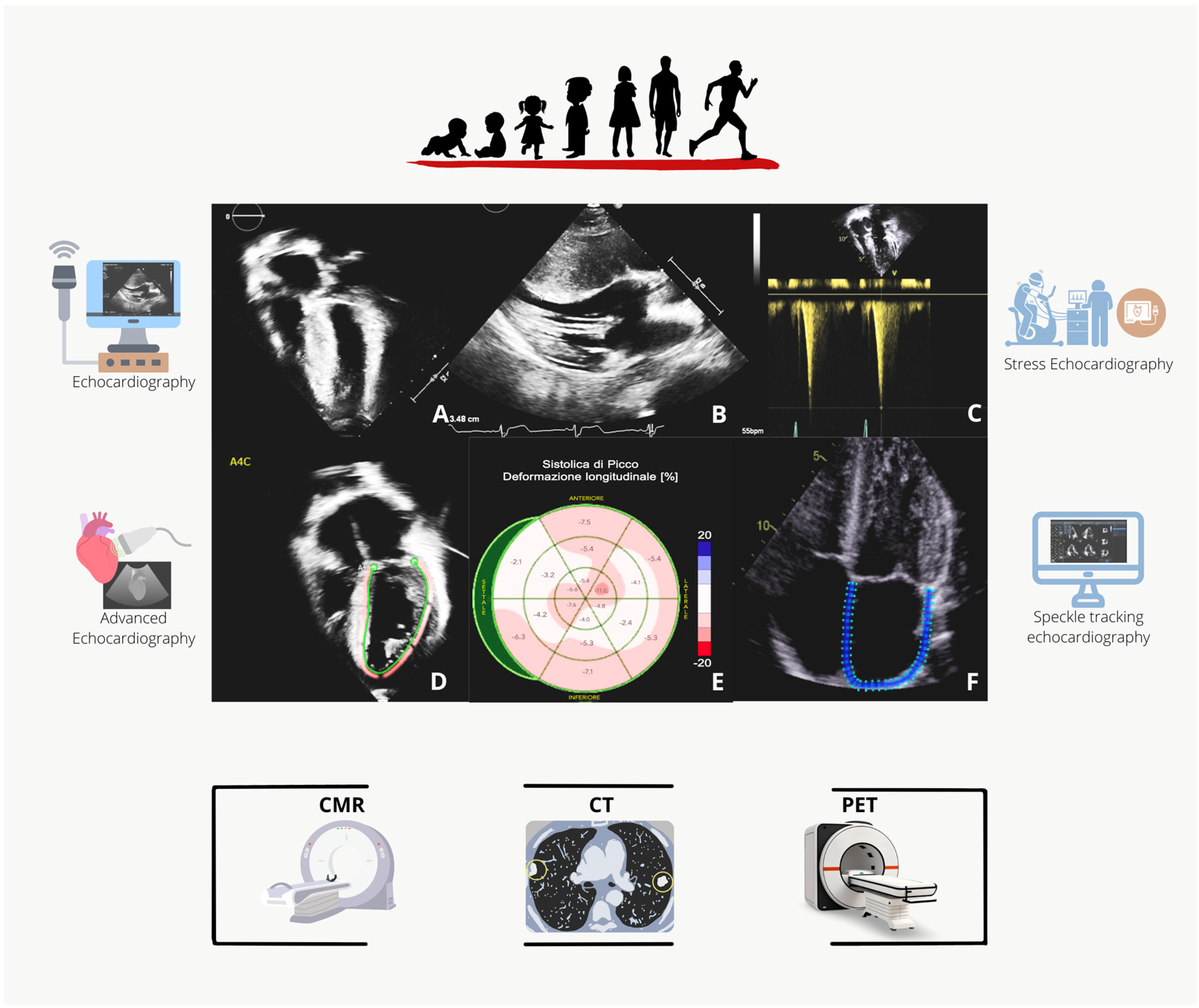
4. Imaging Assessment
- Echocardiography
- Advanced Echocardiography and Stress Echocardiography
- Advanced Imaging
5. The Role of Genetics
6. Treatment Options
6.1. LVOTO
- General measures
- First-line therapy
- Second-line therapy
- Third-line therapy, invasive septal reduction therapy
- Pacing
6.2. Atrial Fibrillation
- Anticoagulation
- Rhythm control
- Rate control
6.3. Heart Failure and Chest Pain
6.4. Prevention of Sudden Cardiac Death
7. Recommendations for Sports
8. Future Directions
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADD | Antiarrhythmic drug |
| AF | Atrial fibrillation |
| ASA | Alcohol septal ablation |
| AS | Aortic stenosis |
| CHD | Congenital heart disease |
| CMR | Cardiac Magnetic Resonance |
| CoA | Coarctation of the aorta |
| CT | Computed tomography |
| DCM | Dilated cardiomyopathy |
| EF | Ejection Fraction |
| EORP | EURObservational Research Programme |
| GLS | Global longitudinal strain |
| HCM | Hypertrophic cardiomyopathy |
| HF | Heart Failure |
| ICD | Implantable Cardioverter Defibrillator |
| IEM | Inborn errors of metabolism |
| LGE | Late gadolinium enhancement |
| LV | Left ventricle |
| LVH | Left ventricular hypertrophy |
| LVOT | Left ventricle outflow tract |
| LVOTO | Left ventricle outflow tract obstruction |
| LVNC | Noncompacted myocardium |
| MH | Myocardial hypertrophy |
| NSVT | Non-sustained ventricular tachycardia |
| RCM | Restrictive cardiomyopathy |
| RV | Right ventricle |
| SAM | Systolic anterior motion |
| SRT | Septal reduction therapies |
| STE | Speckle-tracking echocardiography |
| SCD | Sudden cardiac death |
| TTE | Transthoracic echocardiography |
| TTTS | Twin–twin transfusion syndrome |
References
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy. J. Thorac. Cardiovasc. Surg. 2011, 142, 153–203. [Google Scholar] [CrossRef] [PubMed]
- Paauw, N.D.; Stegeman, R.; de Vroede, M.A.M.J.; Termote, J.U.M.; Freund, M.W.; Breur, J.M.P.J. Neonatal cardiac hypertrophy: The role of hyperinsulinism-a review of literature. Eur. J. Pediatr. 2020, 179, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Aye, C.Y.L.; Lewandowski, A.J.; Lamata, P.; Upton, R.; Davis, E.; Ohuma, E.O.; Kenworthy, Y.; Boardman, H.; Wopperer, S.; Packham, A.; et al. Disproportionate cardiac hypertrophy during early postnatal development in infants born preterm. Pediatr. Res. 2017, 82, 36–46. [Google Scholar] [CrossRef]
- Stegeman, R.; Paauw, N.D.; de Graaf, R.; van Loon, R.L.E.; Termote, J.U.M.; Breur, J.M.P.J. The etiology of cardiac hypertrophy in infants. Sci. Rep. 2021, 11, 10626. [Google Scholar] [CrossRef] [PubMed]
- Colan, S.D.; Lipshultz, S.E.; Lowe, A.M.; Sleeper, L.A.; Messere, J.; Cox, G.F.; Lurie, P.R.; Orav, E.J.; Towbin, J.A. Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: Findings from the Pediatric Cardiomyopathy Registry. Circulation 2007, 115, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Rubino, M.; Lioncino, M.; Di Fraia, F.; Pacileo, R.; Verrillo, F.; Cirillo, A.; Caiazza, M.; Fusco, A.; Esposito, A.; et al. Hypertrophic Cardiomyopathy in Children: Pathophysiology, Diagnosis, and Treatment of Non-sarcomeric Causes. Front. Pediatr. 2021, 9, 632293. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef]
- Moon-Grady, A.J.; Donofrio, M.T.; Gelehrter, S.; Hornberger, L.; Kreeger, J.; Lee, W.; Michelfelder, E.; Morris, S.A.; Peyvandi, S.; Pinto, N.M.; et al. Guidelines and Recommendations for Performance of the Fetal Echocardiogram: An Update from the American Society of Echocardiography. J. Am. Soc. Echocardiogr. 2023, 36, 679–723. [Google Scholar] [CrossRef] [PubMed]
- Pedra, S.R.; Smallhorn, J.F.; Ryan, G.; Chitayat, D.; Taylor, G.P.; Khan, R.; Abdolell, M.; Hornberger, L.K. Fetal cardiomyopathies: Pathogenic mechanisms, hemodynamic findings, and clinical outcome. Circulation 2002, 106, 585–591. [Google Scholar] [CrossRef]
