
Estimating Drug Efficacy with a Diet-Induced NASH Model in Chimeric Mice with Humanized Livers

 and
and
Abstract
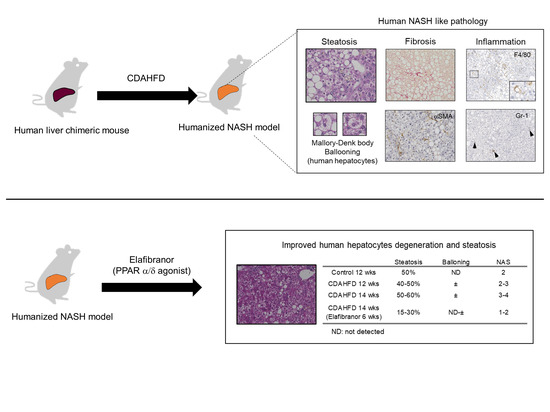
:
1. Introduction
2. Materials and Methods
2.1. Animal and Generation of a CDAHFD-Induced NASH Model
2.2. Elafibranor Efficacy Study for the NASH Model
2.3. Measurement of Biochemical Markers
2.4. Histological Analysis
2.5. Real-Time Quantitative Reverse Transcriptase-PCR (qRT-PCR)
2.6. Measurement of Liver Oxidative Stress
2.7. Statistical Analysis
3. Results
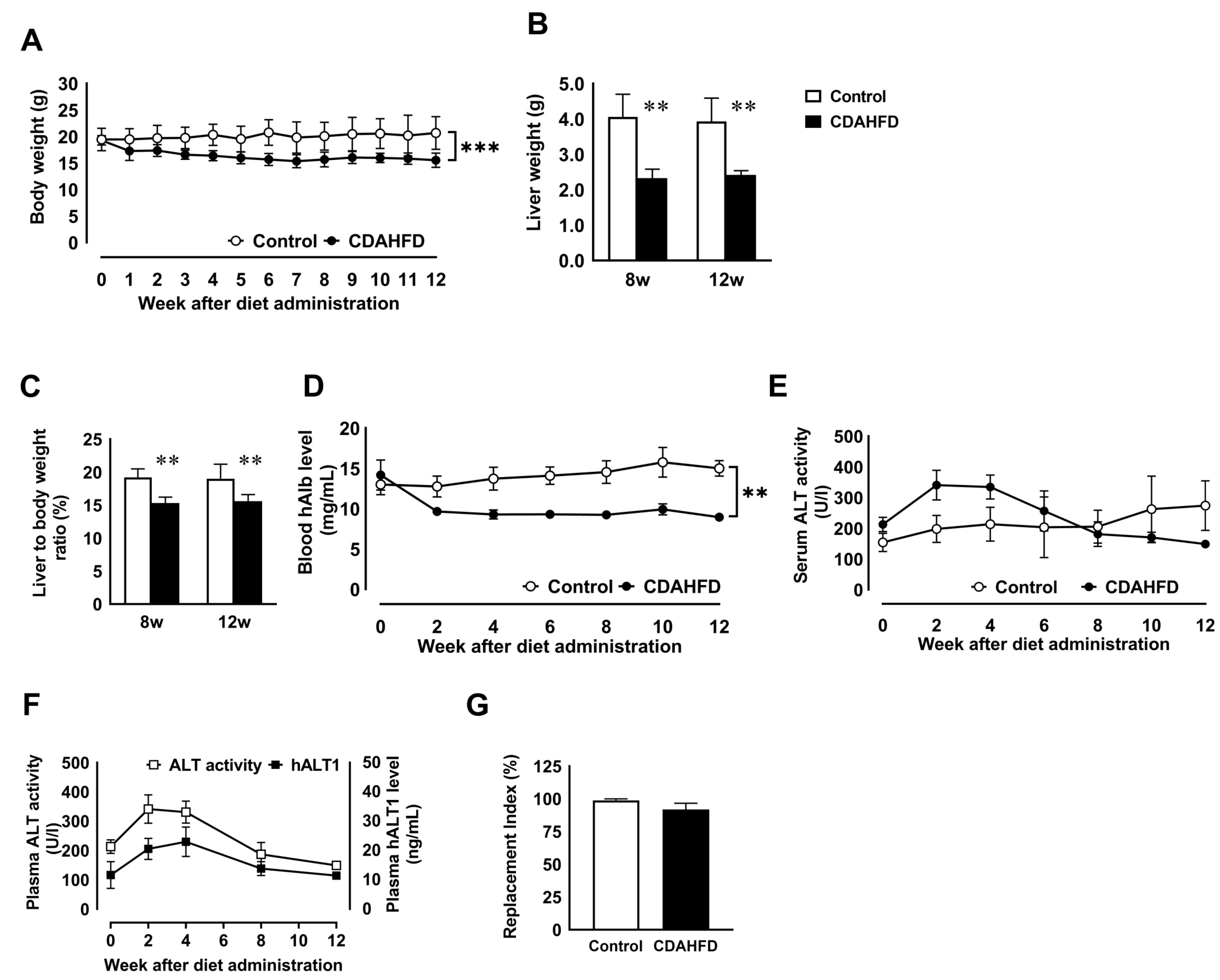
3.1. CDAHFD Feeding Decreased Body and Liver Weights and Deteriorated Liver Function of Chimeric Mice with H-Heps
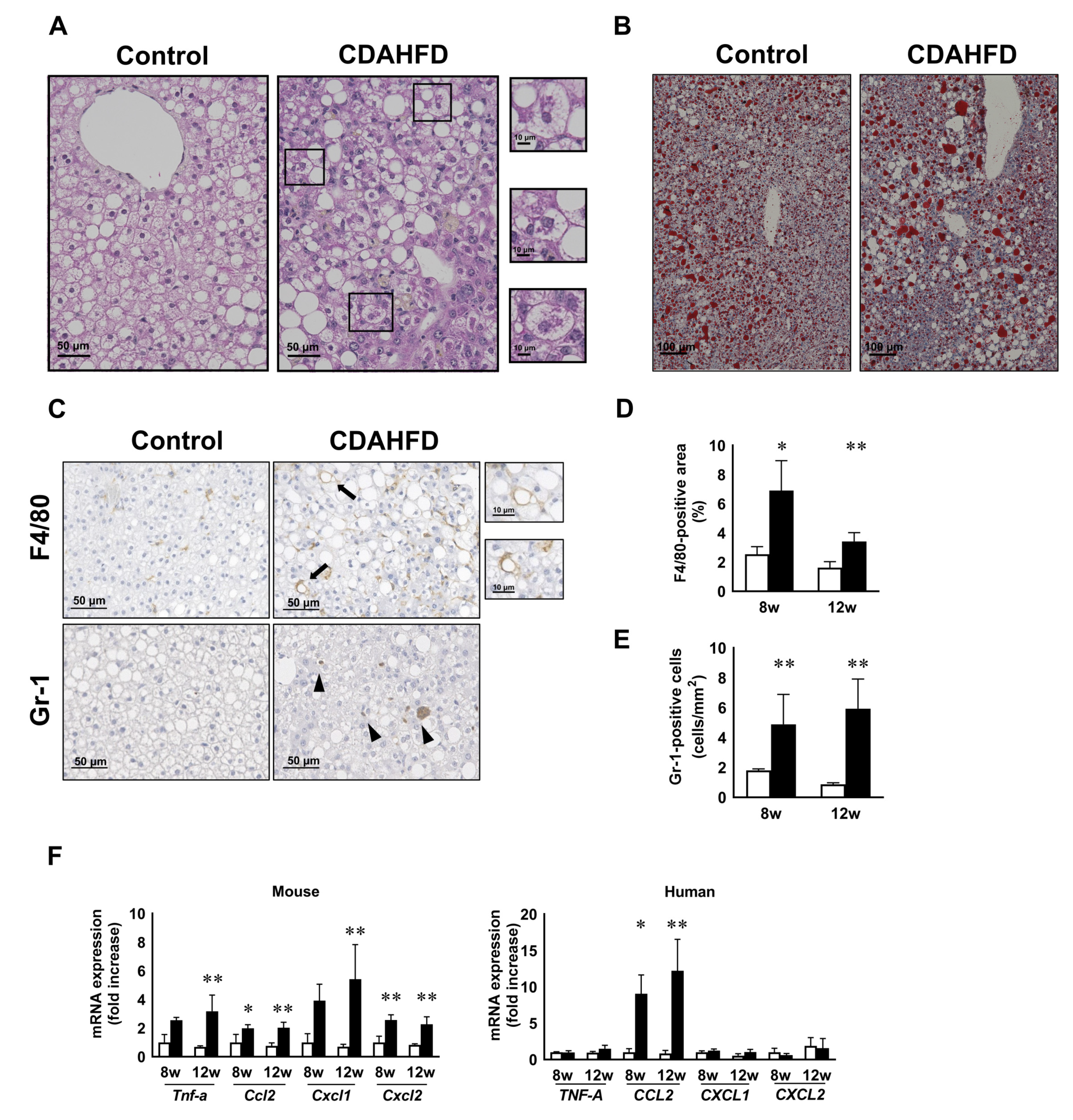
3.2. CDAHFD Feeding Increased H-Hep Ballooning Containing Mallory–Denk Bodies and Inflammatory Cells
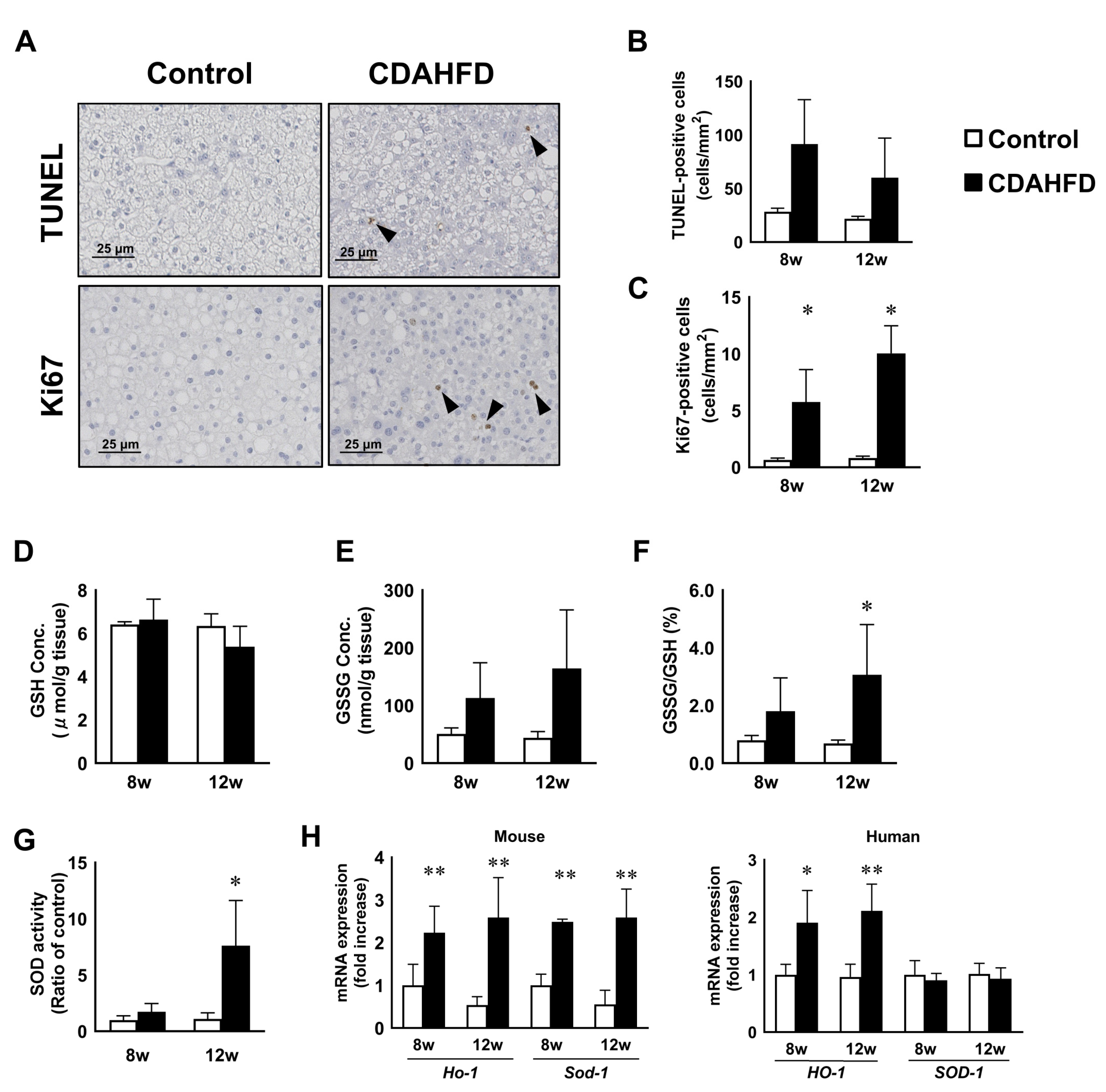
