The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing



Abstract
1. Introduction
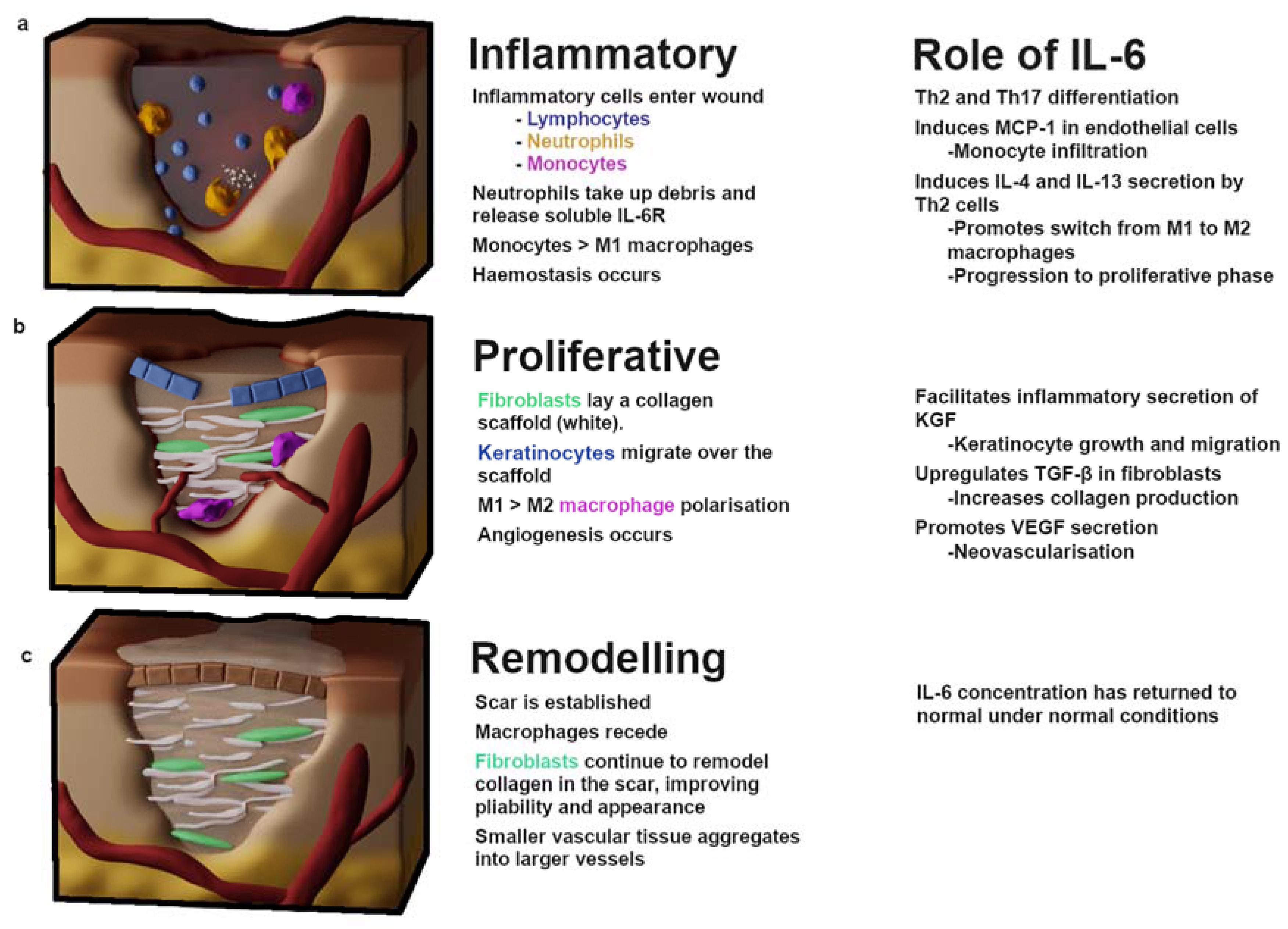
2. Wound Healing
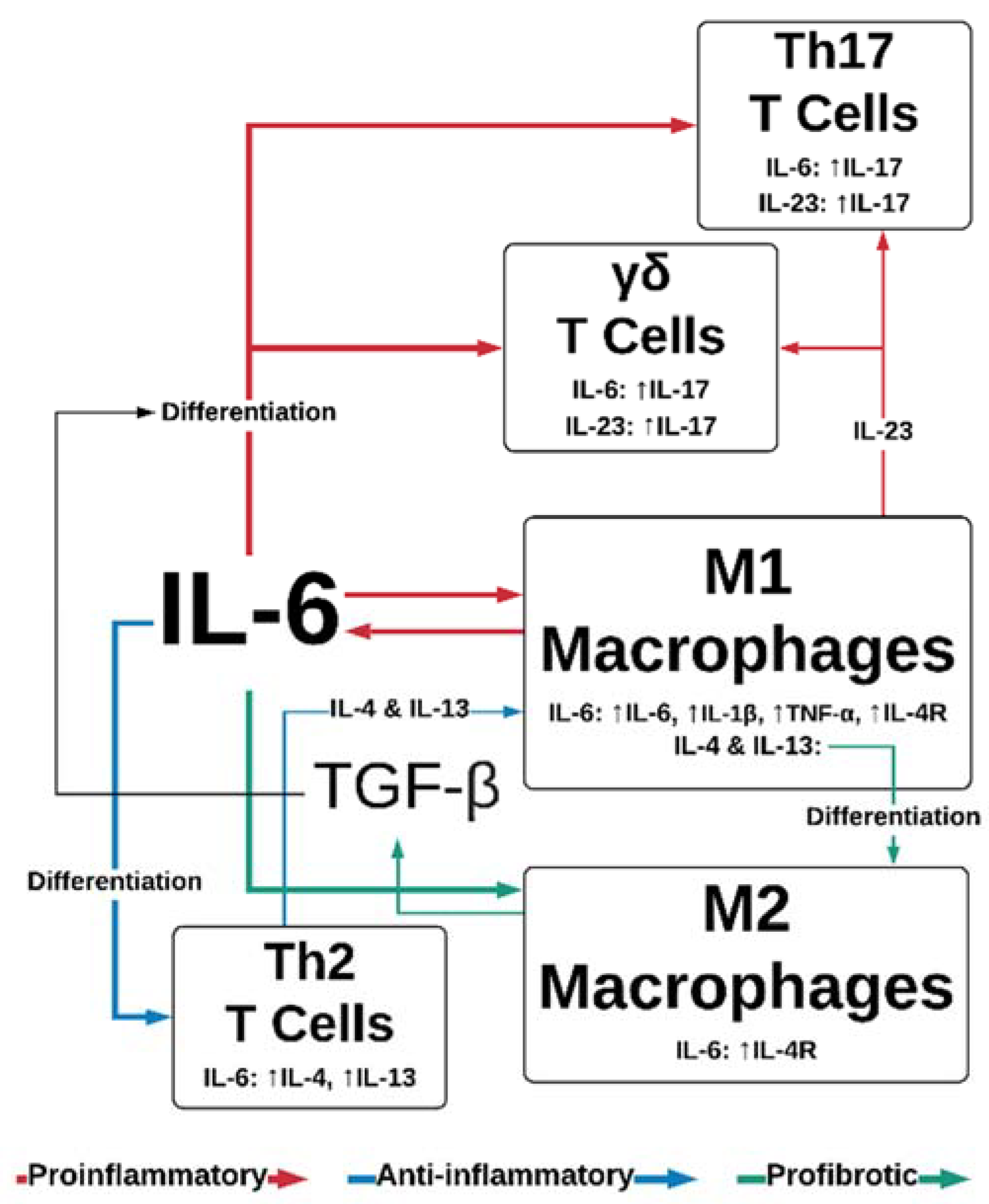
2.1. Inflammation
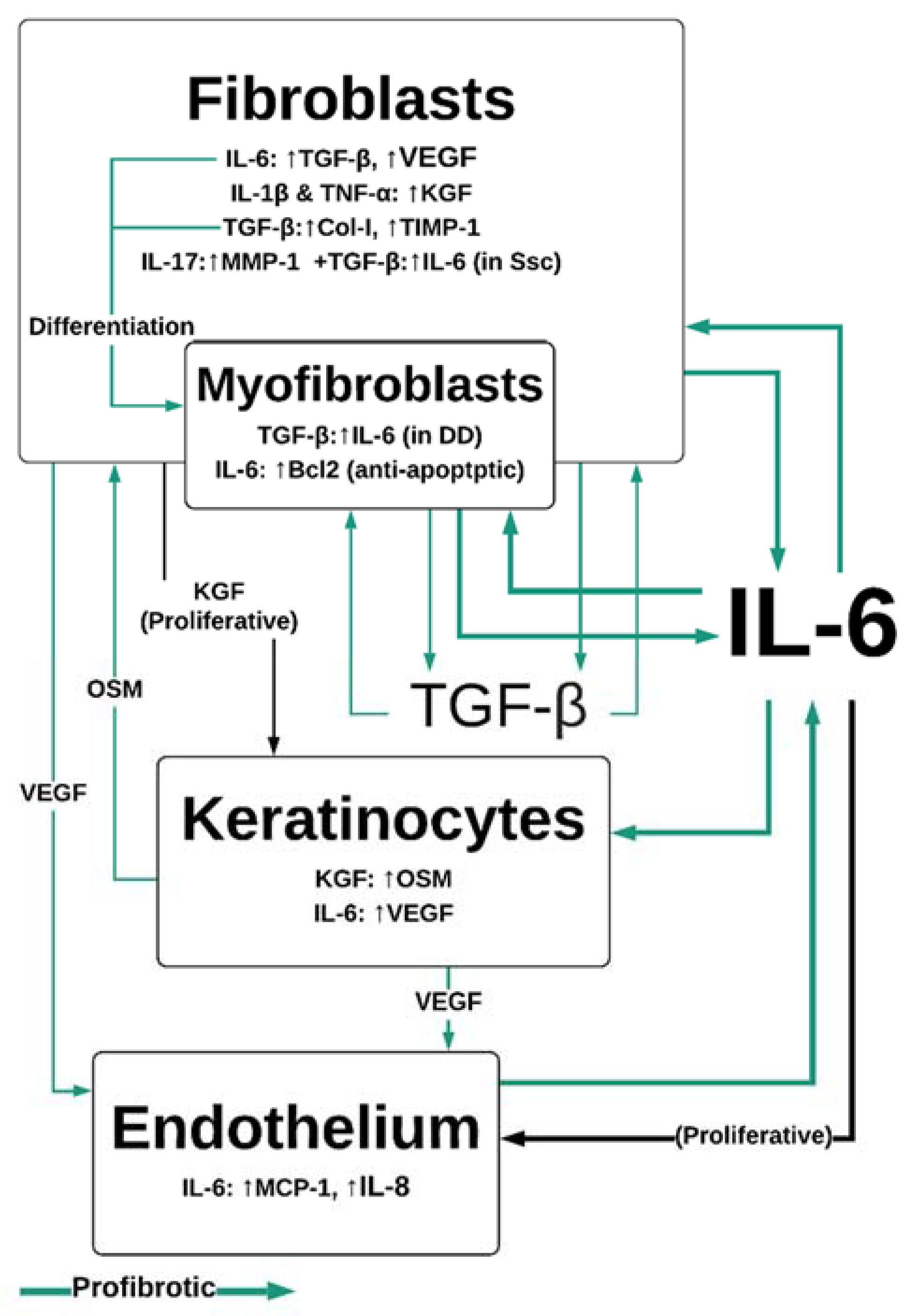
2.2. Proliferation
3. Fibrotic Diseases of the Skin
3.1. Hypertrophic Scarring
3.2. Keloids
3.3. Scleroderma and Systemic Sclerosis
3.4. Dupuytren’s Disease
3.5. Treatments Modulating IL-6 (Cutaneous Fibrosis)
3.5.1. Corticosteroids
3.5.2. Verapamil
3.5.3. IL-6 Blockade
3.5.4. Pirfenidone
4. Age, Obesity, Diabetes and Chronic Wounds
4.1. Age
4.2. Diabetes and Obesity
4.3. Treatments Modulating IL-6 (Wound Healing)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lin, Z.-Q.; Kondo, T.; Ishida, Y.; Takayasu, T.; Mukaida, N. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol. 2003, 73, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T. The biology of interleukin-6. Blood 1989, 74, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Weissenbach, M.; Clahsen, T.; Weber, C.; Spitzer, D.; Wirth, D.; Vestweber, D.; Heinrich, P.C.; Schaper, F. Interleukin-6 is a direct mediator of T cell migration. Eur. J. Immunol. 2004, 34, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Cross, A.L.; Edwards, S.W.; Moots, R.J. Effects of IL-6 and IL-6 blockade on neutrophil function in vitro and in vivo. Rheumatology 2014, 53, 1321–1331. [Google Scholar] [CrossRef]
- Nishikai-Yan Shen, T.; Kanazawa, S.; Kado, M.; Okada, K.; Luo, L.; Hayashi, A.; Mizuno, H.; Tanaka, R. Interleukin-6 stimulates Akt and p38 MAPK phosphorylation and fibroblast migration in non-diabetic but not diabetic mice. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Roytblat, L.; Rachinsky, M.; Fisher, A.; Greemberg, L.; Shapira, Y.; Douvdevani, A.; Gelman, S. Raised Interleukin-6 Levels in Obese Patients. Obes. Res. 2000, 8, 673–675. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, T.W.; Park, Y.E.; Lee, Y.J. Modified Resection Arthroplasty for Infected Non-healing Ulcers with Toe Deformity in Diabetic Patients. Foot Ankle Int. 2008, 29, 493–497. [Google Scholar] [CrossRef]
- Jones, J.; Barr, W.; Robinson, J.; Carlisle, C. Depression in patients with chronic venous ulceration. Br. J. Nurs. 2006, 15, S17–S23. [Google Scholar] [CrossRef]
- Han, G.; Ceilley, R. Chronic Wound Healing: A Review of Current Management and Treatments. Advances 2017, 34, 599–610. [Google Scholar] [CrossRef]
- Dunkin, C.S.J.; Pleat, J.M.; Gillespie, P.H.; Tyler, M.P.H.; Roberts, A.H.N.; McGrouther, D.A. Scarring occurs at a critical depth of skin injury: Precise measurement in a graduated dermal scratch in human volunteers. Plast. Reconstr. Surg. 2007, 119, 1722–1732. [Google Scholar] [CrossRef]
- Bayat, A.; McGrouther, D.A.; Ferguson, M.W.J. Skin scarring. BMJ 2003, 326, 88–92. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, S.T.; O’Shaughnessy, M.; O’Connor, T.P. Aetiology and management of hypertrophic scars and keloids. Ann. R. Coll. Surg. Engl. 1996, 78, 168–175. [Google Scholar] [PubMed]
- Sorkin, M.; Cholok, D.; Levi, B. Scar Management of the Burned Hand. Hand Clin. 2017, 33, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Falder, S.; Browne, A.; Edgar, D.; Staples, E.; Fong, J.; Rea, S.; Wood, F. Core outcomes for adult burn survivors: A clinical overview. Burns 2009, 35, 618–641. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.C.; McKenna, S.P.; Siddhi, K.; McGrouther, D.A.; Bayat, A. The hidden cost of skin scars: Quality of life after skin scarring. J. Plast. Reconstr. Aesthetic Surg. 2008, 61, 1049–1058. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Kondo, T.; Ohshima, T. The dynamics of inflammatory cytokines in the healing process of mouse skin wound: A preliminary study for possible wound age determination. Int. J. Leg. Med. 1996, 108, 231–236. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef]
- Gemperle, C.; Schmid, M.; Herova, M.; Marti-Jaun, J.; Wuest, S.J.A.; Loretz, C.; Hersberger, M. Regulation of the Formyl Peptide Receptor 1 (FPR1) Gene in Primary Human Macrophages. PLoS ONE 2012, 7, e50195. [Google Scholar] [CrossRef]
