
PEX6 Mutation in a Child with Infantile Refsum Disease—A Case Report and Literature Review
 , , , , ,
, , , , , 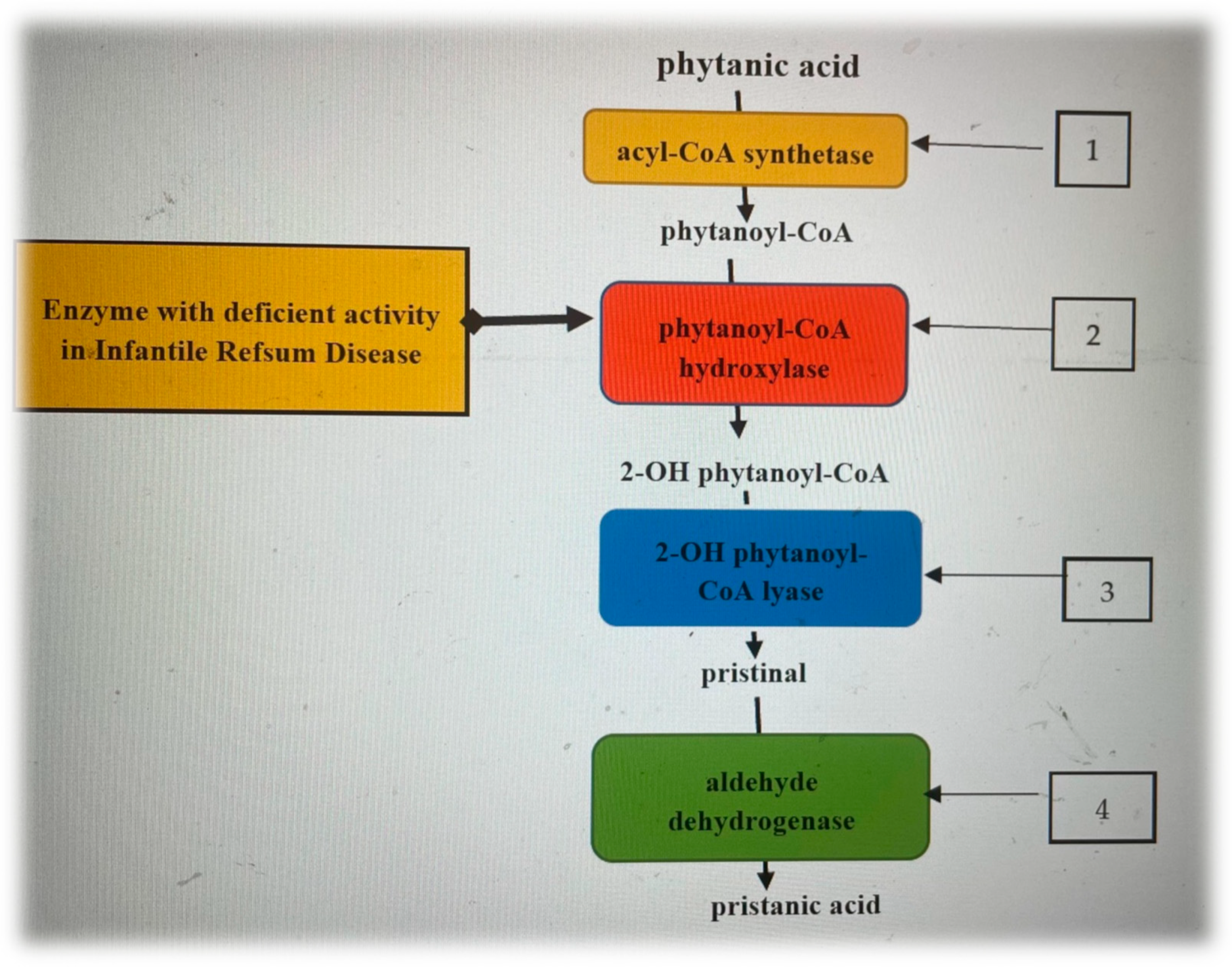{kind=link}
Abstract
1. Introduction
2. Case Report
2.1. Clinical Picture
Neurological Examination
2.2. Investigations
- Brain CT with angiography, then contrast-enhanced MRI, with a right temporal brain tissue hematoma of 4.4/2.6 cm being identified.