3.3. CDAHFD Feeding Caused Apoptosis, Proliferation of H-Heps, and Oxidative Stress
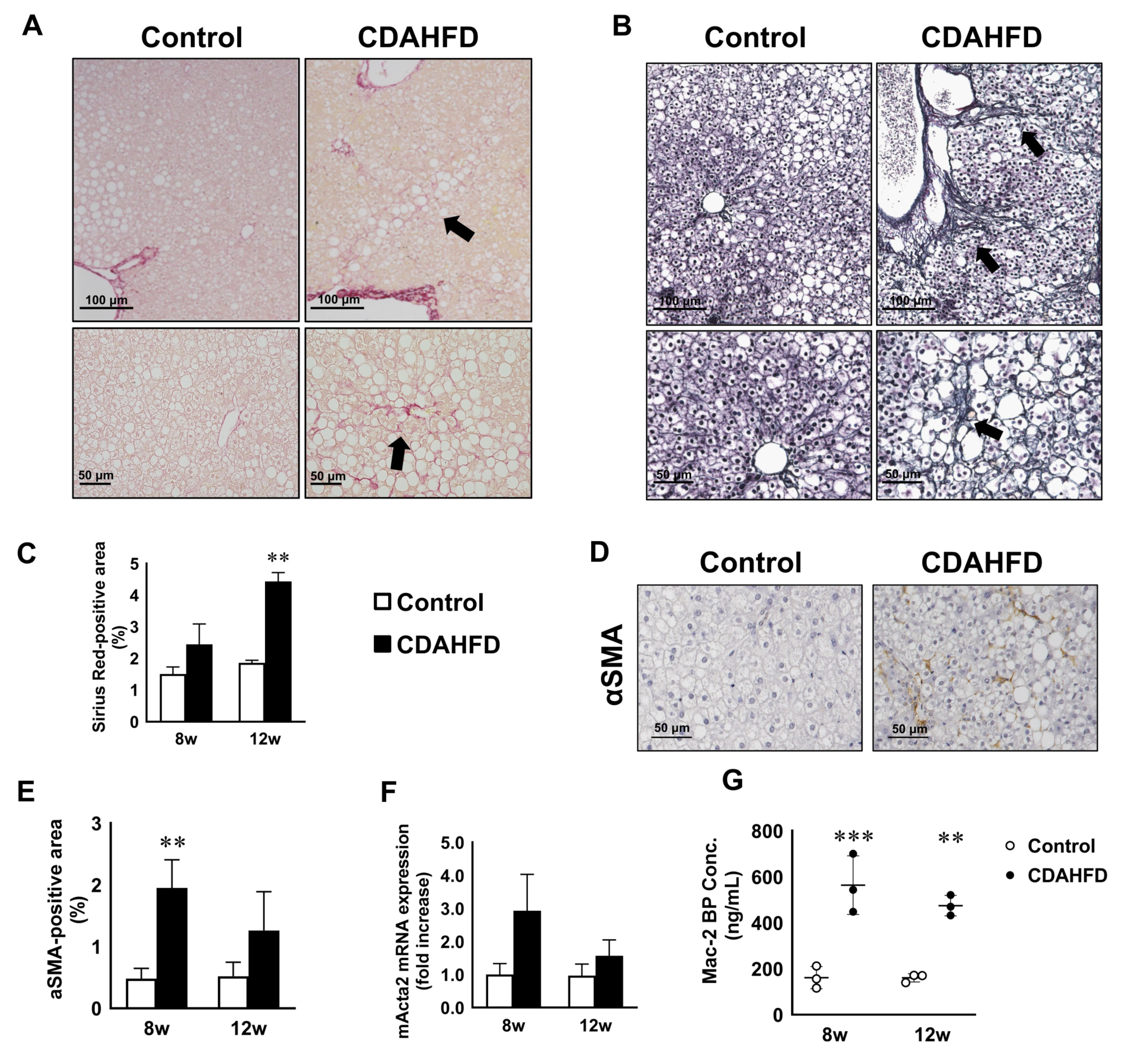
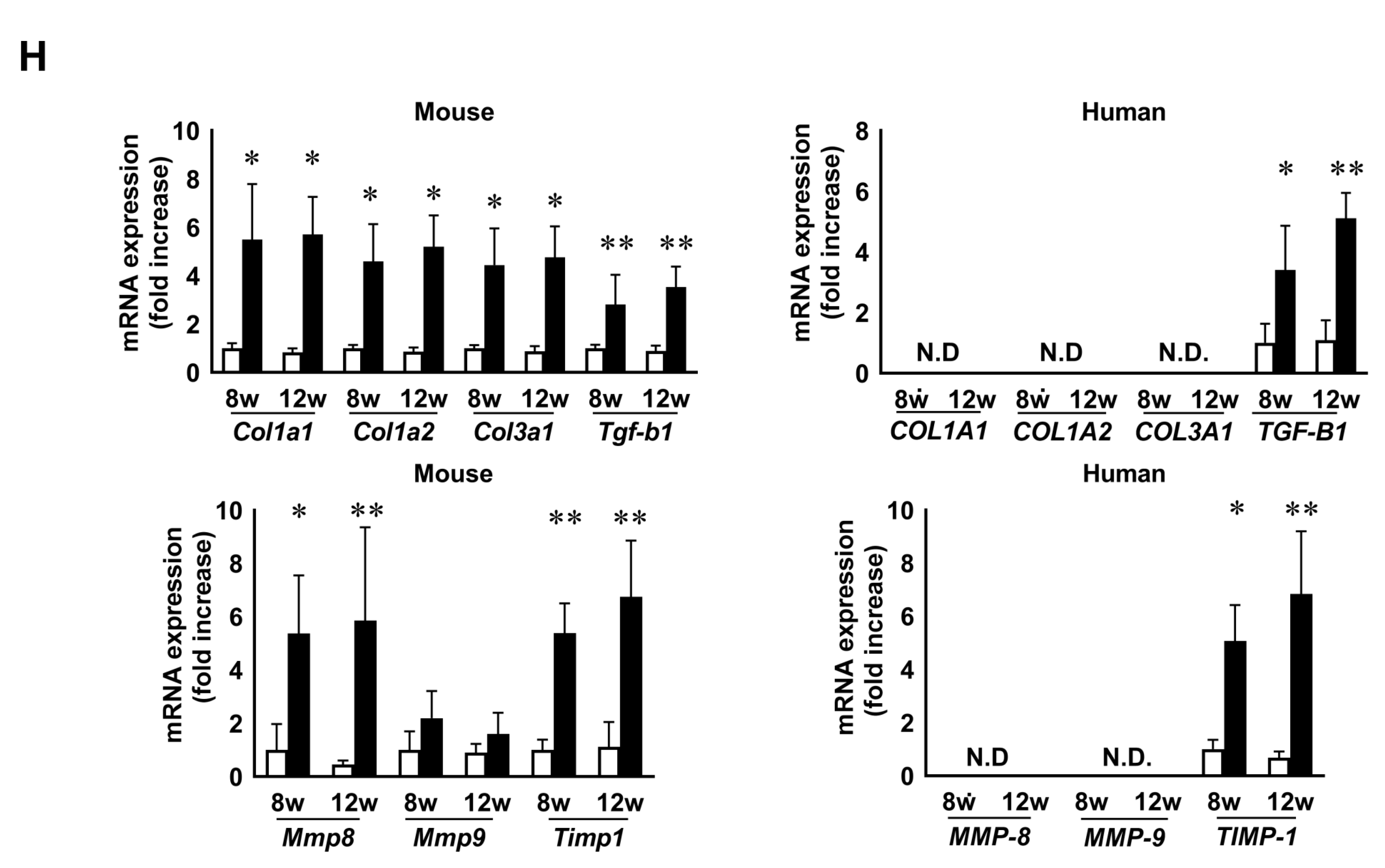
3.4. CDAHFD Feeding Caused Liver Fibrosis Accompanied by Increases of Fibrotic Gene Expression and Mac-2 Binding Protein Concentration in Mouse Sera
3.5. Reversion to a Control Diet Improved Steatosis and H-Hep Ballooning, But Not Fibrosis
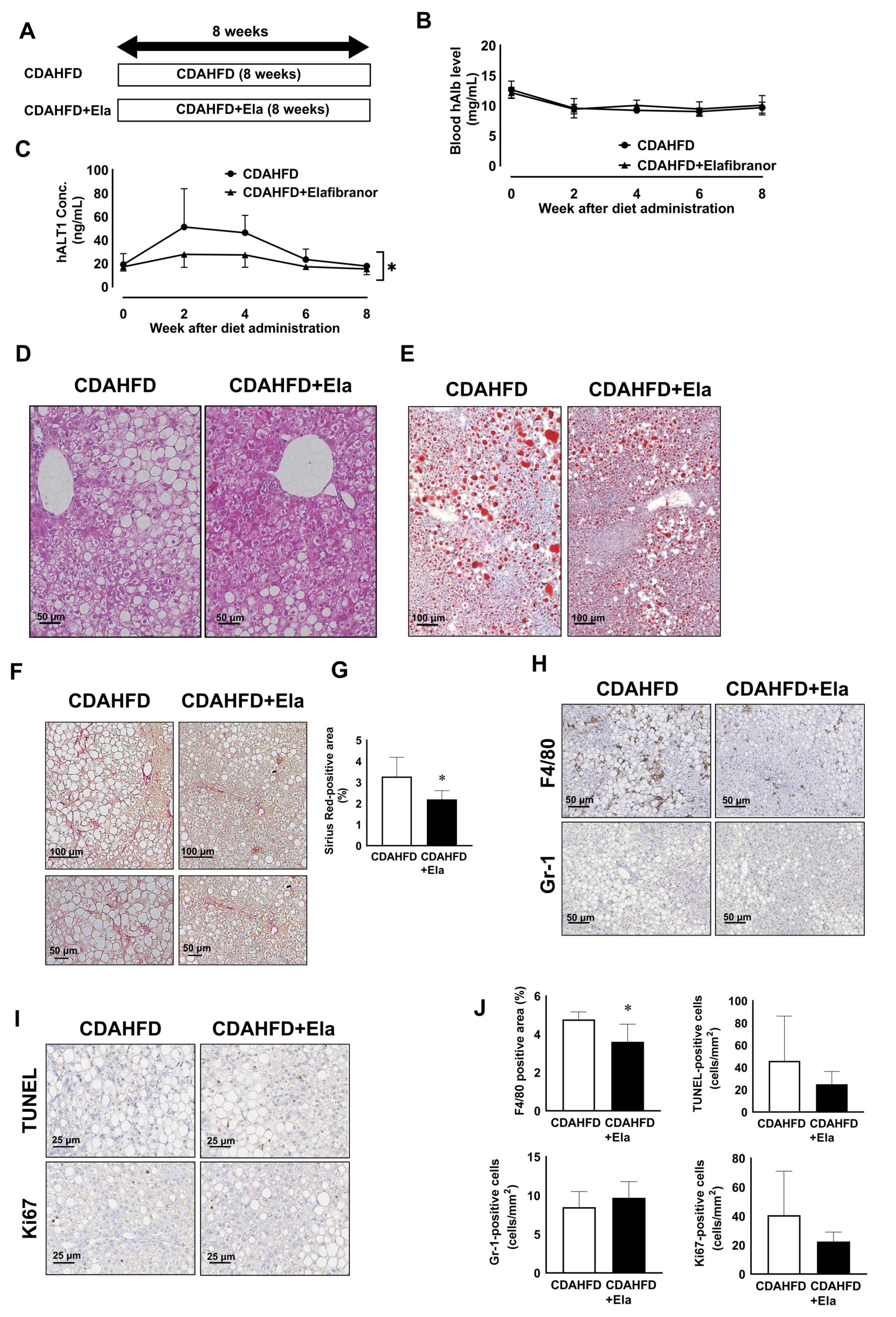
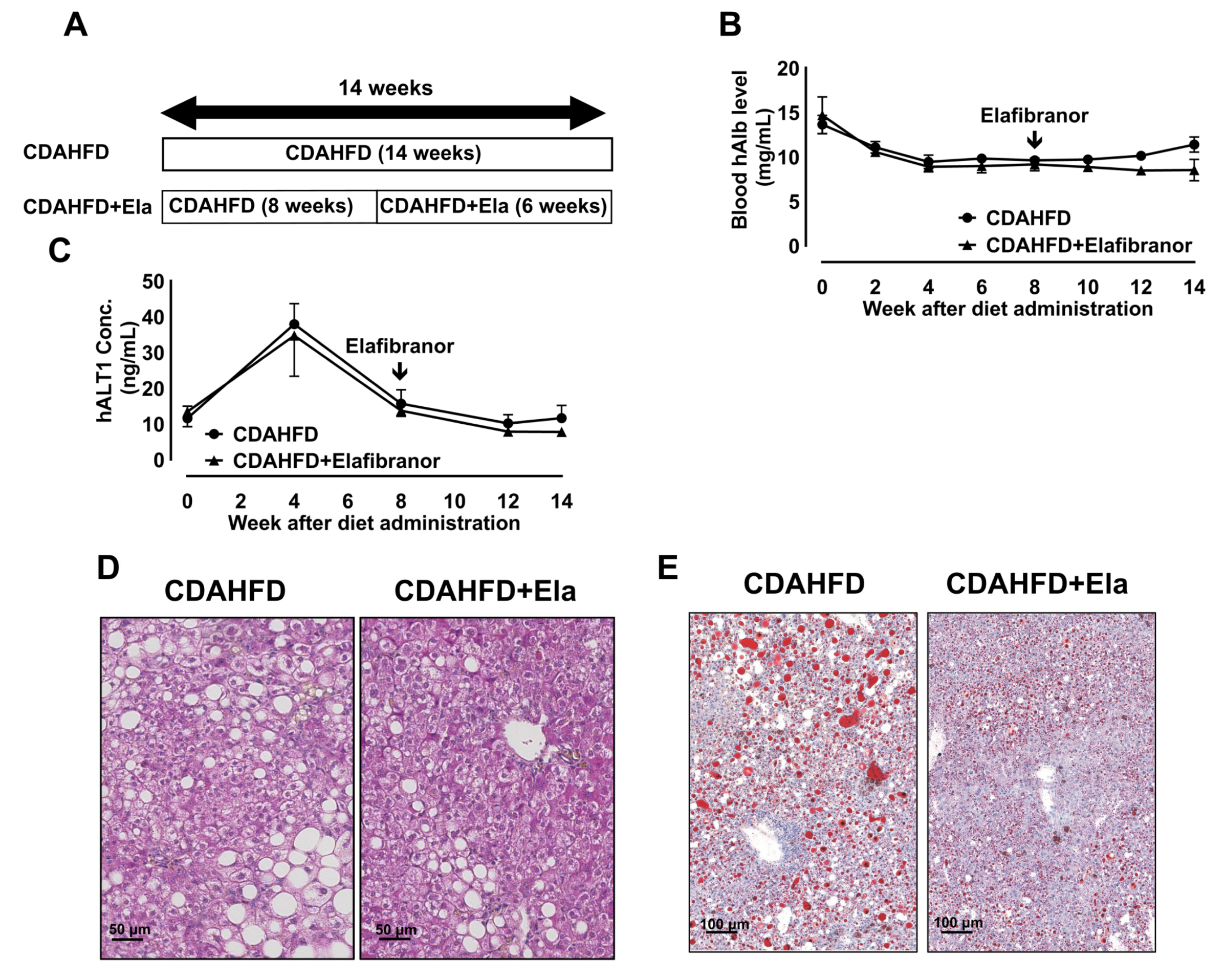
3.6. Prophylactic Treatment of Elafibranor Prevented Steatosis, H-Hep Ballooning, and Fibrosis While the Therapeutic Treatment Improved Steatosis and H-Hep Ballooning, But Not Fibrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mulhall, B.P.; Ong, J.P.; Younossi, Z.M. Non-alcoholic fatty liver disease: An overview. J. Gastroenterol. Hepatol. 