- Zhang, X.; Mosser, D. Macrophage activation by endogenous danger signals. J. Pathol. 2008, 214, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Masters, A.R.; Fortner, K.A.; Champagne, D.P.; Yanguas-Casás, N.; Silberger, D.J.; Weaver, C.T.; Haynes, L.; Rincon, M. IL-6 promotes the differentiation of a subset of naive CD8+ T cells into IL-21–producing B helper CD8+ T cells. J. Exp. Med. 2016, 213, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Nish, S.A.; Schenten, D.; Wunderlich, F.T.; Pope, S.D.; Gao, Y.; Hoshi, N.; Yu, S.; Yan, X.; Lee, H.K.; Pasman, L.; et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Brandacher, G.; Steurer, W.; Kaser, S.; Offner, F.A.; Zoller, H.; Theurl, I.; Widder, W.; Molnar, C.; Ludwiczek, O.; et al. Interleukin-6 stimulates thrombopoiesis through thrombopoietin: Role in inflammatory thrombocytosis. Blood 2001, 98, 2720–2725. [Google Scholar] [CrossRef]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. 2006, 8, S3. [Google Scholar] [CrossRef]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Prame Kumar, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef]
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076. [Google Scholar] [CrossRef]
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. IL-6 Trans-Signaling via the Soluble IL-6 Receptor: Importance for the Pro-Inflammatory Activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, G.; Saler, M.; Villani, L.; Rumolo, A.; Tresoldi, M.M.; Faga, A. Platelet Rich Plasma Enhancement of Skin Regeneration in an ex-vivo Human Experimental Model. Front. Bioeng. Biotechnol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Masuki, H.; Okudera, T.; Watanebe, T.; Suzuki, M.; Nishiyama, K.; Okudera, H.; Nakata, K.; Uematsu, K.; Su, C.-Y.; Kawase, T. Growth factor and pro-inflammatory cytokine contents in platelet-rich plasma (PRP), plasma rich in growth factors (PRGF), advanced platelet-rich fibrin (A-PRF), and concentrated growth factors (CGF). Int. J. Implant. Dent. 2016, 2, 19. [Google Scholar] [CrossRef]
- Varkey, M.; Ding, J.; Tredget, E.E. Fibrotic Remodeling of Tissue-Engineered Skin with Deep Dermal Fibroblasts Is Reduced by Keratinocytes. Tissue Eng. Part A 2013, 716–727. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor Necrosis Factor-α Signaling in Macrophages. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 87. [Google Scholar] [CrossRef]
- Madej, M.P.; Töpfer, E.; Boraschi, D.; Italiani, P. Different Regulation of Interleukin-1 Production and Activity in Monocytes and Macrophages: Innate Memory as an Endogenous Mechanism of IL-1 Inhibition. Front. Pharm. 2017, 8. [Google Scholar] [CrossRef]
- Tang, A.; Gilchrest, B.A. Regulation of keratinocyte growth factor gene expression in human skin fibroblasts. J. Dermatol. Sci. 1996, 11, 41–50. [Google Scholar] [CrossRef]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte–Fibroblast Interactions in Wound Healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef]
- Canady, J.; Arndt, S.; Karrer, S.; Bosserhoff, A.K. Increased KGF Expression Promotes Fibroblast Activation in a Double Paracrine Manner Resulting in Cutaneous Fibrosis. J. Investig. Dermatol. 2013, 133, 647–657. [Google Scholar] [CrossRef]
- Peng, Y.; Wu, S.; Tang, Q.; Li, S.; Peng, C. KGF-1 accelerates wound contraction through the TGF-β1/Smad signaling pathway in a double-paracrine manner. J. Biol. Chem. 2019, 294, 8361–8370. [Google Scholar] [CrossRef] [PubMed]
- Braune, J.; Weyer, U.; Hobusch, C.; Mauer, J.; Brüning, J.C.; Bechmann, I.; Gericke, M. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J. Immunol. 2017, 198, 2927–2934. [Google Scholar] [CrossRef] [PubMed]
- Luckett-Chastain, L.; Calhoun, K.; Schartz, T.; Gallucci, R.M. IL-6 influences the balance between M1 and M2 macrophages in a mouse model of irritant contact dermatitis. J. Immunol. 2016, 196, 196.17. [Google Scholar]
- Chen, L.; Wang, S.; Wang, Y.; Zhang, W.; Ma, K.; Hu, C.; Zhu, H.; Liang, S.; Liu, M.; Xu, N. IL-6 influences the polarization of macrophages and the formation and growth of colorectal tumor. Oncotarget 2018, 9, 17443–17454. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, P6–P13. [Google Scholar] [CrossRef]
