Aptamer-siRNA Chimeras: Discovery, Progress, and Future Prospects

Abstract
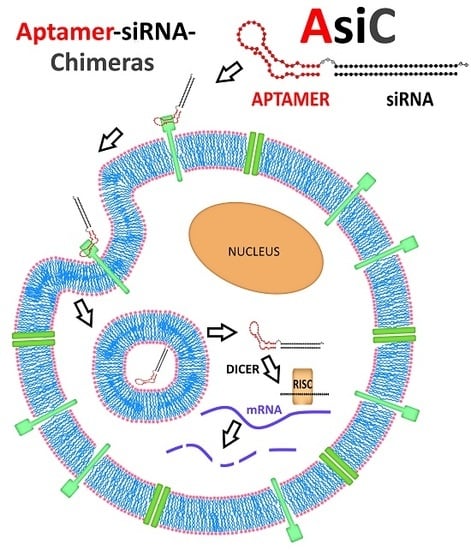
{kind=link}
{kind=link}
{kind=link}
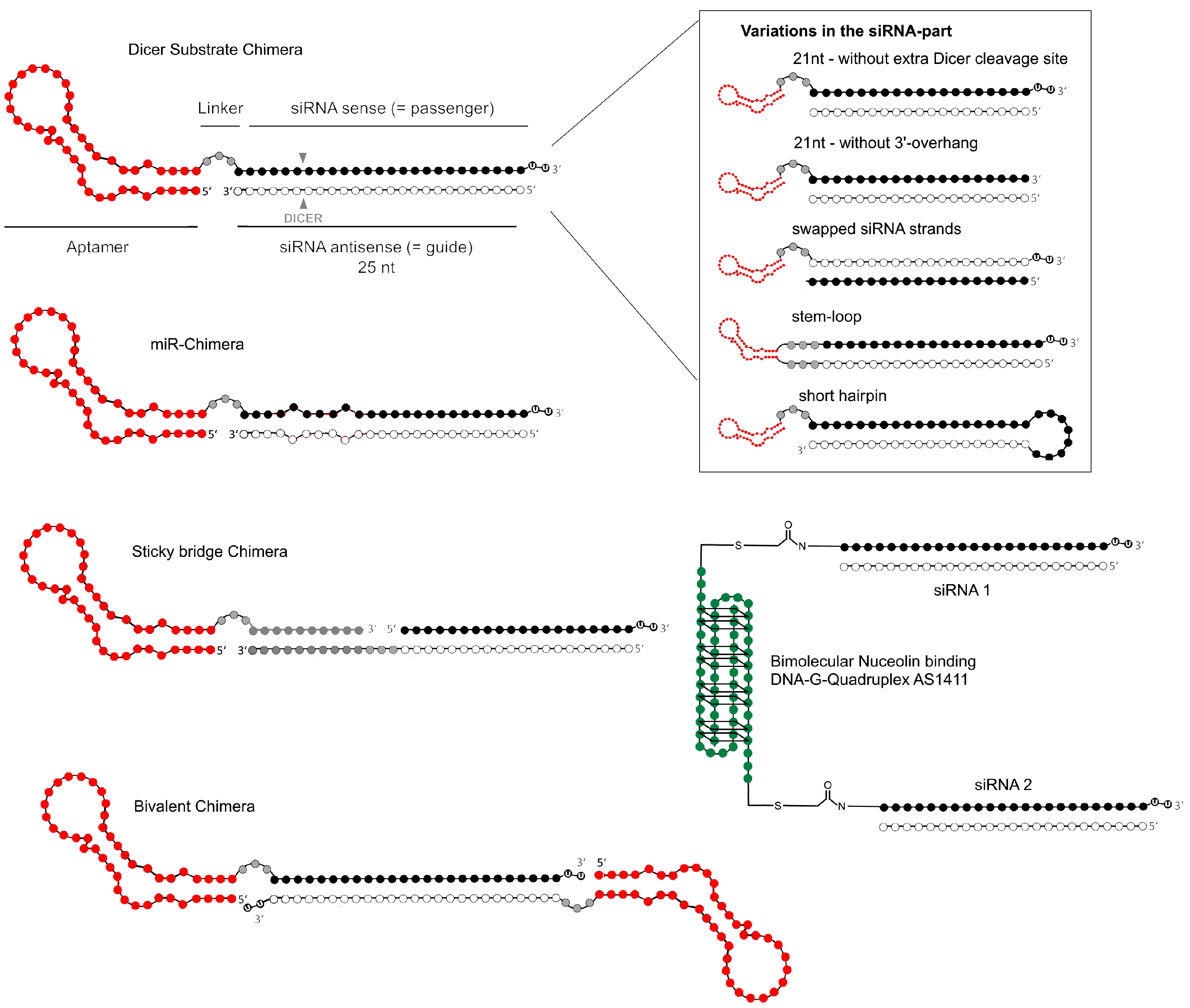{kind=link}
1. Introduction
2. Aptamers as Ligands for the Targeted Delivery of Therapeutic Oligonucleotides
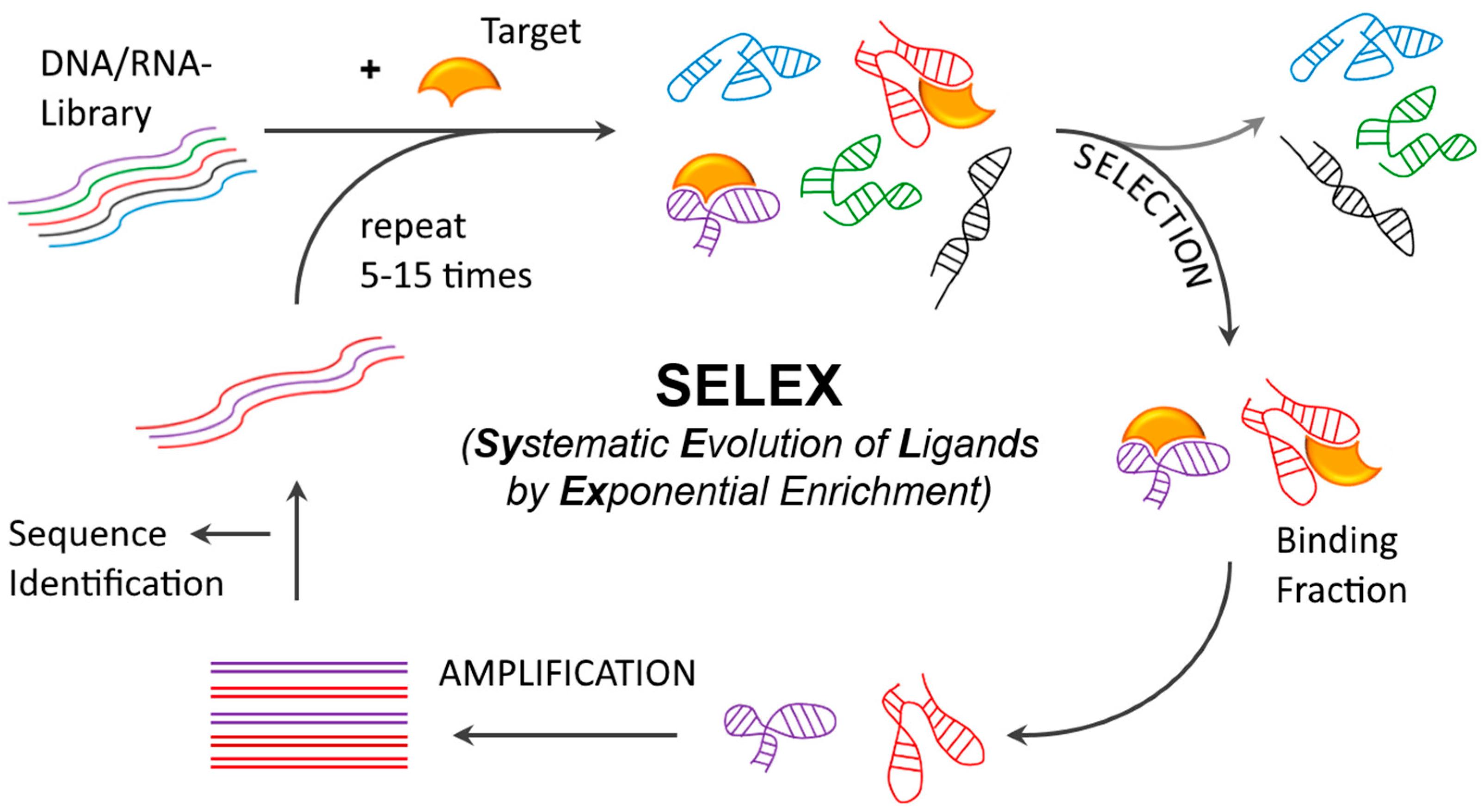
2.1. Aptamer Development and Identification
2.2. Early Aptamer-siRNA Conjugates
2.3. Conjugation Strategies
3. Recent Advances in Aptamer-siRNA Applications
3.1. Cytotoxic Cancer Therapy
3.2. Cancer Stem Cell Therapy
3.3. Cancer-Immunotherapy
3.4. Anti-Viral Therapy
4. Aptamer-siRNA Development: Major Considerations
4.1. Selection of siRNA
4.2. Aptamer Considerations
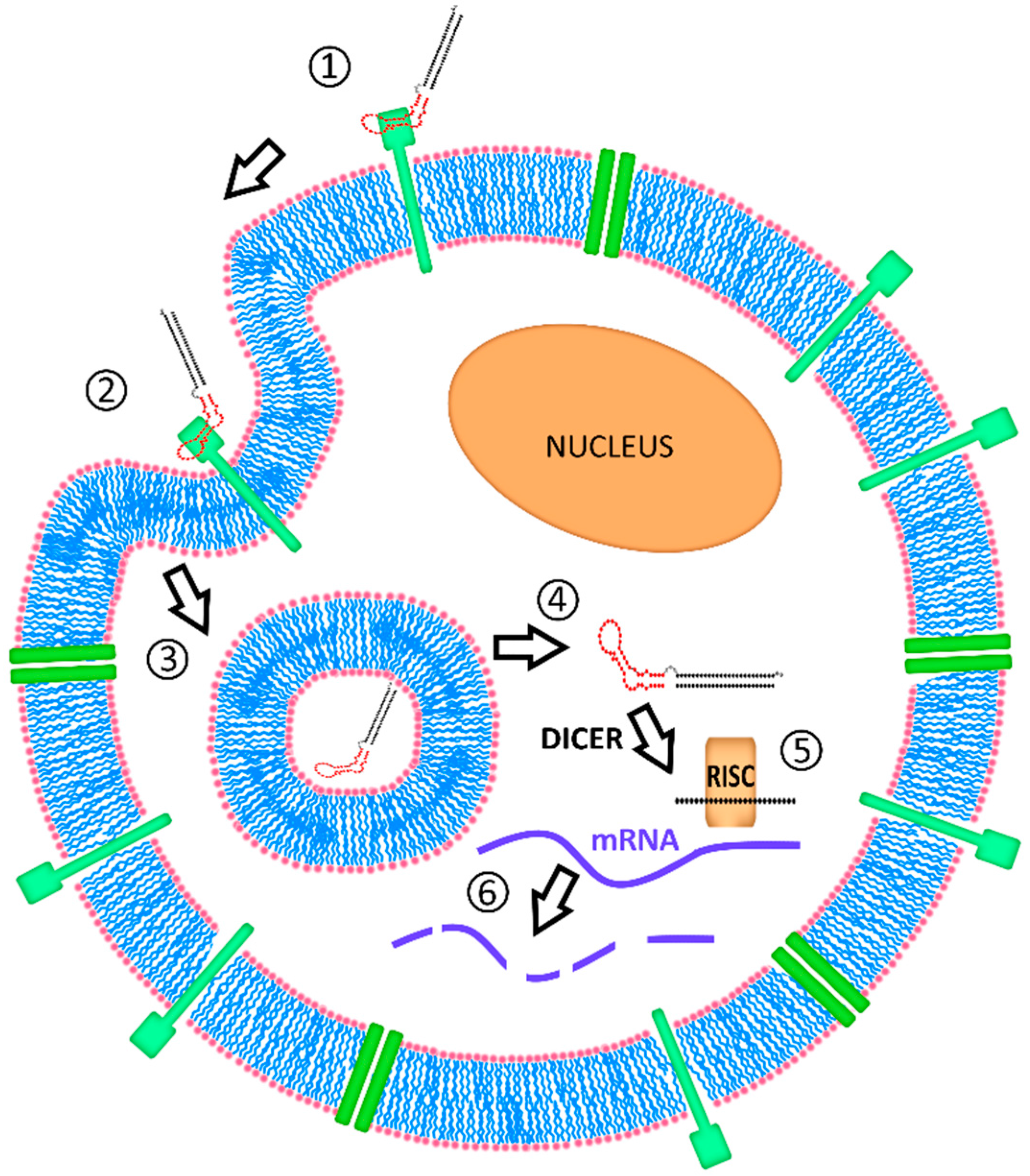
4.3. Intracellular Fate of Aptamer-siRNA Conjugates
5. Conclusions and Future Perspective
Acknowledgments
Conflicts of Interest
References
- Strebhardt, K.; Ullrich, A. Paul ehrlich's magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, K.; Gogtay, N.J. Therapeutic nucleic acids: Current clinical status. Br. J. Clin. Pharmacol. 2016, 82, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R.; et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Margus, H.; Padari, K.; Pooga, M. Cell-penetrating peptides as versatile vehicles for oligonucleotide delivery. Mol. Ther. 2012, 20, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Cully, M. Drug delivery: Nanoparticles improve profile of molecularly targeted cancer drug. Nat. Rev. Drug Discov. 2016, 15, 231. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent n-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P. Delivery of small-interfering RNA (siRNA) to the brain. Expert Opin. Ther. Pat. 2009, 19, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Mager, I.; Meyer, A.H.; Li, J.; Lenter, M.; Hildebrandt, T.; Leparc, G.; Wood, M.J. Targeting blood-brain-barrier transcytosis—Perspectives for drug delivery. Neuropharmacology 2016, 120, 4–7. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.K.; Lizzio, V.; Merkel, O.M. Folate receptor targeted delivery of siRNA and paclitaxel to ovarian cancer cells via folate conjugated triblock copolymer to overcome tlr4 driven chemotherapy resistance. Biomacromolecules 2016, 17, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, J. Delivery systems for sirna drug development in cancer therapy. Asian J. Pharm. Sci. 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Tatiparti, K.; Sau, S.; Kashaw, S.K.; Iyer, A.K. SiRNA delivery strategies: A comprehensive review of recent developments. Nanomaterials 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Foy, J.W.; Rittenhouse, K.; Modi, M.; Patel, M. Local tolerance and systemic safety of pegaptanib sodium in the dog and rabbit. J. Ocul. Pharmacol. Ther. 2007, 23, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Cell-type-specific, aptamer-functionalized agents for targeted disease therapy. Mol. Ther. Nucleic Acids 2014, 3, e169. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage t4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yu, Y.; Jiang, F.; Zhou, J.; Li, Y.; Liang, C.; Dang, L.; Lu, A.; Zhang, G. Development of cell-selex technology and its application in cancer diagnosis and therapy. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using cell-selex. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Parekh, P.; Turner, P.; Moyer, R.W.; Tan, W. Generating aptamers for recognition of virus-infected cells. Clin. Chem. 2009, 55, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Thiel, K.W.; Hernandez, L.I.; Dassie, J.P.; Thiel, W.H.; Liu, X.; Stockdale, K.R.; Rothman, A.M.; Hernandez, F.J.; McNamara, J.O., 2nd; Giangrande, P.H. Delivery of chemo-sensitizing siRNAs to her2+-breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012, 40, 6319–6337. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Bair, T.; Peek, A.S.; Liu, X.; Dassie, J.; Stockdale, K.R.; Behlke, M.A.; Miller, F.J., Jr.; Giangrande, P.H. Rapid identification of cell-specific, internalizing RNA aptamers with bioinformatics analyses of a cell-based aptamer selection. PLoS ONE 2012, 7, e43836. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Maufort, J.P.; Nie, J.; Stewart, R.; McIntosh, B.E.; Conti, L.R.; Ahmad, K.M.; Soh, H.T.; Thomson, J.A. Development of an efficient targeted cell-selex procedure for DNA aptamer reagents. PLoS ONE 2013, 8, e71798. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Sakota, E.; Nakamura, Y. The efficient cell-selex strategy, icell-selex, using isogenic cell lines for selection and counter-selection to generate RNA aptamers to cell surface proteins. Biochimie 2016, 131, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of crispr-cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H. Galaxy workflows for web-based bioinformatics analysis of aptamer high-throughput sequencing data. Mol. Ther. Nucleic Acids 2016, 5, e345. [Google Scholar] [CrossRef] [PubMed]
- Beier, R.; Boschke, E.; Labudde, D. New strategies for evaluation and analysis of selex experiments. Biomed. Res. Int. 2014, 2014, 849743. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.Y.; Byun, J. Nucleic acid aptamers: New methods for selection, stabilization, and application in biomedical science. Biomol. Ther. 2013, 21, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Szeitner, Z.; Andras, J.; Gyurcsanyi, R.E.; Meszaros, T. Is less more? Lessons from aptamer selection strategies. J. Pharm. Biomed. Anal. 2014, 101, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.P.; Somasunderam, A.; Nieves-Alicea, R.; Li, X.; Hu, A.; Sood, A.K.; Ferrari, M.; Gorenstein, D.G.; Tanaka, T. Identification of thioaptamer ligand against e-selectin: Potential application for inflamed vasculature targeting. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Berg, K.; Lange, T.; Mittelberger, F.; Schumacher, U.; Hahn, U. Selection and characterization of an alpha6beta4 integrin blocking DNA aptamer. Mol. Ther. Nucleic Acids 2016, 5, e294. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Thiel, K.W.; Flenker, K.S.; Bair, T.; Dupuy, A.J.; McNamara, J.O., 2nd; Miller, F.J.; Giangrande, P.H. Cell-internalization selex: Method for identifying cell-internalizing RNA aptamers for delivering siRNAs to target cells. Methods Mol. Biol. 2015, 1218, 187–199. [Google Scholar] [PubMed]
- Mu, Q.; Annapragada, A.; Srivastava, M.; Li, X.; Wu, J.; Thiviyanathan, V.; Wang, H.; Williams, A.; Gorenstein, D.; Annapragada, A.; et al. Conjugate-selex: A high-throughput screening of thioaptamer-liposomal nanoparticle conjugates for targeted intracellular delivery of anticancer drugs. Mol. Ther. Nucleic Acids 2016, 5, e382. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, H.; Ding, H.; Huang, Y.; Cao, X.; Yang, G.; Li, J.; Xie, Z.; Meng, Y.; Li, X.; et al. Identification of an aptamer targeting hnrnp a1 by tissue slide-based selex. J. Pathol. 