- EEG while awake and asleep (Neurology Department Obregia Hospital Bucharest)—an asymmetrical baseline pathway with a slower pace and epileptic discharges that were spike-like in the right temporal area was noted.
- Abdominal ultrasound—liver enlargement and an accessory lien were observed.
- Thrombophilia genetic panel—a heterozygote mutation for the MTHFR C677T gene and a homozygote mutation for the PAI-1 4G gene were found.
- Positive antibodies for the cytoplasm of neutrophils (ANCA) were found.
- Antibodies for elastase presented with an increased value.
- Calprotectin presented with an increased value.
- Antithrombin presented with an increased value.
- Protein C and S presented with a low value.
- Coagulation factor X demonstrated a serious reduction of 34% (the existence of Stuart–Prower syndrome was considered). A moderate deficit of the coagulation factors II, V, and VII was identified. The prothrombin time and INR were increased.
2.3. Diagnosis and Associated Conditions
3. Discussion
Antenatal Diagnosis and Gene Therapy
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braverman, N.E.; D’Agostino, M.D.; Maclean, G.E. Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives. Dev. Disabil. Res. Rev. 2013, 17, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Masih, S.; Moirangthem, A.; Phadke, S.R. Twins with PEX7 related intellectual disability and cataract: Highlighting phenotypes of peroxisome biogenesis disorder 9B. Am. J. Med. Genet. A. 2021, 185, 1504–1508. [Google Scholar] [CrossRef]
- Aubourg, P.; Wanders, R. Peroxisomal disorders. Handb. Clin. Neurol. 2013, 113, 1593–1609. [Google Scholar] [PubMed]
- Bader, P.I.; Dougherty, S.; Cangany, N.; Raymond, G.; Jackson, C.E. Infantile Refsum disease in four Amish sibs. Am. J. Med. Genet. 2000, 90, 110–114. [Google Scholar] [CrossRef]
- Choksi, V.; Hoeffner, E.; Karaarslan, E.; Yalcinkaya, C.; Cakirer, S. Infantile refsum disease: Case report. AJNR Am. J. Neuroradiol. 2003, 24, 2082–2084. [Google Scholar]
- Wanders, R.J.; Komen, J.; Ferdinandusse, S. Phytanic acid metabolism in health and disease. Biochim. Biophys. Acta 2011, 1811, 498–507. [Google Scholar] [CrossRef]
- Foulon, V.; Asselberghs, S.; Geens, W.; Mannaerts, G.P.; Casteels, M.; Van Veldhoven, P.P. Further studies on the substrate spectrum of phytanoyl-CoA hydroxylase: Implications for Refsum disease? J. Lipid Res. 2003, 44, 2349–2355. [Google Scholar] [CrossRef]
- Horn, M.A.; Van den Brink, D.M.; Wanders, R.J.A.; Duran, M.; Tallaksen, C.M.E.; Stokke, O.H.; Moser, H.; Skjeldal, O.H. Phenotype of adult Refsum disease due to a defect in peroxin 7. Neurology 2007, 68, 698–700. [Google Scholar] [CrossRef]
- Jansen, G.A.; Waterham, H.R.; Wanders, R.J. Molecular basis of Refsum disease: Sequence variations in phytanoyl-CoA hydroxylase (PHYH) and the PTS2 receptor (PEX7). Hum. Mutat. 2004, 23, 209–218. [Google Scholar] [CrossRef]
- Nakanishi, T.; Kagamizono, K.; Yokoyama, S.; Suzuki, R.; Sakakibara, H.; Erickson, L.; Kawahara, S. Effects of dietary phytol on tissue accumulation of phytanic acid and pristanic acid and on the tissue lipid profiles in mice. Anim. Sci. J. 2020, 91, e13424. [Google Scholar] [CrossRef]