- Dufour, A.M.; Alvarez, M.; Russo, B.; Chizzolini, C. Interleukin-6 and Type-I Collagen Production by Systemic Sclerosis Fibroblasts Are Differentially Regulated by Interleukin-17A in the Presence of Transforming Growth Factor-Beta 1. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-Smooth Muscle Actin Expression Upregulates Fibroblast Contractile Activity. MBoC 2001, 12, 2730–2741. [Google Scholar] [CrossRef]
- Gallucci, R.M.; Lee, E.G.; Tomasek, J.J. IL-6 Modulates Alpha-Smooth Muscle Actin Expression in Dermal Fibroblasts from IL-6-Deficient Mice. J. Investig. Dermatol. 2006, 126, 561–568. [Google Scholar] [CrossRef]
- Catar, R.; Witowski, J.; Zhu, N.; Lücht, C.; Soria, A.D.; Fernandez, J.U.; Chen, L.; Jones, S.A.; Fielding, C.A.; Rudolf, A.; et al. IL-6 Trans–Signaling Links Inflammation with Angiogenesis in the Peritoneal Membrane. JASN 2017, 28, 1188–1199. [Google Scholar] [CrossRef]
- Hou, T.; Tieu, B.C.; Ray, S.; Recinos III, A.; Cui, R.; Tilton, R.G.; Brasier, A.R. Roles of IL-6-gp130 Signaling in Vascular Inflammation. Curr. Cardiol. Rev. 2008, 4, 179–192. [Google Scholar] [CrossRef]
- Fee, D.; Grzybicki, D.; Dobbs, M.; Ihyer, S.; Clotfelter, J.; Macvilay, S.; Hart, M.N.; Sandor, M.; Fabry, Z. Interleukin 6 promotes vasculogenesis of murine brain microvessel endothelial cells. Cytokine 2000, 12, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.F.; Yeo, K.T.; Berse, B.; Yeo, T.K.; Senger, D.R.; Dvorak, H.F.; van de Water, L. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J. Exp. Med. 1992, 176, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lee, K.; Park, S.W.; Chung, H.; Jung, D.; Na, Y.R.; Quan, H.; Cho, C.S.; Che, J.-H.; Kim, J.H.; et al. Lactic Acid Upregulates VEGF Expression in Macrophages and Facilitates Choroidal Neovascularization. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3747–3754. [Google Scholar] [CrossRef] [PubMed]
- Kwak, N.; Okamoto, N.; Wood, J.M.; Campochiaro, P.A. VEGF is major stimulator in model of choroidal neovascularization. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3158–3164. [Google Scholar]
- Streit, M.; Velasco, P.; Riccardi, L.; Spencer, L.; Brown, L.F.; Janes, L.; Lange-Asschenfeldt, B.; Yano, K.; Hawighorst, T.; Iruela-Arispe, L.; et al. Thrombospondin-1 suppresses wound healing and granulation tissue formation in the skin of transgenic mice. Embo. J. 2000, 19, 3272–3282. [Google Scholar] [CrossRef]
- Yoo, M.G.; Kim, I.-H. Keloids and Hypertrophic Scars: Characteristic Vascular Structures Visualized by Using Dermoscopy. Ann. Derm. 2014, 26, 603–609. [Google Scholar] [CrossRef]
- Shchudlo, N.; Varsegova, T.; Stupina, T.; Dolganova, T.; Shchudlo, M.; Shihaleva, N.; Kostin, V. Assessment of palmar subcutaneous tissue vascularization in patients with Dupuytren’s contracture. World J. Orthop. 2018, 9, 130–137. [Google Scholar] [CrossRef]
- Ogawa, R. Keloid and Hypertrophic Scars Are the Result of Chronic Inflammation in the Reticular Dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef]
- Deitch, E.A.; Wheelahan, T.M.; Rose, M.P.; Clothier, J.; Cotter, J. Hypertrophic burn scars: Analysis of variables. J. Trauma 1983, 23, 895–898. [Google Scholar] [CrossRef]
- Ogawa, R.; Akaishi, S. Endothelial dysfunction may play a key role in keloid and hypertrophic scar pathogenesis – Keloids and hypertrophic scars may be vascular disorders. Med. Hypotheses 2016, 96, 51–60. [Google Scholar] [CrossRef]
- Moulin, V.; Larochelle, S.; Langlois, C.; Thibault, I.; Lopez-Vallé, C.A.; Roy, M. Normal skin wound and hypertrophic scar myofibroblasts have differential responses to apoptotic inductors. J. Cell. Physiol. 2004, 198, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Minn, K.W. The effect of myofibroblast on contracture of hypertrophic scar. Plast. Reconstr. Surg. 2004, 113, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Moodley, Y.P.; Misso, N.L.A.; Scaffidi, A.K.; Fogel-Petrovic, M.; McAnulty, R.J.; Laurent, G.J.; Thompson, P.J.; Knight, D.A. Inverse Effects of Interleukin-6 on Apoptosis of Fibroblasts from Pulmonary Fibrosis and Normal Lungs. Am. J. Respir. Cell Mol. Biol. 2003, 29, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Muragaki, Y.; Ooshima, A. Upregulation of transforming growth factor-β1 and vascular endothelial growth factor in cultured keloid fibroblasts: Relevance to angiogenic activity. Arch. Derm. Res. 2005, 297, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Al-Attar, A.; Mess, S.; Thomassen, J.M.; Kauffman, C.L.; Davison, S.P. Keloid Pathogenesis and Treatment. Plast. Reconstr. Surg. 2006, 117, 286. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, M.; Tosa, M.; Shimizu, H.; Hyakusoku, H.; Kawanami, O. Functional Implications of the IL-6 Signaling Pathway in Keloid Pathogenesis. J. Investig. Dermatol. 2007, 127, 98–105. [Google Scholar] [CrossRef]