2009, 218, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, X.; Volk, D.E.; Lokesh, G.L.; Elizondo-Riojas, M.A.; Li, L.; Nick, A.M.; Sood, A.K.; Rosenblatt, K.P.; Gorenstein, D.G. Morph-x-select: Morphology-based tissue aptamer selection for ovarian cancer biomarker discovery. Biotechniques 2016, 61, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Liu, Y.; Rabbani, Z.N.; Yang, Z.; Urban, J.H.; Sullenger, B.A.; Clary, B.M. In vivo selection of tumor-targeting RNA motifs. Nat. Chem. Biol. 2010, 6, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Chen, Y.H.; Lennox, K.A.; Behlke, M.A.; Davidson, B.L. In vivo selex for identification of brain-penetrating aptamers. Mol. Ther. Nucleic Acids 2013, 2, e67. [Google Scholar] [CrossRef] [PubMed]
- Chu, T.C.; Twu, K.Y.; Ellington, A.D.; Levy, M. Aptamer mediated siRNA delivery. Nucleic Acids Res. 2006, 34, e73. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O., 2nd; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O., 2nd; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of psma-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Dassie, J.P.; Giangrande, P.H. Current progress on aptamer-targeted oligonucleotide therapeutics. Ther. Deliv. 2013, 4, 1527–1546. [Google Scholar] [CrossRef] [PubMed]
- Wullner, U.; Neef, I.; Eller, A.; Kleines, M.; Tur, M.K.; Barth, S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr. Cancer Drug Targets 2008, 8, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, A.; Brenneman, R.; Bajgelman, M.; Seales, D.; Gilboa, E. Thermal stability of siRNA modulates aptamer- conjugated siRNA inhibition. Mol. Ther. Nucleic Acids 2012, 1, e51. [Google Scholar] [CrossRef] [PubMed]
- Bagalkot, V.; Gao, X. SiRNA-aptamer chimeras on nanoparticles: Preserving targeting functionality for effective gene silencing. ACS Nano 2011, 5, 8131–8139. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Jung, Y.; Choi, H.; Yang, J.; Suh, J.S.; Huh, Y.M.; Kim, K.; Haam, S. Prostate cancer cell death produced by the co-delivery of bcl-xl shRNA and doxorubicin using an aptamer-conjugated polyplex. Biomaterials 2010, 31, 4592–4599. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 440. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Swiderski, P.; Li, H.; Zhang, J.; Neff, C.P.; Akkina, R.; Rossi, J.J. Selection, characterization and application of new RNA hiv gp 120 aptamers for facile delivery of dicer substrate siRNAs into hiv infected cells. Nucleic Acids Res. 2009, 37, 3094–3109. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Tschammer, N.; Mohammed, S.; Guo, P. Specific delivery of therapeutic RNAs to cancer cells via the dimerization mechanism of phi29 motor pRNA. Hum. Gene Ther. 2005, 16, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Khaled, A.; Guo, S.; Li, F.; Guo, P. Controllable self-assembly of nanoparticles for specific delivery of multiple therapeutic molecules to cancer cells using RNA nanotechnology. Nano Lett. 2005, 5, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Bachelet, I.; Church, G.M. A logic-gated nanorobot for targeted transport of molecular payloads. Science 2012, 335, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Passaro, D.; Longobardo, I.; Condorelli, G.; Marotta, P.; Affuso, A.; de Franciscis, V.; Cerchia, L. A neutralizing RNA aptamer against egfr causes selective apoptotic cell death. PLoS ONE 2011, 6, e24071. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Nguyen, H.H.; Byrom, M.; Ellington, A.D. Inhibition of cell proliferation by an anti-egfr aptamer. PLoS ONE 2011, 6, e20299. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, D.; Cao, Z.; Meng, L.; Mallikaratchy, P.; Sefah, K.; Wang, H.; Li, Y.; Tan, W. Cell-specific aptamer probes for membrane protein elucidation in cancer cells. J. Proteome Res. 2008, 7, 2133–2139. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.F.; Tur, M.K.; Barth, S. An aptamer-siRNA chimera silences the eukaryotic elongation factor 2 gene and induces apoptosis in cancers expressing alphavbeta3 integrin. Nucleic Acid Ther. 2013, 23, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, C.; Zhao, J.; Chen, Y. Reversal of paclitaxel resistance in epithelial ovarian carcinoma cells by a muc1 aptamer-let-7i chimera. Cancer Investig. 2012, 30, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Hicke, B.J.; Stephens, A.W.; Gould, T.; Chang, Y.F.; Lynott, C.K.; Heil, J.; Borkowski, S.; Hilger, C.S.; Cook, G.; Warren, S.; et al. Tumor targeting by an aptamer. J. Nucl. Med. 2006, 47, 668–678. [Google Scholar] [PubMed]
- Bates, P.J.; Kahlon, J.B.; Thomas, S.D.; Trent, J.O.; Miller, D.M. Antiproliferative activity of g-rich oligonucleotides correlates with protein binding. J. Biol. Chem. 1999, 274, 26369–26377. [Google Scholar] [CrossRef] [PubMed]
- Kruspe, S.; Mittelberger, F.; Szameit, K.; Hahn, U. Aptamers as drug delivery vehicles. ChemMedChem 2014, 9, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.Y.; Wang, W.Y.; Chang, Y.C.; Chang, C.J.; Yang, P.C.; Peck, K. Synergistic inhibition of lung cancer cell invasion, tumor growth and angiogenesis using aptamer-siRNA chimeras. Biomaterials 2014, 35, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L. Four-stranded nucleic acids: Structure, function and targeting of g-quadruplexes. Chem. Soc. Rev. 2008, 37, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Kruspe, S.; Giangrande, P.H. Design and preparation of aptamer-siRNA chimeras (asics) for targeted cancer therapy. In Methods in Molecular Biology; Springer Science and Business Media: Dordrecht, The Netherlands, 2017. [Google Scholar]
- Esposito, C.L.; Cerchia, L.; Catuogno, S.; De Vita, G.; Dassie, J.P.; Santamaria, G.; Swiderski, P.; Condorelli, G.; Giangrande, P.H.; de Franciscis, V. Multifunctional aptamer-miRNA conjugates for targeted cancer therapy. Mol. Ther. 2014, 22, 1151–1163. [Google Scholar] [CrossRef] [PubMed]
- Iaboni, M.; Russo, V.; Fontanella, R.; Roscigno, G.; Fiore, D.; Donnarumma, E.; Esposito, C.L.; Quintavalle, C.; Giangrande, P.H.; de Franciscis, V.; et al. Aptamer-miRNA-212 conjugate sensitizes nsclc cells to trail. Mol. Ther. Nucleic Acids 2016, 5, e289. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Nuzzo, S.; Kumar, S.A.; Rienzo, A.; Lawrence, C.L.; Pallini, R.; Shaw, L.; Alder, J.E.; Ricci-Vitiani, L.; Catuogno, S.; et al. A combined microRNA-based targeted therapeutic approach to eradicate glioblastoma stem-like cells. J. Control. Release 2016, 238, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Neff, C.P.; Swiderski, P.; Li, H.; Smith, D.D.; Aboellail, T.; Remling-Mulder, L.; Akkina, R.; Rossi, J.J. Functional in vivo delivery of multiplexed anti-hiv-1 siRNAs via a chemically synthesized aptamer with a sticky bridge. Mol. Ther. 2013, 21, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Otake, Y.; Soundararajan, S.; Sengupta, T.K.; Kio, E.A.; Smith, J.C.; Pineda-Roman, M.; Stuart, R.K.; Spicer, E.K.; Fernandes, D.J. Overexpression of nucleolin in chronic lymphocytic leukemia cells induces stabilization of bcl2 mRNA. Blood 2007, 109, 3069–3075. [Google Scholar] [PubMed]