- Weber, R.; Kantor, P.; Chitayat, D.; Friedberg, M.K.; Golding, F.; Mertens, L.; Nield, L.E.; Ryan, G.; Seed, M.; Yoo, S.J.; et al. Spectrum and outcome of primary cardiomyopathies diagnosed during fetal life. JACC Heart Fail. 2014, 2, 403–411. [Google Scholar] [CrossRef]
- Chimenea, A.; Calderón, A.M.; Antiñolo, G.; Moreno-Reina, E.; García-Díaz, L. Predictive Value of Maternal HbA1c Levels for Fetal Hypertrophic Cardiomyopathy in Pregestational Diabetic Pregnancies. Children 2025, 12, 312. [Google Scholar] [CrossRef] [PubMed]
- Depla, A.L.; De Wit, L.; Steenhuis, T.J.; Slieker, M.G.; Voormolen, D.N.; Scheffer, P.G.; De Heus, R.; Van Rijn, B.B.; Bekker, M.N. Effect of maternal diabetes on fetal heart function on echocardiography: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2021, 57, 539–550. [Google Scholar] [CrossRef]
- Gutgesell, H.P.; Speer, M.E.; Rosenberg, H.S. Characterization of the cardiomyopathy in infants of diabetic mothers. Circulation 1980, 61, 441–450. [Google Scholar] [CrossRef]
- Monda, E.; Verrillo, F.; Altobelli, I.; Lioncino, M.; Caiazza, M.; Rubino, M.; Cirillo, A.; Fusco, A.; Esposito, A.; Di Fraia, F.; et al. Natural history of left ventricular hypertrophy in infants of diabetic mothers. Int. J. Cardiol. 2022, 350, 77–82. [Google Scholar] [CrossRef]
- Battistoni, G.; Montironi, R.; Di Giuseppe, J.; Giannella, L.; Delli Carpini, G.; Baldinelli, A.; Pozzi, M.; Ciavattini, A. Foetal ductus arteriosus constriction unrelated to non-steroidal anti-Inflammatory drugs: A case report and literature review. Ann. Med. 2021, 53, 860–873. [Google Scholar] [CrossRef]
- Gewillig, M.; Brown, S.C.; De Catte, L.; Debeer, A.; Eyskens, B.; Cossey, V.; Van Schoubroeck, D.; Van Hole, C.; Devlieger, R. Premature foetal closure of the arterial duct: Clinical presentations and outcome. Eur. Heart J. 2009, 30, 1530–1536. [Google Scholar] [CrossRef]
- Miller, J.L. Twin to twin transfusion syndrome. Transl. Pediatr. 2021, 10, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Mäkikallio, K.; McElhinney, D.B.; Levine, J.C.; Marx, G.R.; Colan, S.D.; Marshall, A.C.; Lock, J.E.; Marcus, E.N.; Tworetzky, W. Fetal aortic valve stenosis and the evolution of hypoplastic left heart syndrome: Patient selection for fetal intervention. Circulation 2006, 113, 1401–1405. [Google Scholar] [CrossRef] [PubMed]
- Daubeney, P.E.; Sharland, G.K.; Cook, A.C.; Keeton, B.R.; Anderson, R.H.; Webber, S.A. Pulmonary atresia with intact ventricular septum: Impact of fetal echocardiography on incidence at birth and postnatal outcome. UK and Eire Collaborative Study of Pulmonary Atresia with Intact Ventricular Septum. Circulation 1998, 98, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Di Giosaffatte, N.; Pinna, V.; Daniele, P.; Corno, S.; D’Ambrosio, V.; Andreucci, E.; Marozza, A.; Sirchia, F.; Tortora, G.; et al. When to test fetuses for RASopathies? Proposition from a systematic analysis of 352 multicenter cases and a postnatal cohort. Genet. Med. 2021, 23, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Nardi, E.; Silvano, A.; Ammar, O.; Gensini, F.; Marozza, A.; Pasquini, L.; Castiglione, F.; Seravalli, V. Foetal cardiac rhabdomyoma due to paternal TSC1 Mutation: A case report and literature review. Pathologica 2025, 117, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, A.; Pateman, A.; Chalmers, R.; Coman, D.; Menahem, S. Prenatal cardiac ultrasound finding in congenital disorder of glycosylation type 1a. Fetal Diagn Ther. 2009, 25, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Dubucs, C.; Aziza, J.; Sartor, A.; Heitz, F.; Sevely, A.; Sternberg, D.; Jardel, C.; Cavallé-Garrido, T.; Albrecht, S.; Bernard, C.; et al. Severe Antenatal Hypertrophic Cardiomyopathy Secondary to ACAD9-Related Mitochondrial Complex I Deficiency. Mol. Syndromol. 2023, 14, 101–108. [Google Scholar] [CrossRef]