- Jones, L.R.; Young, W.; Divine, G.; Datta, I.; Chen, K.M.; Ozog, D.; Worsham, M.J. Genome-Wide Scan for Methylation Profiles in Keloids. Dis. Markers 2015. [Google Scholar] [CrossRef]
- Yu, L.; Hébert, M.C.; Zhang, Y.E. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. Embo J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef]
- Jin, Q.; Gui, L.; Niu, F.; Yu, B.; Lauda, N.; Liu, J.; Mao, X.; Chen, Y. Macrophages in keloid are potent at promoting the differentiation and function of regulatory T cells. Exp. Cell Res. 2018, 362, 472–476. [Google Scholar] [CrossRef]
- Steen, V.D.; Medsger, T.A. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000, 43, 2437–2444. [Google Scholar] [CrossRef]
- Meijs, J.; Schouffoer, A.A.; Marsan, N.A.; Stijnen, T.; Putter, H.; Ninaber, M.K.; Huizinga, T.W.J.; de Vries-Bouwstra, J.K. A prediction model for progressive disease in systemic sclerosis. Rmd. Open 2015, 1, e000113. [Google Scholar] [CrossRef]
- Xing, X.; Yang, J.; Yang, X.; Wei, Y.; Zhu, L.; Gao, D.; Li, M. IL-17A Induces Endothelial Inflammation in Systemic Sclerosis via the ERK Signaling Pathway. PLoS ONE 2013, 8, e85032. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Zhao, C.; Qin, F.; He, Z.-Y.; Wang, X.; Zhong, X.-N. Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin. Exp. Rheumatol. 2016, 34 (Suppl. 100), 14–22. [Google Scholar] [PubMed]
- Rolla, G.; Fusaro, E.; Nicola, S.; Bucca, C.; Peroni, C.; Parisi, S.; Cassinis, M.C.; Ferraris, A.; Angelino, F.; Heffler, E.; et al. Th-17 cytokines and interstitial lung involvement in systemic sclerosis. J. Breath Res. 2016, 10, 046013. [Google Scholar] [CrossRef] [PubMed]
- Robak, E.; Gerlicz-Kowalczuk, Z.; Dziankowska-Bartkowiak, B.; Wozniacka, A.; Bogaczewicz, J. Serum concentrations of IL-17A, IL-17B, IL-17E and IL-17F in patients with systemic sclerosis. Arch. Med. Sci. 2019, 15, 706–712. [Google Scholar] [CrossRef]
- Park, M.-J.; Moon, S.-J.; Lee, E.-J.; Jung, K.-A.; Kim, E.-K.; Kim, D.-S.; Lee, J.-H.; Kwok, S.-K.; Min, J.-K.; Park, S.-H.; et al. IL-1-IL-17 Signaling Axis Contributes to Fibrosis and Inflammation in Two Different Murine Models of Systemic Sclerosis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Brembilla, N.C.; Montanari, E.; Truchetet, M.-E.; Raschi, E.; Meroni, P.; Chizzolini, C. Th17 cells favor inflammatory responses while inhibiting type I collagen deposition by dermal fibroblasts: Differential effects in healthy and systemic sclerosis fibroblasts. Arthritis Res. Ther. 2013, 15, R151. [Google Scholar] [CrossRef]
- Uehara, A.; Motegi, S.; Yamada, K.; Uchiyama, A.; Perera, B.; Toki, S.; Ogino, S.; Yokoyama, Y.; Takeuchi, Y.; Ishikawa, O. Mechanistic insight into the norepinephrine-induced fibrosis in systemic sclerosis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- DiBenedetti, D.B.; Nguyen, D.; Zografos, L.; Ziemiecki, R.; Zhou, X. Prevalence, incidence, and treatments of Dupuytren’s disease in the United States: Results from a population-based study. Hand 2011, 6, 149–158. [Google Scholar] [CrossRef]
- Major, M.; Freund, M.K.; Burch, K.S.; Mancuso, N.; Ng, M.; Furniss, D.; Pasaniuc, B.; Ophoff, R.A. Integrative analysis of Dupuytren’s disease identifies novel risk locus and reveals a shared genetic etiology with BMI. Genet. Epidemiol. 2019, 43, 629–645. [Google Scholar] [CrossRef]