- Qiu, W.; Zhou, F.; Zhang, Q.; Sun, X.; Shi, X.; Liang, Y.; Wang, X.; Yue, L. Overexpression of nucleolin and different expression sites both related to the prognosis of gastric cancer. Apmis 2013, 121, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Tian, B.; Liu, J.; Yang, L.; Zeng, L.; Chen, T.; Hong, A.; Wang, X. Nucleolin-targeted extracellular vesicles as a versatile platform for biologics delivery to breast cancer. Theranostics 2017, 7, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the g-rich oligonucleotide as1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Yu, X.; Liu, H.; Wu, D.; She, J.X. Co-targeting egfr and survivin with a bivalent aptamer-dual siRNA chimera effectively suppresses prostate cancer. Sci. Rep. 2016, 6, 30346. [Google Scholar] [CrossRef] [PubMed]
- Schlomm, T.; Kirstein, P.; Iwers, L.; Daniel, B.; Steuber, T.; Walz, J.; Chun, F.H.; Haese, A.; Kollermann, J.; Graefen, M.; et al. Clinical significance of epidermal growth factor receptor protein overexpression and gene copy number gains in prostate cancer. Clin. Cancer Res. 2007, 13, 6579–6584. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Chen, W.Y.; Yin, J.J.; Sheppard-Tillman, H.; Huang, J.; Liu, Y.N. Egf receptor promotes prostate cancer bone metastasis by downregulating mir-1 and activating twist1. Cancer Res. 2015, 75, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.M.; Vader, G.; Medema, R.H. The case for survivin as mitotic regulator. Curr. Opin. Cell Biol. 2006, 18, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.; Lieberman, J. Dysregulation of microRNA biogenesis and gene silencing in cancer. Sci. Signal. 2015, 8, re3. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, M.; Croce, C.M. Role of microRNAs in maintaining cancer stem cells. Adv. Drug Deliv. Rev. 2015, 81, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, L.; Esposito, C.L.; Camorani, S.; Rienzo, A.; Stasio, L.; Insabato, L.; Affuso, A.; de Franciscis, V. Targeting axl with an high-affinity inhibitory aptamer. Mol. Ther. 2012, 20, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Kanlikilicer, P.; Ozpolat, B.; Aslan, B.; Bayraktar, R.; Gurbuz, N.; Rodriguez-Aguayo, C.; Bayraktar, E.; Denizli, M.; Gonzalez-Villasana, V.; Ivan, C.; et al. Therapeutic targeting of axl receptor tyrosine kinase inhibits tumor growth and intraperitoneal metastasis in ovarian cancer models. Mol. Ther. Nucleic Acids 2017. [Google Scholar] [CrossRef]
- Iaboni, M.; Fontanella, R.; Rienzo, A.; Capuozzo, M.; Nuzzo, S.; Santamaria, G.; Catuogno, S.; Condorelli, G.; de Franciscis, V.; Esposito, C.L. Targeting insulin receptor with a novel internalizing aptamer. Mol. Ther. Nucleic Acids 2016, 5, e365. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, D.R.; Weinberg, R.A. Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discov. 2014, 13, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Hung, L.Y.; Wang, C.H.; Che, Y.J.; Fu, C.Y.; Chang, H.Y.; Wang, K.; Lee, G.B. Screening of aptamers specific to colorectal cancer cells and stem cells by utilizing on-chip cell-selex. Sci. Rep. 2015, 5, 10326. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Shigdar, S.; Qiao, G.; Zhou, S.F.; Li, Y.; Wei, M.Q.; Qiao, L.; Shamaileh, H.A.; Zhu, Y.; Zheng, C.; et al. Aptamer-mediated cancer gene therapy. Curr. Gene Ther. 2015, 15, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Hamblin, T.J.; Orchard, J.A.; Ibbotson, R.E.; Davis, Z.; Thomas, P.W.; Stevenson, F.K.; Oscier, D.G. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood 2002, 99, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, E.; Malavasi, F. The metamorphosis of a molecule: From soluble enzyme to the leukocyte receptor CD38. J. Leukoc. Biol. 1999, 65, 151–161. [Google Scholar] [PubMed]