- Ghi, T.; Sotiriadis, A.; Calda, P.; Da Silva Costa, F.; Raine-Fenning, N.; Alfirevic, Z.; McGillivray, G.; International Society of Ultrasound in Obstetrics and Gynecology (ISUOG). ISUOG Practice Guidelines: Invasive procedures for prenatal diagnosis. Ultrasound Obstet. Gynecol. 2016, 48, 256–268. [Google Scholar] [CrossRef]
- Girolami, F.; Gozzini, A.; Pálinkás, E.D.; Ballerini, A.; Tomberli, A.; Baldini, K.; Marchi, A.; Zampieri, M.; Passantino, S.; Porcedda, G.; et al. Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions. J. Clin. Med. 2023, 12, 2489. [Google Scholar] [CrossRef]
- Townsend, M.; Jeewa, A.; Khoury, M.; Cunningham, C.; George, K.; Conway, J. Unique Aspects of Hypertrophic Cardiomyopathy in Children. Can. J. Cardiol. 2024, 40, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Stöllberger, C. Neuromuscular disorders and hypertrophic cardiomyopathy. Neurol. Int. 2021, 13, 99–115. [Google Scholar] [CrossRef]
- Norrish, G.; Field, E.; Mcleod, K.; Ilina, M.; Stuart, G.; Bhole, V.; Uzun, O.; Brown, E.; Daubeney, P.E.F.; Lota, A.; et al. Clinical presentation and survival of childhood hypertrophic cardiomyopathy: A retrospective study in United Kingdom. Eur. Heart J. 2019, 40, 986–993. [Google Scholar] [CrossRef]
- Kaski, J.P.; Norrish, G.; Gimeno Blanes, J.R.; Charron, P.; Elliott, P.; Tavazzi, L.; Tendera, M.; Laroche, C.; Maggioni, A.P.; Baban, A.; et al. Cardiomyopathies in children and adolescents: Aetiology, management, and outcomes in the European Society of Cardiology EURObservational Research Programme Cardiomyopathy and Myocarditis Registry. Eur. Heart J. 2024, 45, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Norrish, G.; Kolt, G.; Cervi, E.; Field, E.; Dady, K.; Ziółkowska, L.; Olivotto, I.; Favilli, S.; Passantino, S.; Limongelli, G.; et al. Clinical presentation and long-term outcomes of infantile hypertrophic cardiomyopathy: A European multicentre study. ESC Heart Fail. 2021, 8, 5057–5067. [Google Scholar] [CrossRef]
- Helms, A.S.; Thompson, A.D.; Day, S.M. Translation of new and emerging therapies for genetic cardiomyopathies. JACC Basic Transl. Sci. 2021, 7, 70–83. [Google Scholar] [CrossRef]
- Andelfinger, G.; Marquis, C.; Raboisson, M.J.; Théoret, Y.; Waldmüller, S.; Wiegand, G.; Gelb, B.D.; Zenker, M.; Delrue, M.A.; Hofbeck, M. Hypertrophic cardiomyopathy in Noonan syndrome treated by MEK-inhibition. J. Am. Coll. Cardiol. 2019, 73, 2237–2239. [Google Scholar] [CrossRef]
- Norrish, G.; Qu, C.; Field, E.; Cervi, E.; Khraiche, D.; Klaassen, S.; Ojala, T.H.; Sinagra, G.; Yamazawa, H.; Marrone, C.; et al. External validation of the HCM Risk-Kids model for predicting sudden cardiac death in childhood hypertrophic cardiomyopathy. Eur. J. Prev. Cardiol. 2022, 29, 678–686. [Google Scholar] [CrossRef]
- Norrish, G.; Protonotarios, A.; Stec, M.; Boleti, O.; Field, E.; Cervi, E.; Elliott, P.M.; Kaski, J.P. Performance of the PRIMaCY sudden death risk prediction model for childhood hypertrophic cardiomyopathy: Implications for implantable cardioverter-defibrillator decision-making. EP Eur. 2023, 25, 330. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, C.; Jichi, F.; Pavlou, M.; Monserrat, L.; Anastasakis, A.; Rapezzi, C.; Biagini, E.; Gimeno, J.R.; Limongelli, G.; McKenna, W.J.; et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur. Heart J. 2014, 35, 2010–2020. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.; Saurers, D.L.; Barker, P.C.A.; Cohen, M.S.; Colan, S.D.; Dwyer, J.; Forsha, D.; Friedberg, M.K.; Lai, W.W.; Printz, B.F.; et al. Guidelines for Performing a Comprehensive Pediatric Transthoracic Echocardiogram: Recommendations from the American Society of Echocardiography. J. Am. Soc. Echocardiogr. 2024, 37, 119–170. [Google Scholar] [CrossRef]