- Verjee, L.S.; Verhoekx, J.S.N.; Chan, J.K.K.; Krausgruber, T.; Nicolaidou, V.; Izadi, D.; Davidson, D.; Feldmann, M.; Midwood, K.S.; Nanchahal, J. Unraveling the signaling pathways promoting fibrosis in Dupuytren’s disease reveals TNF as a therapeutic target. Proc. Natl. Acad. Sci. USA 2013, 110, E928–E937. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Ratkaj, I.; Markova-Car, E.; Jurišić, D.; Horvatić, A.; Vučinić, S.; Lerga, J.; Baus-Lončar, M.; Pavelić, K.; Kraljević Pavelić, S. Inflammatory Gene Expression Upon TGF-β1-Induced p38 Activation in Primary Dupuytren’s Disease Fibroblasts. Front. Mol. Biosci. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Andrew, J.G.; Andrew, S.M.; Ash, A.; Turner, B. An investigation into the role of inflammatory cells in dupuytren’s disease. J. Hand Surg. Br. Eur. Vol. 1991, 16, 267–271. [Google Scholar] [CrossRef]
- Kant, S.B.; van den Kerckhove, E.; Colla, C.; Tuinder, S.; van der Hulst, R.R.W.J.; Piatkowski de Grzymala, A.A. A new treatment of hypertrophic and keloid scars with combined triamcinolone and verapamil: A retrospective study. Eur. J. Plast. Surg. 2018, 41, 69–80. [Google Scholar] [CrossRef]
- On, H.R.; Lee, S.H.; Lee, Y.S.; Chang, H.-S.; Park, C.; Roh, M.R. Evaluating hypertrophic thyroidectomy scar outcomes after treatment with triamcinolone injections and copper bromide laser therapy. Lasers Surg. Med. 2015, 47, 479–484. [Google Scholar] [CrossRef]
- Park, J.H.; Chun, J.Y.; Lee, J.H. Laser-assisted topical corticosteroid delivery for the treatment of keloids. Lasers Med. Sci. 2017, 32, 601–608. [Google Scholar] [CrossRef]
- Ketchum, L.D.; Donahue, T.K. The injection of nodules of Dupuytren’s disease with triamcinolone acetonide. J. Hand Surg. 2000, 25, 1157–1162. [Google Scholar] [CrossRef]
- Yin, C.-Y.; Yu, H.-H.M.; Wang, J.-P.; Huang, Y.-C.; Huang, T.-F.; Chang, M.-C. Long-term follow-up of Dupuytren disease after injection of triamcinolone acetonide in Chinese patients in Taiwan. J. Hand Surg. Eur. Vol. 2017, 42, 678–682. [Google Scholar] [CrossRef]
- Giugliano, G.; Pasquali, D.; Notaro, A.; Brongo, S.; Nicoletti, G.; D’Andrea, F.; Bellastella, A.; Sinisi, A.A. Verapamil inhibits interleukin-6 and vascular endothelial growth factor production in primary cultures of keloid fibroblasts. Br. J. Plast. Surg. 2003, 56, 804–809. [Google Scholar] [CrossRef]
- Wang, R.; Mao, Y.; Zhang, Z.; Li, Z.; Chen, J.; Cen, Y. Role of verapamil in preventing and treating hypertrophic scars and keloids. Int. Wound J. 2016, 13, 461–468. [Google Scholar] [CrossRef]
- Abedini, R.; Sasani, P.; Mahmoudi, H.R.; Nasimi, M.; Teymourpour, A.; Shadlou, Z. Comparison of intralesional verapamil versus intralesional corticosteroids in treatment of keloids and hypertrophic scars: A randomized controlled trial. Burns 2018, 44, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Danielsen, P.L.; Rea, S.M.; Wood, F.M.; Fear, M.W.; Viola, H.M.; Hool, L.C.; Gankande, T.U.; Alghamdi, M.; Stevenson, A.W.; Manzur, M.; et al. Verapamil is Less Effective than Triamcinolone for Prevention of Keloid Scar Recurrence After Excision in a Randomized Controlled Trial. Acta Derm. Venereol. 2016, 96, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Denton, C.P.; Lin, C.J.F.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: Results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann. Rheum. Dis. 2018, 77, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Kaldas, M.; Khanna, P.P.; Furst, D.E.; Clements, P.J.; Kee Wong, W.; Seibold, J.R.; Postlethwaite, A.E.; Khanna, D. Sensitivity to change of the modified Rodnan skin score in diffuse systemic sclerosis—assessment of individual body sites in two large randomized controlled trials. Rheumatology 2009, 48, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Denton, C.P.; Ong, V.H.; Xu, S.; Chen-Harris, H.; Modrusan, Z.; Lafyatis, R.; Khanna, D.; Jahreis, A.; Siegel, J.; Sornasse, T. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: Insights from the faSScinate clinical trial in systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 1362–1371. [Google Scholar] [CrossRef]
- Zhou, C.; Zeldin, Y.; Baratz, M.E.; Kathju, S.; Satish, L. Investigating the effects of Pirfenidone on TGF-β1 stimulated non-SMAD signaling pathways in Dupuytren’s disease -derived fibroblasts. BMC Musculoskelet Disord. 2019, 20, 135. [Google Scholar] [CrossRef]