- Guan, X.H.; Liu, X.H.; Hong, X.; Zhao, N.; Xiao, Y.F.; Wang, L.F.; Tang, L.; Jiang, K.; Qian, Y.S.; Deng, K.Y.; et al. CD38 deficiency protects the heart from ischemia/reperfusion injury through activating sirt1/foxos-mediated antioxidative stress pathway. Oxid. Med. Cell. Longev. 2016, 2016, 7410257. [Google Scholar] [CrossRef] [PubMed]
- Reyes, L.A.; Boslett, J.; Varadharaj, S.; De Pascali, F.; Hemann, C.; Druhan, L.J.; Ambrosio, G.; El-Mahdy, M.; Zweier, J.L. Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc. Natl. Acad. Sci. USA 2015, 112, 11648–11653. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Liu, Y.; Zhu, T.; Zhu, J.; Dimeco, F.; Vescovi, A.L.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Lubman, D.M. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol. Cell. Proteom. 2012, 11, M111.010744. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Ngai, P.; Ho, D.W.; Yu, W.C.; Ng, M.N.; Lau, C.K.; Li, M.L.; Tam, K.H.; Lam, C.T.; Poon, R.T.; et al. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology 2008, 47, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.C.; Gao, L.; He, J.; Long, Y.; Liao, S.; Wang, H.; Li, X.; Yi, W.; Pei, Z.; Wu, M.; et al. CD90 is upregulated in gastric cancer tissues and inhibits gastric cancer cell apoptosis by modulating the expression level of sparc protein. Oncol. Rep. 2015, 34, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Donnenberg, V.S.; Landreneau, R.J.; Donnenberg, A.D. Tumorigenic stem and progenitor cells: Implications for the therapeutic index of anti-cancer agents. J. Control. Release 2007, 122, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Qiao, L.; Zhou, S.F.; Xiang, D.; Wang, T.; Li, Y.; Lim, L.Y.; Kong, L.; Li, L.; Duan, W. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013, 330, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Chen, H.; Yu, C.; Zhang, Y.; Chen, M.; Tian, S.; Sun, C. The promotion of salinomycin delivery to hepatocellular carcinoma cells through egfr and CD133 aptamers conjugation by plga nanoparticles. Nanomedicine 2015, 10, 1863–1879. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zeng, W.; Sun, J.; Yang, M.; Li, L.; Zhou, J.; Wu, Y.; Sun, J.; Liu, G.; Tang, R.; et al. Construction of an aptamer-siRNA chimera-modified tissue-engineered blood vessel for cell-type-specific capture and delivery. ACS Nano 2015, 9, 6069–6076. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Qian, C.; Lv, L.; Pu, C.; Li, Y.; Li, L.; Marappan, M.; Lin, J.; Wang, L.; Duan, W. The use of sensitive chemical antibodies for diagnosis: Detection of low levels of epcam in breast cancer. PLoS ONE 2013, 8, e57613. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Gantier, M.P.; Xiang, D.; Bean, A.G.; Bruce, M.; Zhou, S.F.; Khasraw, M.; Ward, A.; Wang, L.; Wei, M.Q.; et al. Epcam aptamer-mediated survivin silencing sensitized cancer stem cells to doxorubicin in a breast cancer model. Theranostics 2015, 5, 1456–1472. [Google Scholar] [CrossRef] [PubMed]
