

Involvement of the AKT Pathway in Resistance to Erlotinib and Cabozantinib in Triple-Negative Breast Cancer Cell Lines
 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Breast Cancer Therapy Resistance Models
2.2. Colony-Forming Assays
2.3. Cell Migration and Invasion Assays
2.4. Immunoblotting
2.5. SILAC Media Formulation and Incorporation
2.6. LC-MS/MS Data Acquisition and Processing
2.7. Bioinformatics–Kinase Activity Analysis
2.8. Drug Combination Synergy Assays
2.9. Statistical Analysis
3. Results
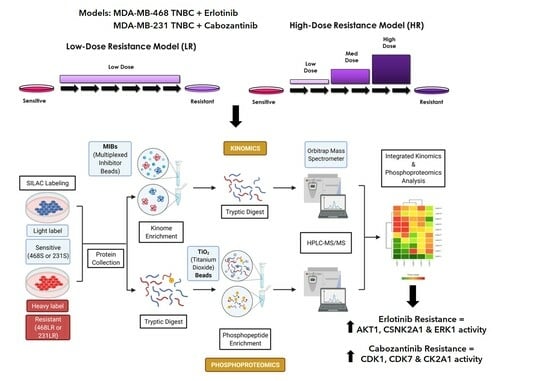
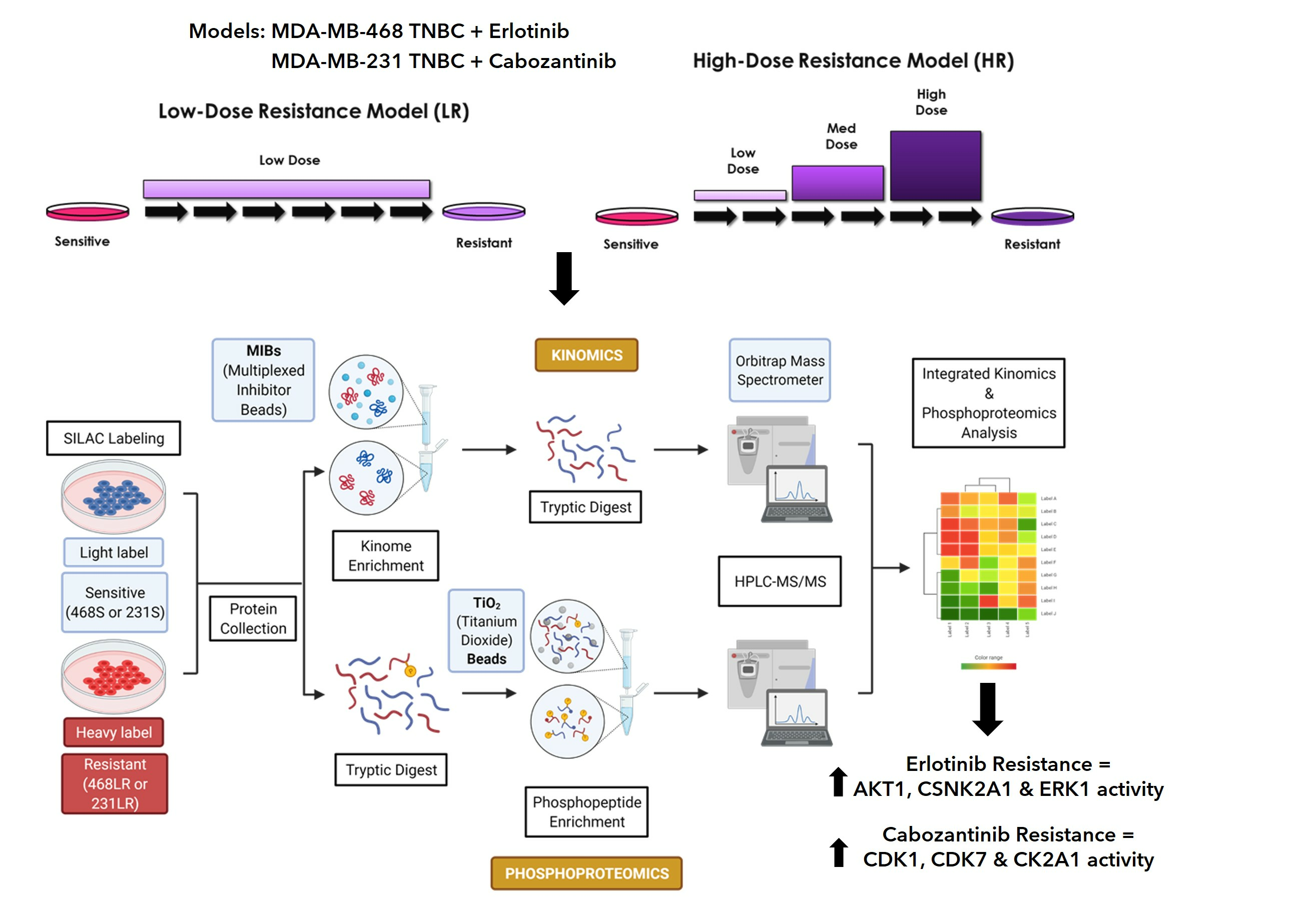
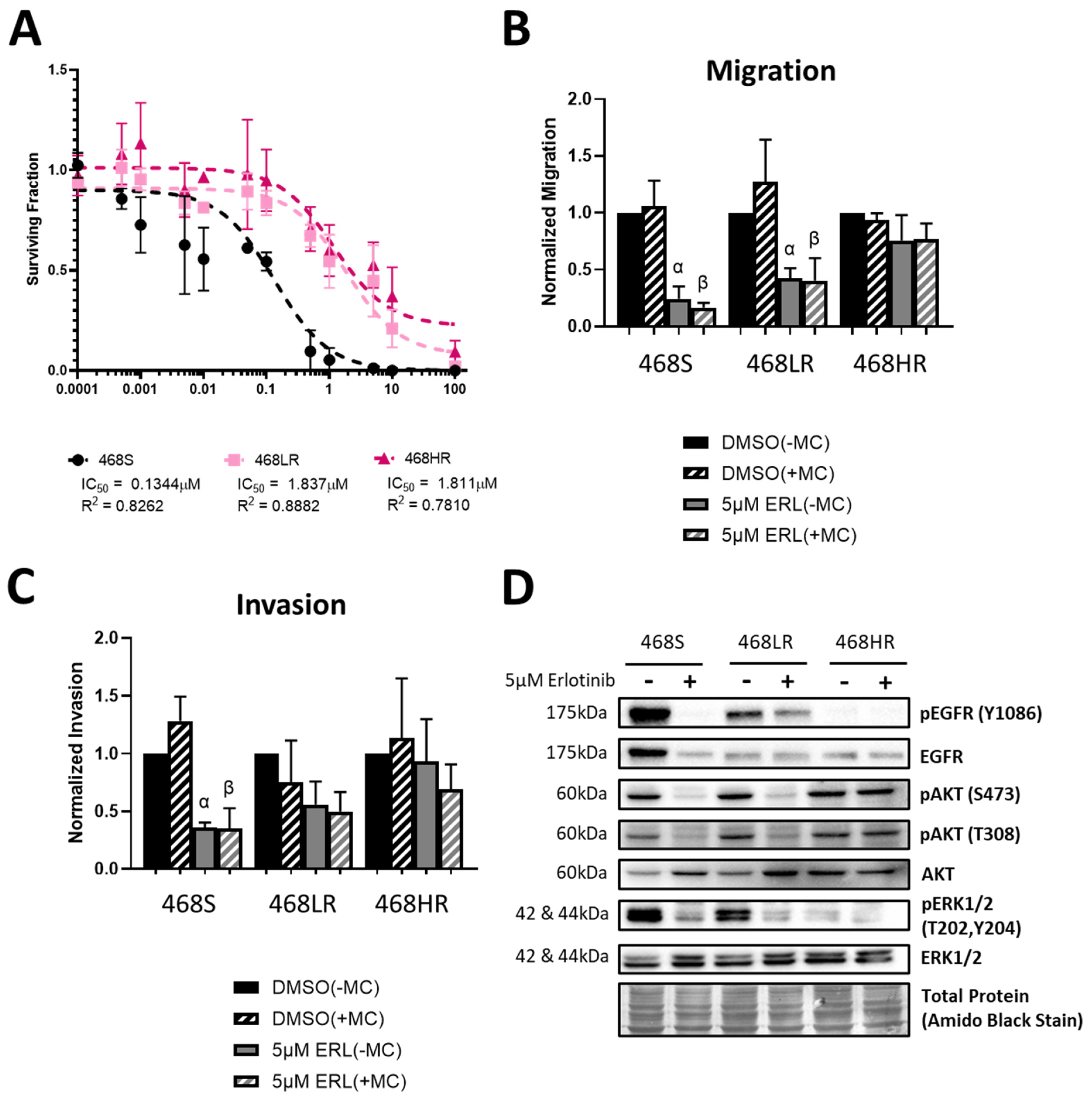