- Alday, L.E.; Moreyra, E. Secondary hypertrophic cardiomyopathy in infancy and childhood. Am. Heart J. 1984, 108, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Compton, G.; Nield, L.; Dragulescu, A.; Benson, L.; Grosse-Wortmann, L. Echocardiography as a Screening Test for Myocardial Scarring in Children with Hypertrophic Cardiomyopathy. Int. J. Pediatr. 2016, 2016, 1980636. [Google Scholar] [CrossRef]
- Maron, M.S.; Maron, B.J.; Harrigan, C.; Buros, J.; Gibson, C.M.; Olivotto, I.; Biller, L.; Lesser, J.R.; Udelson, J.E.; Manning, W.J.; et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J. Am. Coll. Cardiol. 2009, 54, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy: Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, M.D.; Du, W.; Skeens, M.E.; Humes, R.A. Regression equations for calculation of z scores of cardiac structures in a large cohort of healthy infants, children, and adolescents: An echocardiographic study. J. Am. Soc. Echocardiogr. 2008, 21, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Norrish, G.; Jager, J.; Field, E.; Quinn, E.; Fell, H.; Lord, E.; Cicerchia, M.N.; Ochoa, J.P.; Cervi, E.; Elliott, P.M.; et al. Yield of Clinical Screening for Hypertrophic Cardiomyopathy in Child First-Degree Relatives. Circulation 2019, 140, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.W.; Daubeney, P.E.; Chondros, P.; Carlin, J.B.; Colan, S.D.; Cheung, M.; Davis, A.M.; Chow, C.W.; Weintraub, R.G.; National Australian Childhood Cardiomyopathy Study. Clinical Features and Outcomes of Childhood Hypertrophic Cardiomyopathy. Circulation 2005, 112, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Finocchiaro, G.; Merlo, M.; Sheikh, N.; De Angelis, G.; Papadakis, M.; Olivotto, I.; Rapezzi, C.; Carr-White, G.; Sharma, S.; Mestroni, L.; et al. The electrocardiogram in the diagnosis and management of patients with dilated cardiomyopathy. Eur. J. Heart Fail. 2020, 22, 1097–1107. [Google Scholar] [CrossRef]
- Captur, G.; Manisty, C.H.; Raman, B.; Marchi, A.; Wong, T.C.; Ariga, R.; Bhuva, A.; Ormondroyd, E.; Lobascio, I.; Camaioni, C.; et al. Maximal Wall Thickness Measurement in Hypertrophic Cardiomyopathy: Biomarker Variability and its Impact on Clinical Care. JACC Cardiovasc. Imaging 2021, 14, 2123–2134. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Sleeper, L.A.; Towbin, J.A.; Lowe, A.M.; Orav, E.J.; Cox, G.F.; Lurie, P.R.; McCoy, K.L.; McDonald, M.A.; Messere, J.E.; et al. The Incidence of Pediatric Cardiomyopathy in Two Regions of the United States. N. Engl. J. Med. 2003, 348, 1647–1655. [Google Scholar] [CrossRef]
- Sinha, M.D.; Azukaitis, K.; Sladowska-Kozłowska, J.; Bårdsen, T.; Merkevicius, K.; Karlsen Sletten, I.S.; Obrycki, Ł.; Pac, M.; Fernández-Aranda, F.; Bjelakovic, B.; et al. Prevalence of left ventricular hypertrophy in children and young people with primary hypertension: Meta-analysis and meta-regression. Front. Cardiovasc. Med. 2022, 9, 993513. [Google Scholar] [CrossRef]
- Lopes, L.R.; Ho, C.Y.; Elliott, P.M. Genetics of hypertrophic cardiomyopathy: Established and emerging implications for clinical practice. Eur. Heart J. 2024, 45, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- McMahon, C.J.; Nagueh, S.F.; Pignatelli, R.H.; Denfield, S.W.; Dreyer, W.J.; Price, J.F.; Clunie, S.; Bezold, L.I.; Hays, A.L.; Towbin, J.A.; et al. Characterization of left ventricular diastolic function by tissue Doppler imaging and clinical status in children with hypertrophic cardiomyopathy. Circulation 2004, 109, 1756–1762. [Google Scholar] [CrossRef]
- Menon, S.C.; Eidem, B.W.; Dearani, J.A.; Ommen, S.R.; Ackerman, M.J.; Miller, D. Diastolic dysfunction and its histopathological correlation in obstructive hypertrophic cardiomyopathy in children and adolescents. J. Am. Soc. Echocardiogr. 2009, 22, 1327–1334. [Google Scholar] [CrossRef]