- Gilboa-Geffen, A.; Hamar, P.; Le, M.T.; Wheeler, L.A.; Trifonova, R.; Petrocca, F.; Wittrup, A.; Lieberman, J. Gene knockdown by epcam aptamer-siRNA chimeras suppresses epithelial breast cancers and their tumor-initiating cells. Mol. Cancer Ther. 2015, 14, 2279–2291. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Kanwar, J.R.; Kanwar, R.K.; Sreemanthula, J.; Biswas, J.; Khetan, V.; Krishnakumar, S. Epcam aptamer-siRNA chimera targets and regress epithelial cancer. PLoS ONE 2015, 10, e0132407. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Kanwar, J.R.; Athalya, P.K.; Janakiraman, N.; Khetan, V.; Kanwar, R.K.; Eluchuri, S.; Krishnakumar, S. Epcam aptamer mediated cancer cell specific delivery of epcam siRNA using polymeric nanocomplex. J. Biomed. Sci. 2015, 22, 4. [Google Scholar] [CrossRef] [PubMed]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA aptamers that inhibit ctla-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar] [PubMed]
- Walker, L.S.; Sansom, D.M. The emerging role of ctla4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Swiderski, P.; Herrmann, A.; Wang, L.; Kowolik, C.; Kujawski, M.; Lee, H.; Scuto, A.; Liu, Y.; Yang, C.; et al. In vivo delivery of siRNA to immune cells by conjugation to a tlr9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009, 27, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Priceman, S.J.; Swiderski, P.; Kujawski, M.; Xin, H.; Cherryholmes, G.A.; Zhang, W.; Zhang, C.; Lahtz, C.; Kowolik, C.; et al. Ctla4 aptamer delivers stat3 siRNA to tumor-associated and malignant T cells. J. Clin. Investig. 2014, 124, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Bartkowiak, T.; Curran, M.A. 4–1bb agonists: Multi-potent potentiators of tumor immunity. Front. Oncol. 2015, 5, 117. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O.; Kolonias, D.; Pastor, F.; Mittler, R.S.; Chen, L.; Giangrande, P.H.; Sullenger, B.; Gilboa, E. Multivalent 4–1bb binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Investig. 2008, 118, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Berezhnoy, A.; Castro, I.; Levay, A.; Malek, T.R.; Gilboa, E. Aptamer-targeted inhibition of mtor in T cells enhances antitumor immunity. J. Clin. Investig. 2014, 124, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, A.; Berezhnoy, A.; Schrand, B.; Puplampu-Dove, Y.; Gilboa, E. Aptamer-targeted attenuation of il-2 signaling in CD8+ T cells enhances antitumor immunity. Mol. Ther. 2017, 25, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, H.; Li, S.; Zaia, J.; Rossi, J.J. Novel dual inhibitory function aptamer-siRNA delivery system for HIV-1 therapy. Mol. Ther. 2008, 16, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Neff, C.P.; Zhou, J.; Remling, L.; Kuruvilla, J.; Zhang, J.; Li, H.; Smith, D.D.; Swiderski, P.; Rossi, J.J.; Akkina, R. An aptamer-siRNA chimera suppresses HIV-1 viral loads and protects from helper CD4(+) T cell decline in humanized mice. Sci. Transl. Med. 2011, 3, 66ra6. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.A.; Trifonova, R.; Vrbanac, V.; Basar, E.; McKernan, S.; Xu, Z.; Seung, E.; Deruaz, M.; Dudek, T.; Einarsson, J.I.; et al. Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. J. Clin. Investig. 2011, 121, 2401–2412. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.A.; Lin, Y.; Abrams, B.; Jayasena, S.D. Staining of cell surface human CD4 with 2′-f-pyrimidine-containing RNA aptamers for flow cytometry. Nucleic Acids Res. 1998, 26, 3915–3924. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Shibata, T.; Kabashima, T.; Kai, M. Inhibition of HIV-1 protease expression in T cells owing to DNA aptamer-mediated specific delivery of siRNA. Eur. J. Med. Chem. 2012, 56, 396–399. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Burnett, J.C.; Rossi, J.J. Aptamer-siRNA chimeras for HIV. Adv. Exp. Med. Biol. 2015, 848, 211–234. [Google Scholar] [PubMed]