2002, 17, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.L.; Chen, H.; Wang, C.L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Mikolasevic, I.; Milic, S.; Turk Wensveen, T.; Grgic, I.; Jakopcic, I.; Stimac, D.; Wensveen, F.; Orlic, L. Nonalcoholic fatty liver disease—A multisystem disease? World J. Gastroenterol. 2016, 22, 9488–9505. [Google Scholar] [CrossRef]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Torgerson, S.; Hayashi, P.H. The natural history of nonalcoholic fatty liver disease: A clinical histopathological study. Am. J. Gastroenterol. 2003, 98, 2042–2047. [Google Scholar] [CrossRef]
- Bertola, A. Rodent models of fatty liver diseases. Liver Res. 2018, 2, 3–13. [Google Scholar] [CrossRef]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; de Almeida, T.P.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulmasov, B.; Noritake, H.; Carmichael, P.; Oshima, K.; Griggs, D.W.; Neuschwander-Tetri, B.A. An Inhibitor of Arginine-Glycine-Aspartate-Binding Integrins Reverses Fibrosis in a Mouse Model of Nonalcoholic Steatohepatitis. Hepatol. Commun. 2019, 3, 246. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.M.; Vance, D.E. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J. Biol. Chem. 1988, 263, 2998–3004. [Google Scholar] [CrossRef]
- Ikawa-Yoshida, A.; Matsuo, S.; Kato, A.; Ohmori, Y.; Higashida, A.; Kaneko, E.; Matsumoto, M. Hepatocellular carcinoma in a mouse model fed a choline-deficient, L-amino acid-defined, high-fat diet. Int. J. Exp. Pathol. 2017, 98, 221–233. [Google Scholar] [CrossRef]