- Dragulescu, A.; Mertens, L.; Friedberg, M.K. Interpretation of Left Ventricular Diastolic Dysfunction in Children with Cardiomyopathy by Echocardiography. Circ. Cardiovasc. Imaging 2013, 6, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Dorobantu, D.M.; Wadey, C.A.; Amir, N.H.; Stuart, A.G.; Williams, C.A.; Pieles, G.E. The Role of Speckle Tracking Echocardiography in the Evaluation of Common Inherited Cardiomyopathies in Children and Adolescents: A Systematic Review. Diagnostics 2021, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Forsey, J.; Benson, L.; Rozenblyum, E.; Friedberg, M.K.; Mertens, L. Early changes in apical rotation in genotype positive children with hypertrophic cardiomyopathy mutations without hypertrophic changes on two-dimensional imaging. J. Am. Soc. Echocardiogr. 2014, 27, 215–221. [Google Scholar] [CrossRef]
- Sabatino, J.; Di Salvo, G.; Prota, C.; Bucciarelli, V.; Josen, M.; Paredes, J.; Borrelli, N.; Sirico, D.; Prasad, S.; Indolfi, C.; et al. Left Atrial Strain to Identify Diastolic Dysfunction in Children with Cardiomyopathies. J. Clin. Med. 2019, 8, 1243. [Google Scholar] [CrossRef] [PubMed]
- El Assaad, I.; Gauvreau, K.; Rizwan, R.; Margossian, R.; Colan, S.; Chen, M.H. Value of Exercise Stress Echocardiography in Children with Hypertrophic Cardiomyopathy. J. Am. Soc. Echocardiogr. 2020, 33, 888–894. [Google Scholar] [CrossRef]
- Ciliberti, P.; McLeod, I.; Cairello, F.; Kaski, J.P.; Fenton, M.; Giardini, A.; Marek, J. Semi-supine Exercise Stress Echocardiography in Children and Adolescents: Feasibility and Safety. Pediatr. Cardiol. 2015, 36, 633–639. [Google Scholar] [CrossRef]
- Baessato, F.; Romeo, C.; Rabbat, M.G.; Pontone, G.; Meierhofer, C. A Comprehensive Assessment of Cardiomyopathies through Cardiovascular Magnetic Resonance: Focus on the Pediatric Population. Diagnostics 2022, 12, 1022. [Google Scholar] [CrossRef]
- Spaapen, T.O.M.; Bohte, A.E.; Slieker, M.G.; Grotenhuis, H.B. Cardiac MRI in diagnosis, prognosis, and follow-up of hypertrophic cardiomyopathy in children: Current perspectives. Br. J. Radiol. 2024, 97, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Fogel, M.A.; Anwar, S.; Broberg, C.; Browne, L.; Chung, T.; Johnson, T.; Muthurangu, V.; Taylor, M.; Valsangiacomo-Buechel, E.; Wilhelm, C. Society for Cardiovascular Magnetic Resonance/European Society of Cardiovascular Imaging/American Society of Echocardiography/Society for Pediatric Radiology/North American Society for Cardiovascular Imaging Guidelines for the Use of Cardiac Magnetic Resonance in Pediatric Congenital and Acquired Heart Disease: Endorsed by The American Heart Association. Circ. Cardiovasc. Imaging 2022, 15, 014415. [Google Scholar] [CrossRef]
- Moscatelli, S.; Leo, I.; Lisignoli, V.; Boyle, S.; Bucciarelli-Ducci, C.; Secinaro, A.; Montanaro, C. Cardiovascular Magnetic Resonance from Fetal to Adult Life-Indications and Challenges: A State-of-the-Art Review. Children 2023, 10, 763. [Google Scholar] [CrossRef]
- Rowin, E.J.; Maron, M.S. The Role of Cardiac MRI in the Diagnosis and Risk Stratification of Hypertrophic Cardiomyopathy. Arrhythmia Electrophysiol. Rev. 2016, 5, 197–202. [Google Scholar] [CrossRef]
- Ding, W.; Bhushan, S.; Ma, C.; Yan, Y.; Xiao, Z. Right Ventricle Involvement in Hypertrophic Cardiomyopathy and Role of Cardiac Magnetic Resonance in Hypertrophic Cardiomyopathy: Review Article. Heart Surg. Forum. 2021, 24, E746–E750. [Google Scholar] [CrossRef] [PubMed]
- Rosu, R.O.; Lupsor, A.; Necula, A.; Cismaru, G.; Cainap, S.S.; Iacob, D.; Lazea, C.; Cismaru, A.; Negru, A.G.; Pop, D.; et al. Anatomical-MRI Correlations in Adults and Children with Hypertrophic Cardiomyopathy. Diagnostics 2022, 12, 489. [Google Scholar] [CrossRef] [PubMed]
- Chaowu, Y.; Shihua, Z.; Jian, L.; Li, L.; Wei, F. Cardiovascular Magnetic Resonance Characteristics in Children with Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2013, 6, 1013–1020. [Google Scholar] [CrossRef]
- Sivalokanathan, S. The Role of Cardiovascular Magnetic Resonance Imaging in the Evaluation of Hypertrophic Cardiomyopathy. Diagnostics 2022, 12, 314. [Google Scholar] [CrossRef]