- Dassie, J.P.; Hernandez, L.I.; Thomas, G.S.; Long, M.E.; Rockey, W.M.; Howell, C.A.; Chen, Y.; Hernandez, F.J.; Liu, X.Y.; Wilson, M.E.; et al. Targeted inhibition of prostate cancer metastases with an RNA aptamer to prostate-specific membrane antigen. Mol. Ther. 2014, 22, 1910–1922. [Google Scholar] [CrossRef] [PubMed]
- Talbot, L.J.; Mi, Z.; Bhattacharya, S.D.; Kim, V.; Guo, H.; Kuo, P.C. Pharmacokinetic characterization of an RNA aptamer against osteopontin and demonstration of in vivo efficacy in reversing growth of human breast cancer cells. Surgery 2011, 150, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, S.H.; Kim, J.H.; Noh, Y.H.; Noh, G.J.; Lee, S.W. Pharmacokinetics of a cholesterol-conjugated aptamer against the hepatitis c virus (hcv) ns5b protein. Mol. Ther. Nucleic Acids 2015, 4, e254. [Google Scholar] [CrossRef] [PubMed]
- Haupenthal, J.; Baehr, C.; Kiermayer, S.; Zeuzem, S.; Piiper, A. Inhibition of RNAse a family enzymes prevents degradation and loss of silencing activity of siRNAs in serum. Biochem. Pharmacol. 2006, 71, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Bonner, G.; Patra, D.; Lafer, E.M.; Sousa, R. Mutations in t7 RNA polymerase that support the proposal for a common polymerase active site structure. EMBO J. 1992, 11, 3767–3775. [Google Scholar] [PubMed]
- Behlke, M.A. Chemical modification of siRNAs for in vivo use. Oligonucleotides 2008, 18, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Gottfert, F.; Pleiner, T.; Heine, J.; Westphal, V.; Gorlich, D.; Sahl, S.J.; Hell, S.W. Strong signal increase in sted fluorescence microscopy by imaging regions of subdiffraction extent. Proc. Natl. Acad. Sci. USA 2017, 114, 2125–2130. [Google Scholar] [CrossRef] [PubMed]
- Wegel, E.; Gohler, A.; Lagerholm, B.C.; Wainman, A.; Uphoff, S.; Kaufmann, R.; Dobbie, I.M. Imaging cellular structures in super-resolution with sim, sted and localisation microscopy: A practical comparison. Sci. Rep. 2016, 6, 27290. [Google Scholar] [CrossRef] [PubMed]
- Wittrup, A.; Ai, A.; Liu, X.; Hamar, P.; Trifonova, R.; Charisse, K.; Manoharan, M.; Kirchhausen, T.; Lieberman, J. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat. Biotechnol. 2015, 33, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Gao, X. A universal protein tag for delivery of siRNA-aptamer chimeras. Sci. Rep. 2013, 3, 3129. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.W. Iss-n1 makes the first fda-approved drug for spinal muscular atrophy. Transl. Neurosci. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. Fda approves eteplirsen for duchenne muscular dystrophy: The next chapter in the eteplirsen saga. Nucleic Acid Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Lachelt, U.; Wagner, E. Nucleic acid therapeutics using polyplexes: A journey of 50 years (and beyond). Chem. Rev. 2015, 115, 11043–11078. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruspe, S.; Giangrande, P.H. Aptamer-siRNA Chimeras: Discovery, Progress, and Future Prospects. Biomedicines 2017, 5, 45. https://doi.org/10.3390/biomedicines5030045
Kruspe S, Giangrande PH. Aptamer-siRNA Chimeras: Discovery, Progress, and Future Prospects. Biomedicines. 2017; 5(3):45. https://doi.org/10.3390/biomedicines5030045
Chicago/Turabian StyleKruspe, Sven, and Paloma H. Giangrande. 2017. "Aptamer-siRNA Chimeras: Discovery, Progress, and Future Prospects" Biomedicines 5, no. 3: 45. https://doi.org/10.3390/biomedicines5030045
APA StyleKruspe, S., & Giangrande, P. H. (2017). Aptamer-siRNA Chimeras: Discovery, Progress, and Future Prospects. Biomedicines, 5(3), 45. https://doi.org/10.3390/biomedicines5030045