- Tateno, C.; Kawase, Y.; Tobita, Y.; Hamamura, S.; Ohshita, H.; Yokomichi, H.; Sanada, H.; Kakuni, M.; Shiota, A.; Kojima, Y.; et al. Generation of novel chimeric mice with humanized livers by using hemizygous cDNA-uPA/SCID mice. PLoS ONE 2015, 10, e0142145. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Matsui, T.; Okumura, H.; Nakajima, M.; Nishimura, M.; Naito, S.; Tateno, C.; Yoshizato, K.; Yokoi, T. Expression of human phase II enzymes in chimeric mice with humanized liver. Drug Metab. Dispos. 2005, 33, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Okumura, H.; Katoh, M.; Sawada, T.; Nakajima, M.; Soeno, Y.; Yabuuchi, H.; Ikeda, T.; Tateno, C.; Yoshizato, K.; Yokoi, T. Humanization of excretory pathway in chimeric mice with humanized liver. Toxicol. Sci. 2007, 97, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Yoshizato, K.; Tateno, C. A mouse with humanized liver as an animal model for predicting drug effects and for studying hepatic viral infection: Where to next? Expert Opin. Drug Metab. Toxicol. 2013, 9, 1419–1435. [Google Scholar] [CrossRef] [PubMed]
- Tateno, C.; Kataoka, M.; Utoh, R.; Tachibana, A.; Itamoto, T.; Asahara, T.; Miya, F.; Tsunoda, T.; Yoshizato, K. Growth hormone-dependent pathogenesis of human hepatic steatosis in a novel mouse model bearing a human hepatocyte-repopulated liver. Endocrinology 2011, 152, 1479–1491. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Xu, P.; Zhai, Y. The opportunities and challenges of peroxisome proliferator-activated receptors ligands in clinical drug discovery and development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear Receptors and Nonalcoholic Fatty Liver Disease. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5149456/ (accessed on 5 February 2020).