- Waterhouse, D.F.; Ismail, T.F.; Prasad, S.K.; Wilson, M.G.; O’Hanlon, R. Imaging focal and interstitial fibrosis with cardiovascular magnetic resonance in athletes with left ventricular hypertrophy: Implications for sporting participation. Br. J. Sports Med. 2012, 46, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Goldie, F.C.; Lee, M.M.Y.; Coats, C.J.; Nordin, S. Advances in Multi-Modality Imaging in Hypertrophic Cardiomyopathy. J. Clin. Med. 2024, 13, 842. [Google Scholar] [CrossRef]
- Dorfman, A.L.; Fazel, R.; Einstein, A.J.; Applegate, K.E.; Krumholz, H.M.; Wang, Y.; Christodoulou, E.; Chen, J.; Sanchez, R.; Nallamothu, B.K. Use of Medical Imaging Procedures with Ionizing Radiation in Children: A Population-Based Study. Arch. Pediatr. Adolesc. Med. 2011, 165, 458–464. [Google Scholar] [CrossRef]
- Moscatelli, S.; Leo, I.; Bianco, F.; Surkova, E.; Pezel, T.; Donald, N.A.; Triumbari, E.K.A.; Bassareo, P.P.; Pradhan, A.; Cimini, A.; et al. The Role of Multimodality Imaging in Patients with Congenital Heart Disease and Infective Endocarditis. Diagnostics 2023, 13, 3638. [Google Scholar] [CrossRef] [PubMed]
- Ziolkowska, L.; Boruc, A.; Sobielarska-Lysiak, D.; Grzyb, A.; Petryka-Mazurkiewicz, J.; Mazurkiewicz, Ł.; Brzezinska-Rajszys, G. Prognostic Significance of Myocardial Ischemia Detected by Single-Photon Emission Computed Tomography in Children with Hypertrophic Cardiomyopathy. Pediatr. Cardiol. 2021, 42, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Dahdal, J.; Jukema, R.A.; Harms, H.J.; Cramer, M.J.; Raijmakers, P.G.; Knaapen, P.; Danad, I. PET myocardial perfusion imaging: Trends, challenges, and opportunities. J. Nucl. Cardiol. 2024, 40, 102011. [Google Scholar] [CrossRef] [PubMed]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Ireland, C.G.; Ho, C.Y. Genetic Testing in Hypertrophic Cardiomyopathy. Am. J. Cardiol. 2024, 1, 212. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Desai, M.Y.; Nishimura, R.A.; Spirito, P.; Rakowski, H.; Towbin, J.A.; Rowin, E.J.; Maron, M.S.; Sherrid, M.V. Diagnosis and Evaluation of Hypertrophic Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Ommen, S.R.; Ho, C.Y.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, 1239–1311. [Google Scholar] [CrossRef] [PubMed]
- Authors/Task Force Members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Lafreniere-Roula, M.; Bolkier, Y.; Zahavich, L.; Mathew, J.; George, K.; Wilson, J.; Stephenson, E.A.; Benson, L.N.; Manlhiot, C.; Mital, S. Family screening for hypertrophic cardiomyopathy: Is it time to change practice guidelines? Eur. Heart J. 2019, 40, 3672–3681. [Google Scholar] [CrossRef]
- Camm, A.J.; Lip, G.Y.; De Caterina, R.; Savelieva, I.; Atar, D.; Hohnloser, S.H.; Hindricks, G.; Kirchhof, P.; ESC Committee for Practice Guidelines (CPG). 2012 focused update of the ESC Guidelines for the management of atrial fibrillation. Eur. Heart J. 2012, 33, 2719–2747. [Google Scholar] [CrossRef]
- Dybro, A.M.; Rasmussen, T.B.; Nielsen, R.R.; Ladefoged, B.T.; Andersen, M.J.; Jensen, M.K.; Poulsen, S.H. Effects of Metoprolol on Exercise Hemodynamics in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 79, 1565–1575. [Google Scholar] [CrossRef]
- Möbius-Winkler, M.N.; Laufs, U.; Lenk, K. The Diagnosis and Treatment of Hypertrophic Cardiomyopathy. Dtsch. Arztebl. Int. 2024, 121, 805–811. [Google Scholar] [CrossRef]
- Spicer, R.L.; Rocchini, A.P.; Crowley, D.C.; Vasiliades, J.; Rosenthal, A. Hemodynamic effects of verapamil in children and adolescents with hypertrophic cardiomyopathy. Circulation 1983, 67, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Toshima, H.; Koga, Y.; Nagata, H.; Toyomasu, K.; Itaya, K.; Matoba, T. Comparable effects of oral diltiazem and verapamil in the treatment of hypertrophic cardiomyopathy. Double-blind crossover study. Jpn. Heart J. 1986, 27, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Rosing, D.R.; Idänpään-Heikkilä, U.; Maron, B.J.; Bonow, R.O.; Epstein, S.E. Use of calcium-channel blocking drugs in hypertrophic cardiomyopathy. Am. J. Cardiol. 1985, 55, 185B–195B. [Google Scholar] [CrossRef] [PubMed]