- Sanjuan-Cerveró, R. Prevention of Recurrences in Dupuytren’s Contracture: Are We in the Right Side? Sn Compr. Clin. Med. 2019, 1, 938–943. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, F.; Gallo, P.H.; Baratz, M.E.; Kathju, S.; Satish, L. Anti-fibrotic action of pirfenidone in Dupuytren’s disease-derived fibroblasts. BMC Musculoskelet Disord. 2016, 17, 469. [Google Scholar] [CrossRef][Green Version]
- Clarkson, P. Cortisone as an adjunct to surgery in the treatment of keloids. Lancet 1953, 261, 923–926. [Google Scholar] [CrossRef]
- Khan, M.A.; Bashir, M.M.; Khan, F.A. Intralesional triamcinolone alone and in combination with 5-fluorouracil for the treatment of keloid and hypertrophic scars. J. Pak. Med. Assoc. 2014, 64, 1003–1007. [Google Scholar]
- Distler, O.; Cozzio, A. Systemic sclerosis and localized scleroderma—current concepts and novel targets for therapy. Semin Immunopathol. 2016, 38, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Oddera, S.; Cagnoni, F.; Mangraviti, S.; Giron-Michel, J.; Popova, O.; Canonica, G.W. Effects of triamcinolone acetonide on adult human lung fibroblasts: Decrease in proliferation, surface molecule expression and mediator release. Int. Arch. Allergy Immunol. 2002, 129, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Ebrahem, Q.; Minamoto, A.; Hoppe, G.; Anand-Apte, B.; Sears, J.E. Triamcinolone Acetonide Inhibits IL-6– and VEGF-Induced Angiogenesis Downstream of the IL-6 and VEGF Receptors. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4935–4941. [Google Scholar] [CrossRef] [PubMed]
- Draaijers, L.J.; Tempelman, F.R.H.; Botman, Y.A.M.; Tuinebreijer, W.E.; Middelkoop, E.; Kreis, R.W.; van Zuijlen, P.P.M. The patient and observer scar assessment scale: A reliable and feasible tool for scar evaluation. Plast. Reconstr. Surg. 2004, 113, 1960–1965; discussion 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Baryza, M.J.; Baryza, G.A. The Vancouver Scar Scale: An administration tool and its interrater reliability. J. Burn Care Rehabil. 1995, 16, 535–538. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, S.H. Skin Barrier and Calcium. Ann. Derm. 2018, 30, 265–275. [Google Scholar] [CrossRef]
- Ball, C.; Izadi, D.; Verjee, L.S.; Chan, J.; Nanchahal, J. Systematic review of non-surgical treatments for early dupuytren’s disease. BMC Musculoskelet Disord. 2016, 17, 345. [Google Scholar] [CrossRef]
- Roulet, S.; Bacle, G.; Guéry, J.; Charruau, B.; Marteau, E.; Laulan, J. Outcomes at 7 and 21 years after surgical treatment of Dupuytren’s disease by fasciectomy and open-palm technique. Hand Surg. Rehabil. 2018, 37, 305–310. [Google Scholar] [CrossRef]
- Werlinrud, J.C.; Hansen, K.L.; Larsen, S.; Lauritsen, J. Five-year results after collagenase treatment of Dupuytren disease. J. Hand Surg. Eur. Vol. 2018, 43, 841–847. [Google Scholar] [CrossRef]
- Oku, H.; Shimizu, T.; Kawabata, T.; Nagira, M.; Hikita, I.; Ueyama, A.; Matsushima, S.; Torii, M.; Arimura, A. Antifibrotic action of pirfenidone and prednisolone: Different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 2008, 590, 400–408. [Google Scholar] [CrossRef]
- Taniguchi, H.; Ebina, M.; Kondoh, Y.; Ogura, T.; Azuma, A.; Suga, M.; Taguchi, Y.; Takahashi, H.; Nakata, K.; Sato, A.; et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2010, 35, 821–829. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Nathan, S.D.; Hill, C.; Marshall, J.; Dejonckheere, F.; Thuresson, P.-O.; Maher, T.M. Predicting Life Expectancy for Pirfenidone in Idiopathic Pulmonary Fibrosis. J. Manag. Care Spec. Pharm. 2017, 23, S17–S24. [Google Scholar] [CrossRef] [PubMed]
- Margaritopoulos, G.A.; Trachalaki, A.; Wells, A.U.; Vasarmidi, E.; Bibaki, E.; Papastratigakis, G.; Detorakis, S.; Tzanakis, N.; Antoniou, K.M. Pirfenidone improves survival in IPF: Results from a real-life study. BMC Pulm. Med. 2018, 18, 177. [Google Scholar] [CrossRef] [PubMed]
- Zurkova, M.; Kriegova, E.; Kolek, V.; Lostakova, V.; Sterclova, M.; Bartos, V.; Doubkova, M.; Binkova, I.; Svoboda, M.; Strenkova, J.; et al. Effect of pirfenidone on lung function decline and survival: 5-yr experience from a real-life IPF cohort from the Czech EMPIRE registry. Respir. Res. 2019, 20, 16. [Google Scholar] [CrossRef]