- Motojima, K.; Passilly, P.; Peters, J.M.; Gonzalez, F.J.; Latruffe, N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor α and γ activators in a tissue- and inducer-specific manner. J. Biol. Chem. 1998, 273, 16710–16714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckmans, J.; Natale, A.; Rombaut, M.; Buyl, K.; Rogiers, V.; De Kock, J.; Vanhaecke, T.; Rodrigues, R.M. Anti-NASH Drug Development Hitches a Lift on PPAR Agonism. Cells 2020, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Rakhshandehroo, M.; Hooiveld, G.; Müller, M.; Kersten, S. Comparative analysis of gene regulation by the transcription factor PPARα between mouse and human. PLoS ONE 2009, 4, e0006796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tateno, C.; Yamamoto, T.; Utoh, R.; Yamasaki, C.; Ishida, Y.; Myoken, Y.; Oofusa, K.; Okada, M.; Tsutsui, N.; Yoshizato, K. Chimeric mice with hepatocyte-humanized liver as an appropriate model to study human peroxisome proliferator-activated receptor-α. Toxicol. Pathol. 2015, 43, 233–248. [Google Scholar] [CrossRef]
- de la Rosa Rodriguez, M.A.; Sugahara, G.; Hooiveld, G.J.E.J.; Ishida, Y.; Tateno, C.; Kersten, S. The whole transcriptome effects of the PPARα agonist fenofibrate on livers of hepatocyte humanized mice. BMC Genomics 2018, 19, 443. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Tølbøl, K.S.; Kristiansen, M.N.B.; Hansen, H.H.; Veidal, S.S.; Rigbolt, K.T.G.; Gillum, M.P.; Jelsing, J.; Vrang, N.; Feigh, M. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet-induced obese mouse models of biopsy-confirmed nonalcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 179–194. [Google Scholar] [CrossRef]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Briand, F.; Maupoint, J.; Brousseau, E.; Breyner, N.; Bouchet, M.; Costard, C.; Leste-Lasserre, T.; Petitjean, M.; Chen, L.; Chabrat, A.; et al. Elafibranor improves diet-induced nonalcoholic steatohepatitis associated with heart failure with preserved ejection fraction in Golden Syrian hamsters. Metab. Clin. Exp. 2021, 117, 154707. [Google Scholar] [CrossRef]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator- activated receptor α/δ agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- Cariou, B.; Zaïr, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPARα/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, Y.; Yamasaki, C.; Iwanari, H.; Yamashita, H.; Ogawa, Y.; Yanagi, A.; Furukawa, S.; Kojima, Y.; Chayama, K.; Kamiie, J.; et al. Detection of acute toxicity of aflatoxin B1 to human hepatocytes in vitro and in vivo using chimeric mice with humanized livers. PLoS ONE 2020, 15, e0239540. [Google Scholar] [CrossRef]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201. [Google Scholar] [CrossRef] [Green Version]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Kamada, Y.; Ono, M.; Hyogo, H.; Fujii, H.; Sumida, Y.; Yamada, M.; Mori, K.; Tanaka, S.; Maekawa, T.; Ebisutani, Y.; et al. Use of Mac-2 binding protein as a biomarker for nonalcoholic fatty liver disease diagnosis. Hepatol. Commun. 2017, 1, 780–791. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362. [Google Scholar] [CrossRef] [Green Version]
- Imajo, K.; Yoneda, M.; Kessoku, T.; Ogawa, Y.; Maeda, S.; Sumida, Y.; Hyogo, H.; Eguchi, Y.; Wada, K.; Nakajima, A. Rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Int. J. Mol. Sci. 2013, 14, 21833–21857. [Google Scholar] [CrossRef] [Green Version]
- Tateno, C.; Miya, F.; Wake, K.; Kataoka, M.; Ishida, Y.; Yamasaki, C.; Yanagi, A.; Kakuni, M.; Wisse, E.; Verheyen, F.; et al. Morphological and microarray analyses of human hepatocytes from xenogeneic host livers. Lab. Investig. 2012, 93, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Ohtsuki, S.; Kawakami, H.; Inoue, T.; Nakamura, K.; Tateno, C.; Katsukura, Y.; Obuchi, W.; Uchida, Y.; Kamiie, J.; Horie, T.; et al. Validation of uPA/SCID mouse with humanized liver as a human liver model: Protein quantification of transporters, cytochromes P450, and UDP-glucuronosyltransferases by LC-MS/MS. Drug Metab. Dispos. 2014, 42, 1039–1043. [Google Scholar] [CrossRef]
- Steenbergen, R.H.G.; Joyce, M.A.; Lund, G.; Lewis, J.; Chen, R.; Barsby, N.; Zhu, L.F.; Tyrrell, D.L.J.; Kneteman, N.M. Lipoprotein profiles in SCID/uPA mice transplanted with human hepatocytes become human-like and correlate with HCV infection success. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Okuda, Y.; Kushida, M.; Sumida, K.; Takeuchi, H.; Nagahori, H.; Fukuda, T.; Lake, B.G.; Cohen, S.M.; Kawamura, S. Human Hepatocytes Support the Hypertrophic but not the Hyperplastic Response to the Murine Nongenotoxic Hepatocarcinogen Sodium Phenobarbital in an In Vivo Study Using a Chimeric Mouse with Humanized Liver. Toxicol. Sci. 2014, 142, 137–157. [Google Scholar] [CrossRef] [PubMed]