- Wierzbicki, S. Refsum Disease—A Disorder of Peroxisomal Alpha-oxidation. In Encyclopedia of Movement Disorders; Kompoliti, K., Metman, L.V., Eds.; Academic Press: Cambridge, MA, USA, 2010; pp. 21–25. [Google Scholar]
- Bobe, G.; Zhang, Z.; Kopp, R.; Garzotto, M.; Shannon, J.; Takata, Y. Phytol and its metabolites phytanic and pristanic acids for risk of cancer: Current evidence and future directions. Eur. J. Cancer Prev. 2020, 29, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Brain Lipotoxicity of Phytanic Acid and Very Long-chain Fatty Acids. Harmful Cellular/Mitochondrial Activities in Refsum Disease and X-Linked Adrenoleukodystrophy. Aging Dis. 2016, 7, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Bhagavan, N.V.; Chung-Eun, H. Lipids I: Fatty Acids and Eicosanoids in Essentials of Medical Biochemistry, 2nd ed.; Bhagavan, N.V., Chung-Eun, H., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 269–297. [Google Scholar]
- Zomer, A.W.; van Der Burg, B.; Jansen, G.A.; Wanders, R.J.; Poll-The, B.T.; van Der Saag, P.T. Pristanic acid and phytanic acid: Naturally occurring ligands for the nuclear receptor peroxisome proliferator-activated receptor alpha. J. Lipid Res. 2000, 41, 1801–1807. [Google Scholar] [CrossRef]
- Maciejewska-Skrendo, A.; Massidda, M.; Tocco, F.; Leźnicka, K. The Influence of the Differentiation of Genes Encoding Peroxisome Proliferator-Activated Receptors and Their Coactivators on Nutrient and Energy Metabolism. Nutrients 2022, 14, 5378. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Popa, E.; Zugun-Eloae, F.; Zlei, M.; Traian, M.; Bacusca, A.; Popa, A.E.; Coman, E.A. Peroxisome Proliferator-Activated Receptors—Alpha in Chronic Inflammation -Mini-Review. Int. J. Pharmacol. Phytochem. Ethnomedicine 2019, 12, 1–12. [Google Scholar] [CrossRef]
- Dhaunsi, G.; Alsaeid, M.; Akhtar, S. Phytanic acid attenuates insulin-like growth factor-1 activity via nitric oxide-mediated γ-secretase activation in rat aortic smooth muscle cells: Possible implications for pathogenesis of infantile Refsum disease. Pediatr. Res. 2017, 81, 531–536. [Google Scholar] [CrossRef]
- Zuckerbraun, B.S.; Stoyanovsky, D.A.; Sengupta, R.; Shapiro, R.A.; Ozanich, B.A.; Rao, J.; Barbato, J.E.; Tzeng, E. Nitric oxide-induced inhibition of smooth muscle cell proliferation involves S-nitrosation and inactivation of RhoA. Am. J. Physiol. Cell Physiol. 2007, 292, C824–C831. [Google Scholar] [CrossRef]
- Idel, S.; Ellinghaus, P.; Wolfrum, C.; Nofer, J.R.; Gloerich, J.; Assmann, G.; Spener, F.; Seedorf, U. Branched chain fatty acids induce nitric oxide-dependent apoptosis in vascular smooth muscle cells. J. Biol. Chem. 2002, 277, 49319–49322. [Google Scholar] [CrossRef]
- Sá, M.J.; Rocha, J.C.; Almeida, M.F.; Carmona, C.; Martins, E.; Miranda, V.; Coutinho, M.; Ferreira, R.; Pacheco, S.; Laranjeira, F.; et al. Infantile Refsum Disease: Influence of Dietary Treatment on Plasma Phytanic Acid Levels. JIMD Rep. 2016, 26, 53–60. [Google Scholar]