3.1. Erlotinib-Resistant Human MDA-MB-468 TNBC Cells Demonstrate a Loss of Inhibition of Cell Proliferation, Migration, Invasion, and EGFR/ERK/AKT Signaling
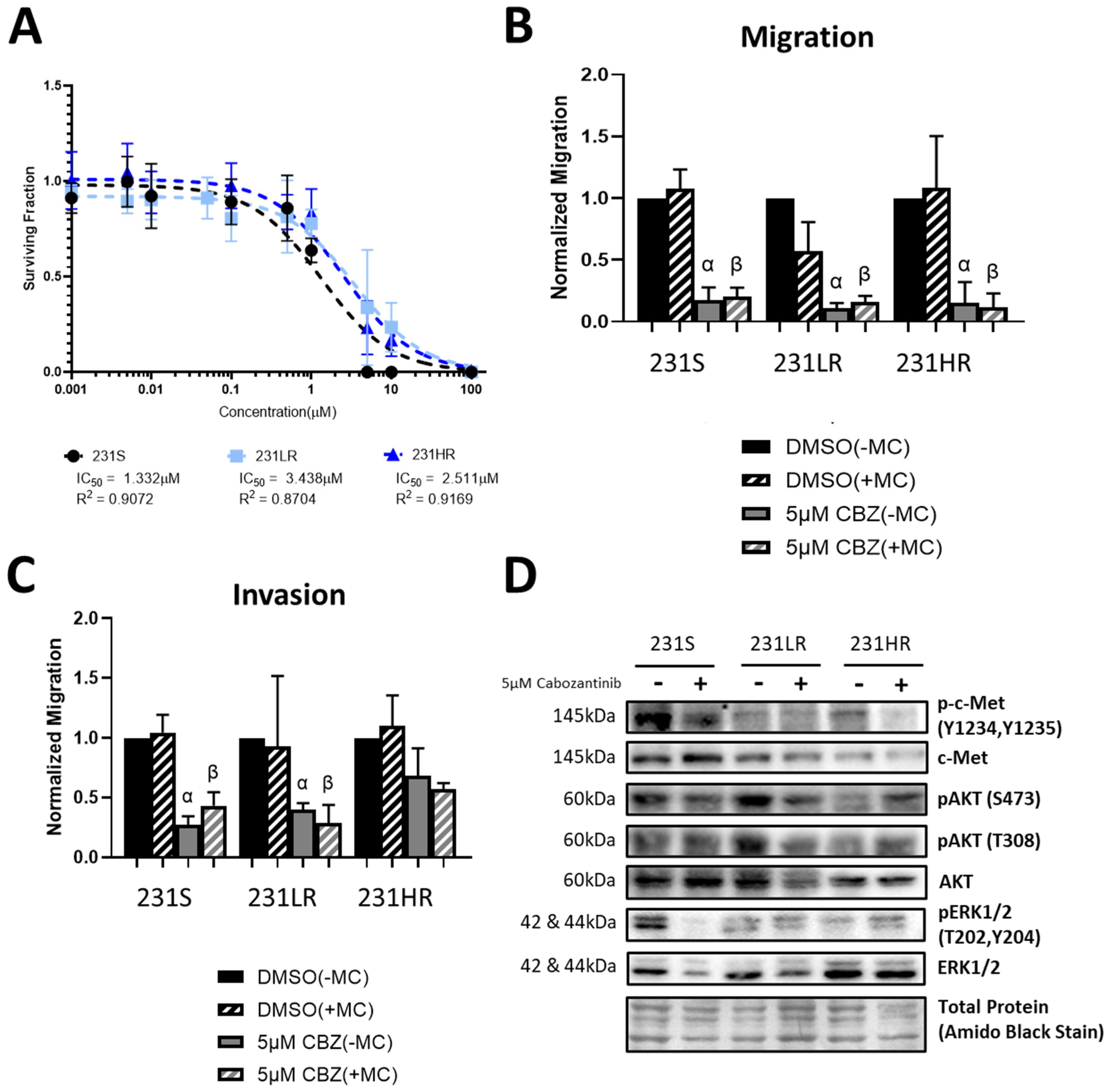
3.2. Cabozantinib-Resistant Human MDA-MB-431 TNBC Cells Demonstrate a Lack of Inhibition of Cell Proliferation, Invasion, and AKT Signaling