- Souza, S.C.; Frick, G.P.; Wang, X.; Kopchick, J.J.; Lobo, R.B.; Goodman, H.M. A single arginine residue determines species specificity of the human growth hormone receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 959–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utoh, R.; Tateno, C.; Kataoka, M.; Tachibana, A.; Masumoto, N.; Yamasaki, C.; Shimada, T.; Itamoto, T.; Asahara, T.; Yoshizato, K. Hepatic hyperplasia associated with discordant xenogeneic parenchymal-nonparenchymal interactions in human hepatocyte-repopulated mice. Am. J. Pathol. 2010, 177, 654–665. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Angulo, P.; Lymp, J.; Li, D.; Satomura, S.; Lindor, K. Hyaluronic acid, an accurate serum marker for severe hepatic fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2005, 25, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse models of nonalcoholic steatohepatitis: Toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Naugler, W.E.; Tarlow, B.D.; Fedorov, L.M.; Taylor, M.; Pelz, C.; Li, B.; Darnell, J.; Grompe, M. Fibroblast growth factor signaling controls liver size in mice with humanized livers. Gastroenterology 2015, 149, 728.e15–740.e15. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Sugawara, H.; Sujaku, K.; Hashimoto, O.; Tsuji, R.; Tamaki, S.; Torimura, T.; Inuzuka, S.; Sata, M.; Tanikawa, K. Therapeutic effects of restricted diet and exercise in obese patients with fatty liver. J. Hepatol. 1997, 27, 103–107. [Google Scholar] [CrossRef]
- Harrison, S.A.; Fecht, W.; Brunt, E.M.; Neuschwander-Tetri, B.A. Orlistat for overweight subjects with nonalcoholic steatohepatitis: A randomized, prospective trial. Hepatology 2009, 49, 80–86. [Google Scholar] [CrossRef]
- Tachibana, K.; Yamasaki, D.; Ishimoto, K.; Doi, T. The Role of PPARs in Cancer. PPAR Res. 2008, 2008, 102737. [Google Scholar] [CrossRef] [Green Version]
- Iwaisako, K.; Haimerl, M.; Paik, Y.H.; Taura, K.; Kodama, Y.; Sirlin, C.; Yu, E.; Yu, R.T.; Downes, M.; Evans, R.M.; et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor δ agonist. Proc. Natl. Acad. Sci. USA 2012, 109, E1369–E1376. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal No. | Steatosis (%) | Ballooning | Lobular Inflammation | NAFLD Activity Score | ||
|---|---|---|---|---|---|---|
| control | 8 w | 1 | 60 | N.D. | ± | 3 |
| 2 | 50 | N.D. | N.D. | 2 | ||
| 3 | 40 | N.D. | + | 3 | ||
| 12 w | 4 | 50 | N.D. | ± | 2 | |
| 5 | 50 | N.D. | N.D. | 2 | ||
| 6 | 50 | N.D. | N.D. | 2 | ||
| 7 | 50 | N.D. | N.D. | 2 | ||
| CDAHFD | 8 w | 8 | 50 | N.D. | N.D. | 2 |
| 9 | 40 | N.D. | ± | 3 | ||
| 10 | 40 | N.D. | N.D. | 2 | ||
| 11 | 40 | N.D. | N.D. | 2 | ||
| 12 w | 12 | 50 | + | ± | 3 | |
| 13 | 50 | + | N.D. | 2 | ||
| 14 | 40 | + | N.D. | 2 |
| Animal No. | Steatosis (%) | Ballooning | Lobular Inflammation | NAFLD Activity Score | |
|---|---|---|---|---|---|
| CDAHFD | A | 60 | ± | ± | 4 |
| B | 60 | ± | N.D. | 3 | |
| C | 60 | + | N.D. | 3 | |
| D | 50 | + | N.D. | 3 | |
| CDAHFD + Elafibranor | E | 30 | + | N.D. | 2 |
| F | 30 | N.D. | N.D. | 1 | |
| G | 15 | N.D. | N.D. | 1 | |
| H | 20 | N.D. | ± | 2 | |
| I | 20 | N.D. | ± | 2 | |
| J | 33 | N.D. | ± | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kisoh, K.; Sugahara, G.; Ogawa, Y.; Furukawa, S.; Ishida, Y.; Okanoue, T.; Kohara, M.; Tateno, C. Estimating Drug Efficacy with a Diet-Induced NASH Model in Chimeric Mice with Humanized Livers. Biomedicines 2021, 9, 1647. https://doi.org/10.3390/biomedicines9111647
Kisoh K, Sugahara G, Ogawa Y, Furukawa S, Ishida Y, Okanoue T, Kohara M, Tateno C. Estimating Drug Efficacy with a Diet-Induced NASH Model in Chimeric Mice with Humanized Livers. Biomedicines. 2021; 9(11):1647. https://doi.org/10.3390/biomedicines9111647
Chicago/Turabian StyleKisoh, Keishi, Go Sugahara, Yuko Ogawa, Suzue Furukawa, Yuji Ishida, Takeshi Okanoue, Michinori Kohara, and Chise Tateno. 2021. "Estimating Drug Efficacy with a Diet-Induced NASH Model in Chimeric Mice with Humanized Livers" Biomedicines 9, no. 11: 1647. https://doi.org/10.3390/biomedicines9111647
APA StyleKisoh, K., Sugahara, G., Ogawa, Y., Furukawa, S., Ishida, Y., Okanoue, T., Kohara, M., & Tateno, C. (2021). Estimating Drug Efficacy with a Diet-Induced NASH Model in Chimeric Mice with Humanized Livers. Biomedicines, 9(11), 1647. https://doi.org/10.3390/biomedicines9111647