- Hall, C.L.; Wells, A.R.; Leung, K.P. Pirfenidone reduces profibrotic responses in human dermal myofibroblasts, in vitro. Lab. Investig. 2018, 98, 640–655. [Google Scholar] [CrossRef]
- Roubenoff, R.; Harris, T.B.; Abad, L.W.; Wilson, P.W.F.; Dallal, G.E.; Dinarello, C.A. Monocyte Cytokine Production in an Elderly Population: Effect of Age and Inflammation. J. Gerontol. A Biol. Sci. Med. Sci. 1998, 53, M20–M26. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Horan, M.A.; Ferguson, M.W. Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab. Investig. 1998, 78, 47–58. [Google Scholar]
- Ashcroft, G.S.; Mills, S.J.; Ashworth, J.J. Ageing and wound healing. Biogerontology 2002, 3, 337–345. [Google Scholar] [CrossRef]
- Fujiwara, T.; Dohi, T.; Maan, Z.N.; Rustad, K.C.; Kwon, S.H.; Padmanabhan, J.; Whittam, A.J.; Suga, H.; Duscher, D.; Rodrigues, M.; et al. Age-associated intracellular superoxide dismutase deficiency potentiates dermal fibroblast dysfunction during wound healing. Exp. Dermatol. 2019, 28, 485–492. [Google Scholar] [CrossRef]
- Saldías, M.P.; Fernández, C.; Morgan, A.; Díaz, C.; Morales, D.; Jaña, F.; Gómez, A.; Silva, A.; Briceño, F.; Oyarzún, A.; et al. Aged blood factors decrease cellular responses associated with delayed gingival wound repair. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Makrantonaki, E.; Wlaschek, M.; Scharffetter-Kochanek, K. Pathogenesis of wound healing disorders in the elderly. J. Der Dtsch. Dermatol. Ges. 2017, 15, 255–275. [Google Scholar] [CrossRef] [PubMed]
- Marcus, J.R.; Tyrone, J.W.; Bonomo, S.; Xia, Y.; Mustoe, T.A. Cellular mechanisms for diminished scarring with aging. Plast. Reconstr. Surg. 2000, 105, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Beharka, A.A.; Meydani, M.; Wu, D.; Leka, L.S.; Meydani, A.; Meydani, S.N. Interleukin-6 Production Does Not Increase With Age. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B81–B88. [Google Scholar] [CrossRef]
- Lee, E.G.; Luckett-Chastain, L.R.; Calhoun, K.N.; Frempah, B.; Bastian, A.; Gallucci, R.M. Interleukin 6 Function in the Skin and Isolated Keratinocytes Is Modulated by Hyperglycemia. J. Immunol. Res. 2019. [Google Scholar] [CrossRef]
- Zhu, B.-M.; Ishida, Y.; Robinson, G.W.; Pacher-Zavisin, M.; Yoshimura, A.; Murphy, P.M.; Hennighausen, L. SOCS3 Negatively Regulates the gp130–STAT3 Pathway in Mouse Skin Wound Healing. J. Investig. Dermatol. 2008, 128, 1821–1829. [Google Scholar] [CrossRef]
- Ban, C.R.; Twigg, S.M. Fibrosis in diabetes complications: Pathogenic mechanisms and circulating and urinary markers. Vasc. Health Risk Manag. 2008, 4, 575–596. [Google Scholar]
- Akchurin, O.; Patino, E.; Dalal, V.; Meza, K.; Bhatia, D.; Brovender, S.; Zhu, Y.-S.; Cunningham-Rundles, S.; Perelstein, E.; Kumar, J.; et al. Interleukin-6 Contributes to the Development of Anemia in Juvenile CKD. Kidney Int. Rep. 2019, 4, 470–483. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.-H.; Zhang, Y.-Y.; Wang, Y.-Z.; Wang, J.; Zhao, Y.; Jin, X.-X.; Xue, G.-L.; Li, P.-H.; Sun, Y.-L.; et al. Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFβ1 and miR-29 pathways. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Das, S.K.; Balakrishnan, V. Role of Cytokines in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Indian J. Clin. Biochem. 2011, 26, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Afek, A.; Derazne, E.; Tzur, D.; Cukierman-Yaffe, T.; Gerstein, H.C.; Tirosh, A. Diabetes Risk Among Overweight and Obese Metabolically Healthy Young Adults. Diabetes Care 2014, 37, 2989–2995. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, S.; Thomas, R.; Shihab, P.; Sriraman, D.; Behbehani, K.; Ahmad, R. Obesity Is a Positive Modulator of IL-6R and IL-6 Expression in the Subcutaneous Adipose Tissue: Significance for Metabolic Inflammation. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Murano, I.; Barbatelli, G.; Parisani, V.; Latini, C.; Muzzonigro, G.; Castellucci, M.; Cinti, S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J. Lipid Res. 2008, 49, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Chylikova, J.; Dvorackova, J.; Tauber, Z.; Kamarad, V. M1/M2 macrophage polarization in human obese adipose tissue. Biomed. Pap. 2018, 162, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid. Redox Signal. 2017, 29, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Rodero, M.P.; Patel, J.; Moi, D.; Mazzieri, R.; Khosrotehrani, K. Interleukin-23 regulates interleukin-17 expression in wounds, and its inhibition accelerates diabetic wound healing through the alteration of macrophage polarization. Faseb J. 2018, 32, 2086–2094. [Google Scholar] [CrossRef]