- Sherrid, M.V.; Barac, I.; McKenna, W.J.; Elliott, P.M.; Dickie, S.; Chojnowska, L.; Casey, S.; Maron, B.J. Multicenter study of the efficacy and safety of disopyramide in obstructive hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2005, 45, 1251–1258. [Google Scholar] [CrossRef]
- Sherrid, M.V.; Shetty, A.; Winson, G.; Kim, B.; Musat, D.; Alviar, C.L.; Homel, P.; Balaram, S.K.; Swistel, D.G. Treatment of obstructive hypertrophic cardiomyopathy symptoms and gradient resistant to first-line therapy with β-blockade or verapamil. Circ. Heart Fail. 2013, 6, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, I.; Oreziak, A.; Barriales-Villa, R.; Abraham, T.P.; Masri, A.; Garcia-Pavia, P.; Saberi, S.; Lakdawala, N.K.; Wheeler, M.T.; Owens, A.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Owens, A.; Geske, J.B.; Wolski, K.; Naidu, S.S.; Smedira, N.G.; Cremer, P.C.; Schaff, H.; McErlean, E.; Sewell, C.; et al. Myosin Inhibition in Patients with Obstructive Hypertrophic Cardiomyopathy Referred for Septal Reduction Therapy. J. Am. Coll. Cardiol. 2022, 80, 95–108. [Google Scholar] [CrossRef]
- Desai, M.Y.; Okushi, Y.; Wolski, K.; Geske, J.B.; Owens, A.; Saberi, S.; Wang, A.; Cremer, P.C.; Sherrid, M.; Lakdawala, N.K.; et al. VALOR-HCM Investigators. Mavacamten-Associated Temporal Changes in Left Atrial Function in Obstructive HCM: Insights From the VALOR-HCM Trial. JACC Cardiovasc. Imaging 2025, 18, 251–262. [Google Scholar] [CrossRef]
- Cremer, P.C.; Geske, J.B.; Owens, A.; Jaber, W.A.; Harb, S.C.; Saberi, S.; Wang, A.; Sherrid, M.; Naidu, S.S.; Schaff, H.V.; et al. Mitral Regurgitation in Obstructive Hypertrophic Cardiomyopathy: Insight from the VALOR-HCM Study. JACC Cardiovasc. Imaging 2024, 17, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.Y.; Wolski, K.; Owens, A.; Geske, J.B.; Saberi, S.; Wang, A.; Sherrid, M.; Cremer, P.C.; Lakdawala, N.K.; Tower-Rader, A.; et al. Correction to: Mavacamten in Patients with Hypertrophic Cardiomyopathy Referred for Septal Reduction: Week 128 Results from VALOR-HCM. Circulation 2025, 151, 1001. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Bilen, O.; Burroughs, M.; Costabel, J.P.; de Barros Correia, E.; Dybro, A.M.; Elliott, P.; Lakdawala, N.K.; Mann, A.; Nair, A.; et al. Aficamten vs Metoprolol for Obstructive Hypertrophic Cardiomyopathy: MAPLE-HCM Rationale, Study Design, and Baseline Characteristics. JACC Heart Fail. 2025, 13, 346–357. [Google Scholar] [CrossRef]
- Maron, M.S.; Masri, A.; Choudhury, L.; Olivotto, I.; Saberi, S.; Wang, A.; Garcia-Pavia, P.; Lakdawala, N.K.; Nagueh, S.F.; Rader, F.; et al. Phase 2 Study of Aficamten in Patients with Obstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2023, 81, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Masri, A.; Nassif, M.E.; Barriales-Villa, R.; Abraham, T.P.; Arad, M.; Cardim, N.; Choudhury, L.; Claggett, B.; Coats, C.J.; et al. Impact of Aficamten on Disease and Symptom Burden in Obstructive Hypertrophic Cardiomyopathy: Results From SEQUOIA-HCM. J. Am. Coll. Cardiol. 2024, 84, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Kirchhof, P.; Camm, A.J.; Goette, A.; Brandes, A.; Eckardt, L.; Elvan, A.; Fetsch, T.; van Gelder, I.C.; Haase, D.; Haegeli, L.M.; et al. Early Rhythm-Control Therapy in Patients with Atrial Fibrillation. N. Engl. J. Med. 2020, 383, 1305–1316. [Google Scholar] [CrossRef] [PubMed]