3.3. Acquisition of Resistance to Erlotinib and Cabozantinib Does Not Alter the Breast Cancer Stem Cell Phenotype
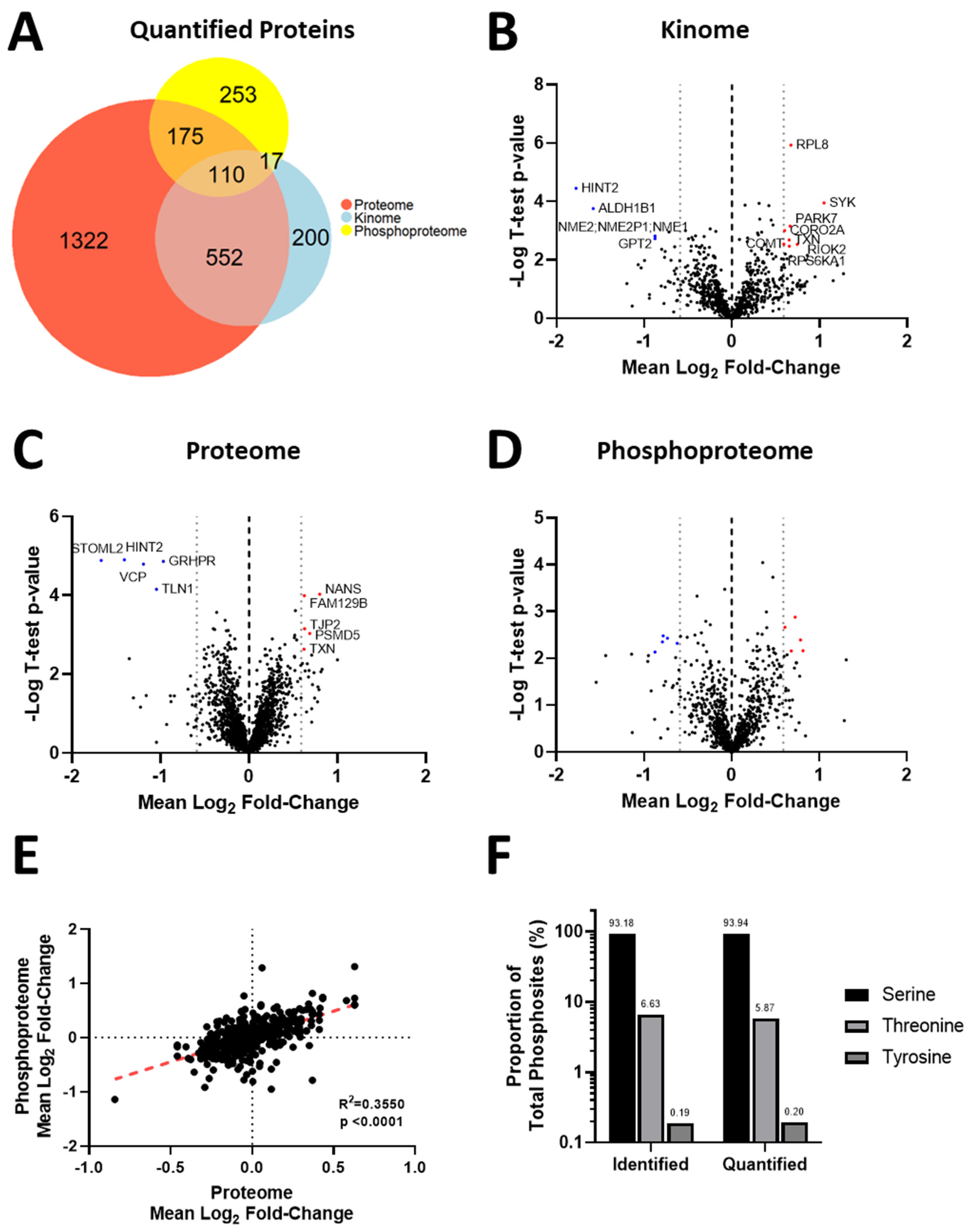
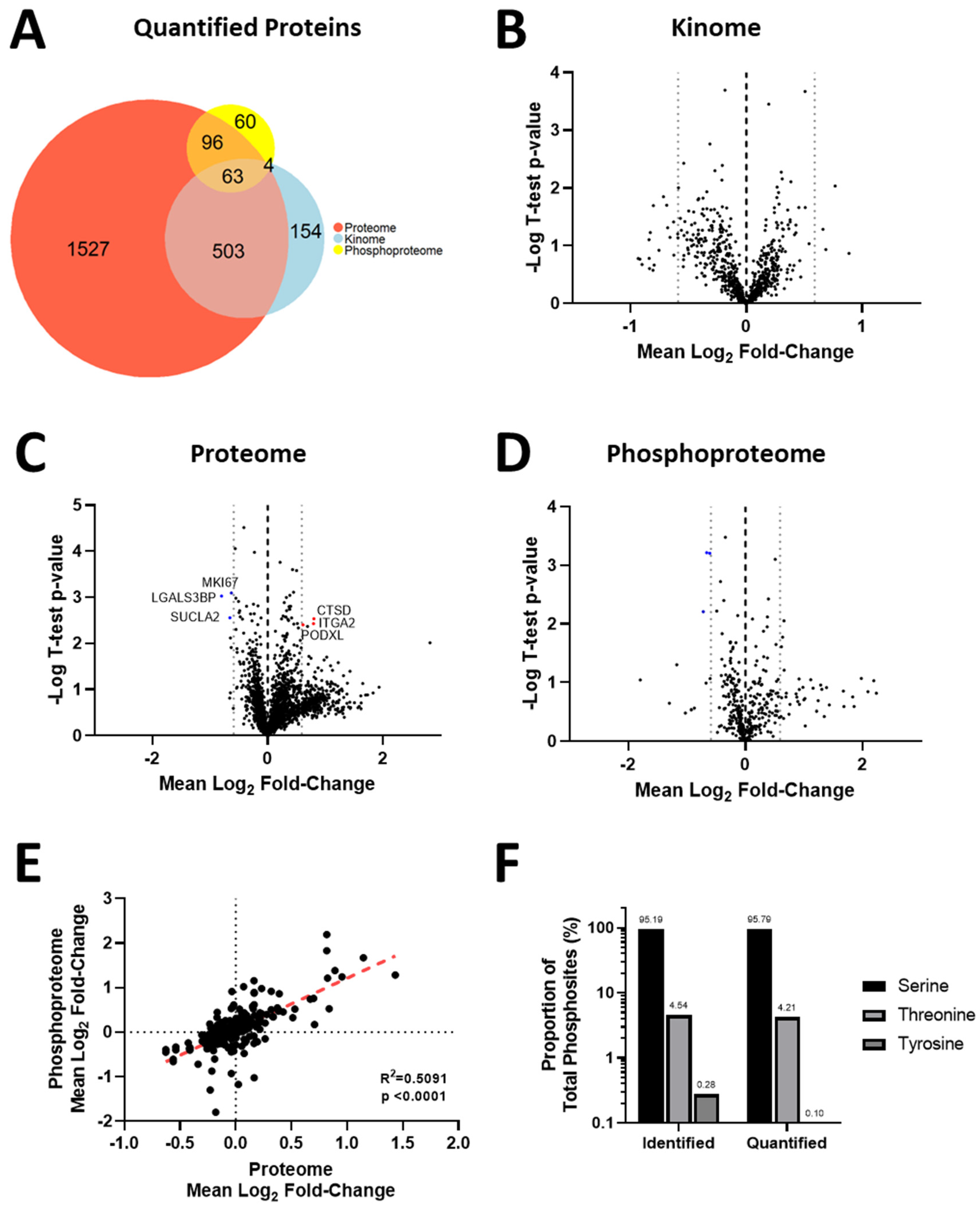
3.4. Acquisition of Resistance to Erlotinib and Cabozantinib Alters the Proteome and Kinome of TNBC Cells with Minimal Change to the Phosphoproteome