- Moser, A.B.; Jones, D.S.; Raymond, G.V.; Moser, H.W. Plasma and red blood cell fatty acids in peroxisomal disorders. Neurochem. Res. 1999, 24, 187–197. [Google Scholar] [CrossRef]
- Kumar, R.; De Jesus, O. Refsum Disease. [Updated 2022 Aug 1]. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Waterham, H.R.; Wanders, R.J.A.; Leroy, B.P. Adult Refsum Disease. 2006 Mar 20 [updated 2021 Sep 30]. In GeneReviews® [Internet]; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Braverman, N.E.; Raymond, G.V.; Rizzo, W.B.; Moser, A.B.; Wilkinson, M.E.; Stone, E.M.; Steinberg, S.J.; Wangler, M.F.; Rush, E.T.; Hacia, J.G.; et al. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol. Genet. Metab. 2016, 117, 313–321. [Google Scholar] [CrossRef]
- Al-Sayed, M.; Al-Hassan, S.; Rashed, M.; Qeba, M.; Coskun, S. Preimplantation genetic diagnosis for Zellweger syndrome. Fertil. Steril. 2007, 87, 146. [Google Scholar] [CrossRef]
- Argyriou, C.; Polosa, A.; Song, J.Y.; Omri, S.; Steele, B.; Cécyre, B.; McDougald, D.S.; Di Pietro, E.; Bouchard, J.F.; Braverman, N.E.; et al. AAV-mediated PEX1 gene augmentation improves visual function in the PEX1-Gly844Asp mouse model for mild Zellweger spectrum disorder. Mol. Ther.-Methods Clin. Dev. 2021, 23, 225–240. [Google Scholar] [CrossRef]
- Rönicke, S.; Kruska, N.; Kahlert, S.; Reiser, G. The influence of the branched-chain fatty acids pristanic acid and Refsum disease-associated phytanic acid on mitochondrial functions and calcium regulation of hippocampal neurons, astrocytes, and oligodendrocytes. Neurobiol. Vol. 2009, 36, 401–410. [Google Scholar] [CrossRef]
- Schwartz, R.A.; Zalewska, A.; Wells, M.J. Refsum Disease. 2021. Available online: https://emedicine.medscape.com/article/1114720-overview#a6 (accessed on 1 January 2023).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Slanina, A.-M.; Coman, A.-E.; Anton-Păduraru, D.-T.; Popa, E.; Barbacariu, C.-L.; Novac, O.; Petroaie, A.D.; Bacușcă, A.-I.; Manole, M.; Cosmescu, A. PEX6 Mutation in a Child with Infantile Refsum Disease—A Case Report and Literature Review. Children 2023, 10, 530. https://doi.org/10.3390/children10030530
Slanina A-M, Coman A-E, Anton-Păduraru D-T, Popa E, Barbacariu C-L, Novac O, Petroaie AD, Bacușcă A-I, Manole M, Cosmescu A. PEX6 Mutation in a Child with Infantile Refsum Disease—A Case Report and Literature Review. Children. 2023; 10(3):530. https://doi.org/10.3390/children10030530
Chicago/Turabian StyleSlanina, Ana-Maria, Adorata-Elena Coman, Dana-Teodora Anton-Păduraru, Elena Popa, Carmen-Liliana Barbacariu, Otilia Novac, Antoneta Dacia Petroaie, Agnes-Iacinta Bacușcă, Mihaela Manole, and Adriana Cosmescu. 2023. "PEX6 Mutation in a Child with Infantile Refsum Disease—A Case Report and Literature Review" Children 10, no. 3: 530. https://doi.org/10.3390/children10030530
APA StyleSlanina, A.-M., Coman, A.-E., Anton-Păduraru, D.-T., Popa, E., Barbacariu, C.-L., Novac, O., Petroaie, A. D., Bacușcă, A.-I., Manole, M., & Cosmescu, A. (2023). PEX6 Mutation in a Child with Infantile Refsum Disease—A Case Report and Literature Review. Children, 10(3), 530. https://doi.org/10.3390/children10030530