- Authors/Task Force Members; McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2022, 24, 4–131. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Bozkurt, B.; Aguilar, D.; Allen, L.A.; Byun, J.J.; Colvin, M.M.; Deswal, A.; Drazner, M.H.; Dunlay, S.M.; Evers, L.R.; et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2022, 145, 895–1032. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Gray, B.; Haugaa, K.H.; Lampert, R.; Sharma, S.; Kovacic, J.C. Athletic Activity for Patients with Hypertrophic Cardiomyopathy and Other Inherited Cardiovascular Diseases: JACC Focus Seminar 3/4. J. Am. Coll. Cardiol. 2022, 80, 1268–1283. [Google Scholar] [CrossRef] [PubMed]
- Masood, I.R.; Edelson, J.B. Exercise and Sports Participation in Children with Cardiomyopathy: A Review. Curr. Treat. Options Cardiovasc. Med. 2023, 25, 543–559. [Google Scholar] [CrossRef]
- Lampert, R.; Olshansky, B.; Heidbuchel, H.; Lawless, C.; Saarel, E.; Ackerman, M.; Calkins, H.; Estes, N.M.; Link, M.S.; Maron, B.J.; et al. Safety of sports for athletes with implantable cardioverter-defibrillators: Results of a prospective, multinational registry. Circulation 2013, 127, 2021–2030. [Google Scholar] [CrossRef]
- Wolf, C.M.; Zenker, M.; Boleti, O.; Norrish, G.; Russell, M.; Meisner, J.K.; Peng, D.M.; Prendiville, T.; Kleinmahon, J.; Kantor, P.F.; et al. Impact of MEK Inhibition on Childhood RASopathy-Associated Hypertrophic Cardiomyopathy. JACC Basic Transl. Sci. 2025, 10, 152–166. [Google Scholar] [CrossRef]
- Neupane, A.; Petrykey, K.; Li, K.; French, J.; Zhou, X.; Wang, J.; Im, C.; Dixon, S.B.; Ehrhardt, M.J.; Mulrooney, D.A.; et al. TTN and BAG3 in Cancer Therapy-Related Cardiomyopathy Among Long-Term Survivors of Childhood Cancer. JAMA Netw. Open 2025, 8, e2515793. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classes | Drugs | Dosages (Children) | Dosages (Adults) |
|---|---|---|---|
| Non-vasodilating beta-blocker | Atenolol | 0.5–1 mg/kg every 12–24 h (max 25–50 mg) | 25–100 mg once daily |
| Metoprolol | 1–2 mg/kg every 6–12 h | 25–100 mg twice daily | |
| Bisoprolol | 0.2–0.4 mg/kg daily | 1.25–20 mg once daily | |
| Propranolol | 0.2–0.5 mg/kg every 6–8 h | 10–25 mg every 6–8 h | |
| Non-dihydropyridine calcium channel blockers | Verapamil | 1–3 mg/kg every 8–12 h | 40 mg every 12 h to 480 mg (extended release) once daily |
| Diltiazem | 1 mg/kg every 8 h | 60 mg every 8 h to 360 mg (extended release) once daily | |
| Class Ia antiarrhythmic drug | Disopyramide | 1.5–4 mg/kg/dose every 6 h | 400–600 mg/day |
| Cardiac myosin ATPase inhibitors | Mavacamten | Not available | Initial dose 5 mg once daily. Maximal dose 15 mg once daily |
| Aficamten | Not available | Initial dose 5 mg once daily. Maximal dose 20 mg once daily |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avesani, M.; Pomiato, E.; Moscatelli, S.; Sabatino, J.; Borrelli, N.; Luedke, L.; De Sarro, R.; Pavesi, S.; Pelaia, G.; Mastellone, C.; et al. Cardiac Hypertrophy: A Comprehensive Review from Prenatal Life to Young Adulthood. Children 2025, 12, 989. https://doi.org/10.3390/children12080989
Avesani M, Pomiato E, Moscatelli S, Sabatino J, Borrelli N, Luedke L, De Sarro R, Pavesi S, Pelaia G, Mastellone C, et al. Cardiac Hypertrophy: A Comprehensive Review from Prenatal Life to Young Adulthood. Children. 2025; 12(8):989. https://doi.org/10.3390/children12080989
Chicago/Turabian StyleAvesani, Martina, Elettra Pomiato, Sara Moscatelli, Jolanda Sabatino, Nunzia Borrelli, Leonie Luedke, Rosalba De Sarro, Sara Pavesi, Giulia Pelaia, Claudio Mastellone, and et al. 2025. "Cardiac Hypertrophy: A Comprehensive Review from Prenatal Life to Young Adulthood" Children 12, no. 8: 989. https://doi.org/10.3390/children12080989
APA StyleAvesani, M., Pomiato, E., Moscatelli, S., Sabatino, J., Borrelli, N., Luedke, L., De Sarro, R., Pavesi, S., Pelaia, G., Mastellone, C., Leo, I., & Di Salvo, G., on behalf of the Working Group on Congenital Heart Disease Cardiovascular Prevention in Paediatric Age of the Italian Society of Cardiology Sic. (2025). Cardiac Hypertrophy: A Comprehensive Review from Prenatal Life to Young Adulthood. Children, 12(8), 989. https://doi.org/10.3390/children12080989