- Malik, S.; Want, M.Y.; Awasthi, A. The Emerging Roles of Gamma–Delta T Cells in Tissue Inflammation in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-Producing γδ T Cells Selectively Expand in Response to Pathogen Products and Environmental Signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef]
- Iwakura, Y.; Ishigame, H. The IL-23/IL-17 axis in inflammation. J. Clin. Investig. 2006, 116, 1218–1222. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Sakamoto, A.; Yamamoto, T.; Narahara, S.; Sugiuchi, H.; Yamaguchi, Y. Differential regulation of IL-23 production in M1 macrophages by TIR8/SIGIRR through TLR4- or TLR7/8-mediated signaling. Cytokine 2017, 99, 310–315. [Google Scholar] [CrossRef]
- Nicoletti, G.; Saler, M.; Tresoldi, M.M.; Scevola, S.; Faga, A. Unrecognized Cell Torpidity as a Risk Factor in Elective Plastic Surgery. Plast Reconstr Surg Glob. Open 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Bosanquet, D.; Rangaraj, A.; Richards, A.; Riddell, A.; Saravolac, V.; Harding, K. Topical steroids for chronic wounds displaying abnormal inflammation. Ann. R Coll. Surg. Engl. 2013, 95, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Nikkhah, D.; Dheansa, B. Topical steroids for chronic wounds displaying abnormal inflammation. Ann. R Coll. Surg. Engl. 2013, 95, 448. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.E.; Foster, D.S.; Longaker, M.T. Management of Chronic Wounds—2018. JAMA 2018, 320, 1481–1482. [Google Scholar] [CrossRef]
- Chereddy, K.K.; Her, C.-H.; Comune, M.; Moia, C.; Lopes, A.; Porporato, P.E.; Vanacker, J.; Lam, M.C.; Steinstraesser, L.; Sonveaux, P.; et al. PLGA nanoparticles loaded with host defense peptide LL37 promote wound healing. J. Control. Release 2014, 194, 138–147. [Google Scholar] [CrossRef]
- Fumakia, M.; Ho, E.A. Nanoparticles Encapsulated with LL37 and Serpin A1 Promotes Wound Healing and Synergistically Enhances Antibacterial Activity. Mol. Pharm. 2016, 13, 2318–2331. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Treatment | Effect | Disease | Outcome |
|---|---|---|---|
| Corticosteroids | ↓IL-6, ↓VEGF, ↓STAT3 | Hypertrophic/keloid scars | Improved scar score [84,85,86] |
| Dupuytren’s disease | Softening of nodules, improved motility [87,88] | ||
| Verapamil (Calcium antagonist) | ↓IL-6, ↓VEGF, ↑Apoptosis (Fibroblasts) | Hypertrophic/keloid scars | Reduced growth, improved scar score [89,90] (disputed efficacy [91,92]) |
| Tocilizumab (IL-6 blockade) | Prevents IL-6-receptor binding | Scleroderma/systemic sclerosis | Improved skin score, reverses TGF-β-dependent phenotype in SSc fibroblasts [93,94,95] |
| Pirfenidone | ↓TGF- β-signaling, ↓IL-6 | Dupuytren’s disease | Preclinical—reduces TGF-β-dependent signaling in DD fibroblasts [96,97,98] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, B.Z.; Stevenson, A.W.; Prêle, C.M.; Fear, M.W.; Wood, F.M. The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing. Biomedicines 2020, 8, 101. https://doi.org/10.3390/biomedicines8050101
Johnson BZ, Stevenson AW, Prêle CM, Fear MW, Wood FM. The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing. Biomedicines. 2020; 8(5):101. https://doi.org/10.3390/biomedicines8050101
Chicago/Turabian StyleJohnson, Blair Z., Andrew W. Stevenson, Cecilia M. Prêle, Mark W. Fear, and Fiona M. Wood. 2020. "The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing" Biomedicines 8, no. 5: 101. https://doi.org/10.3390/biomedicines8050101
APA StyleJohnson, B. Z., Stevenson, A. W., Prêle, C. M., Fear, M. W., & Wood, F. M. (2020). The Role of IL-6 in Skin Fibrosis and Cutaneous Wound Healing. Biomedicines, 8(5), 101. https://doi.org/10.3390/biomedicines8050101