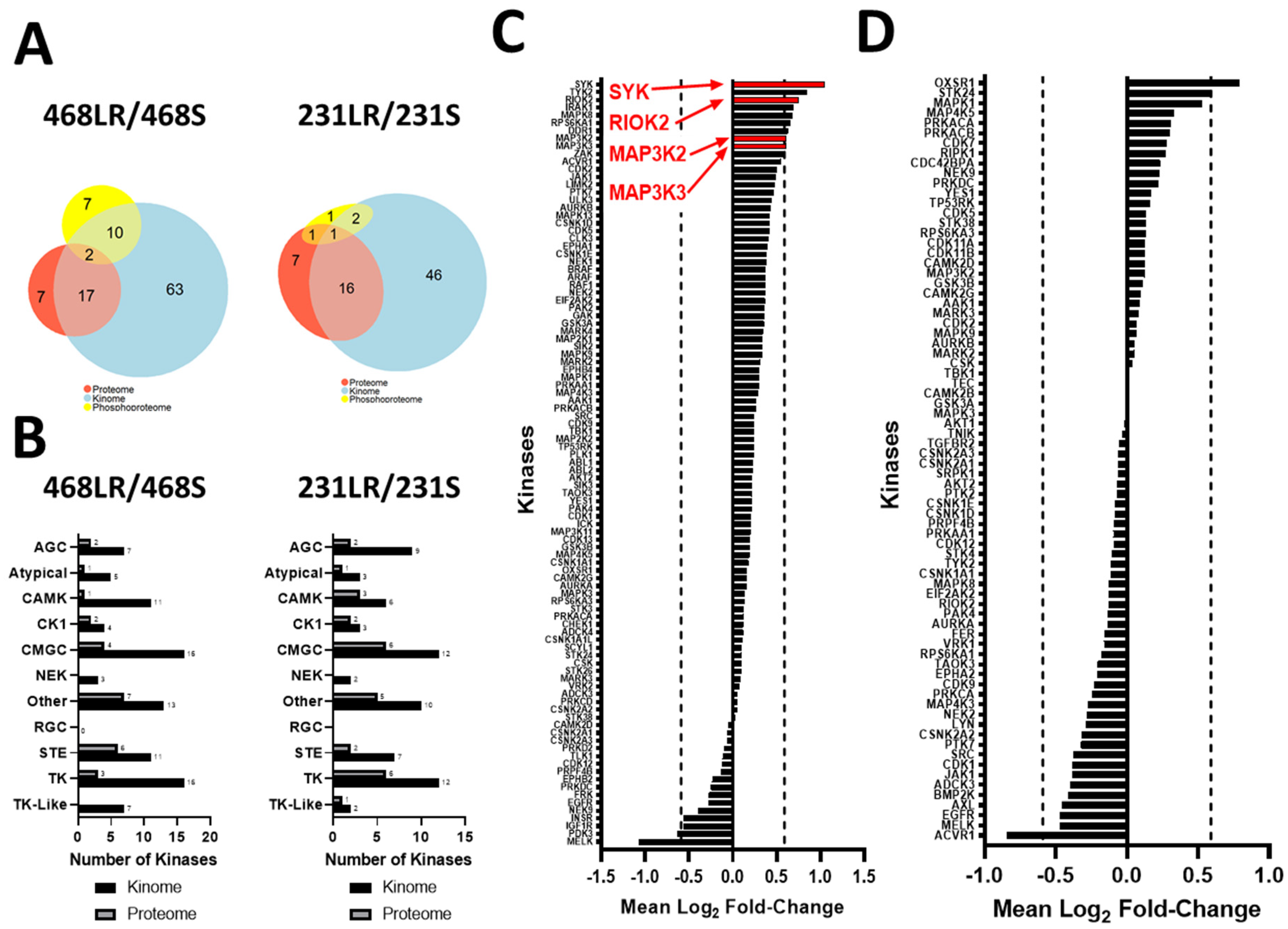
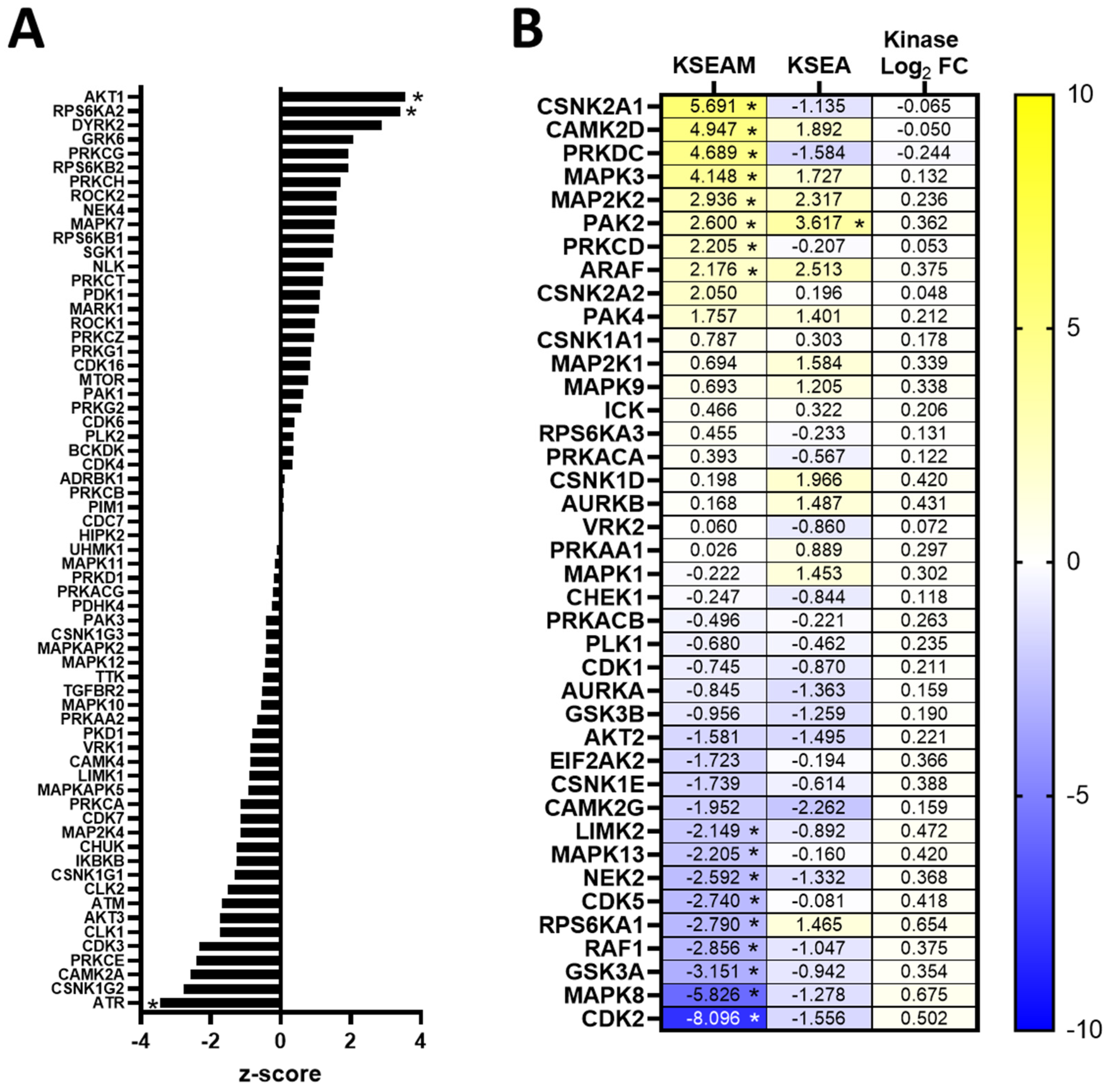
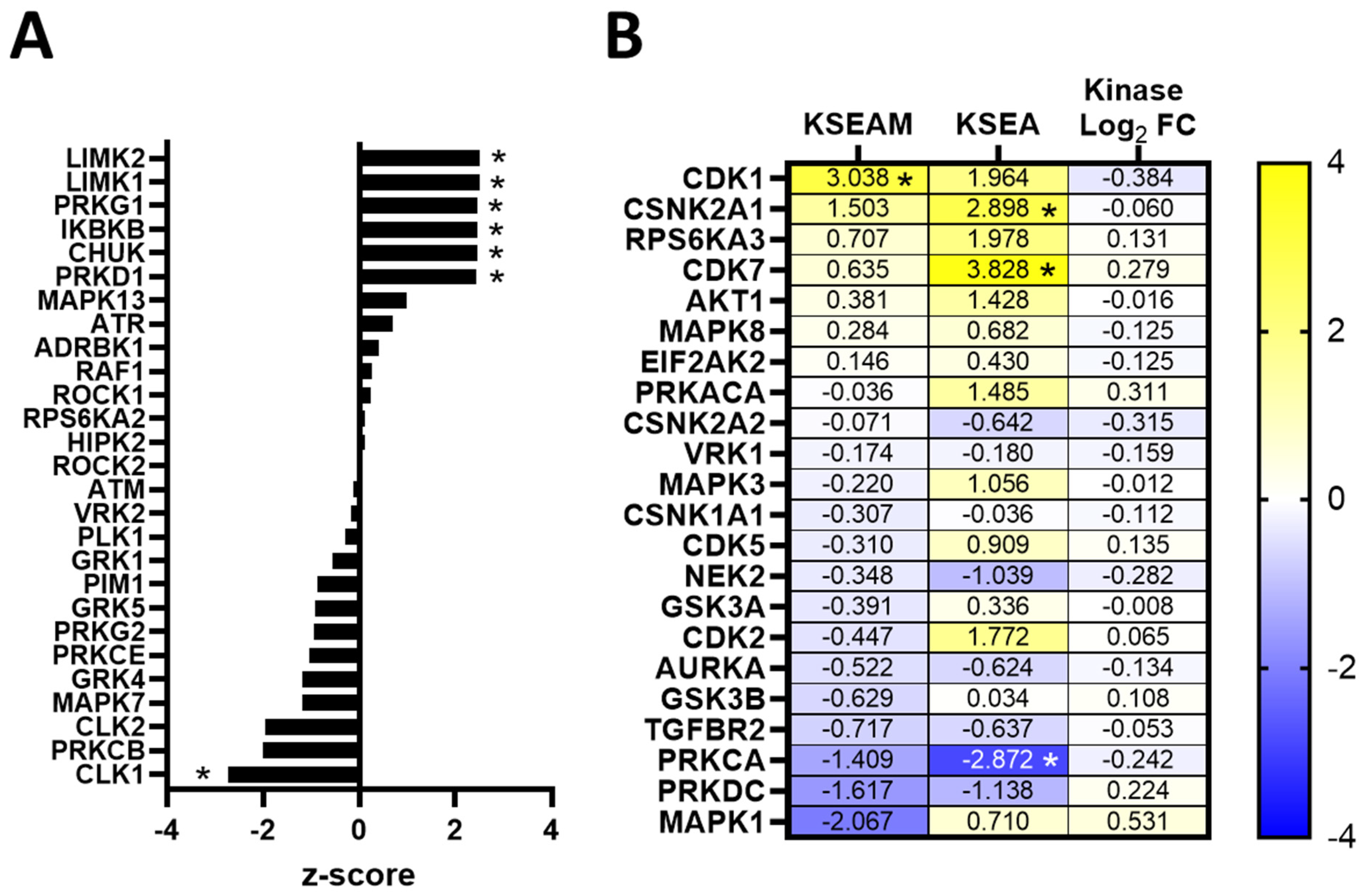
3.5. Kinase Expression and/or Activity Is Elevated in Erlotinib- and Cabozantinib-Resistant TNBC Cells
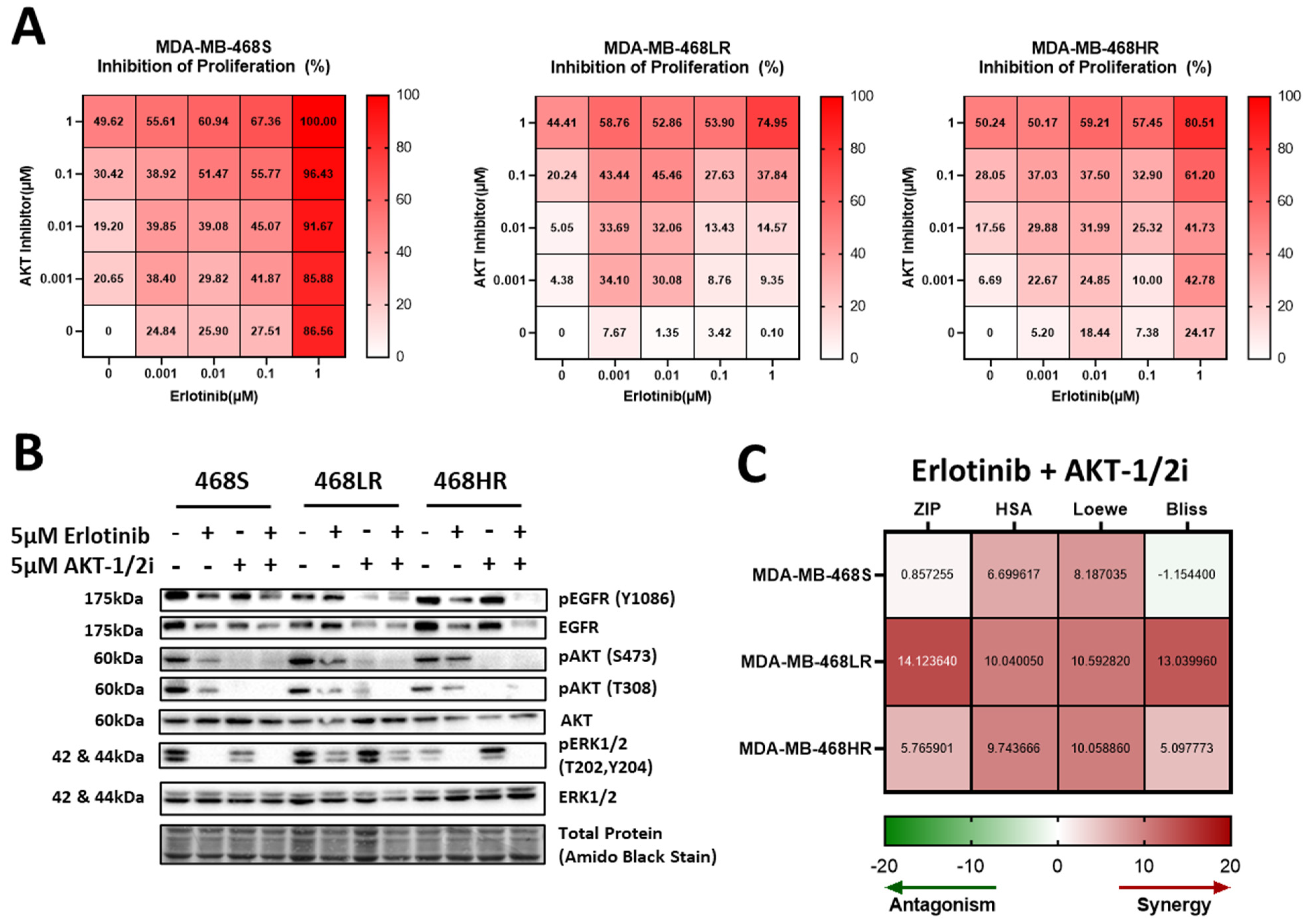
3.6. Resistance to Erlotinib Can Be Reversed in MDA-MB-468 Cells via Synergistic Targeting of AKT Activity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene Expression Patterns of Breast Carcinomas Distinguish Tumor Subclasses with Clinical Implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—Mechanism of Action and Use in Clinical Practice. NEJM 2007, 357, 39–51. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sotiriou, C. Luminal Breast Cancer: From Biology to Treatment. Nat. Rev. Clin. Oncol. 2013, 10, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Zardavas, D.; Baselga, J.; Piccart, M. Emerging Targeted Agents in Metastatic Breast Cancer. Nat. Rev. Clin. Oncol. 2013, 10, 191–210. [Google Scholar] [CrossRef]
- Kawalec, P.; Łopuch, S.; Mikrut, A. Effectiveness of Targeted Therapy in Patients with Previously Untreated Metastatic Breast Cancer: A Systematic Review and Meta-Analysis. Clin. Breast Cancer 2015, 15, 90–100.e1. [Google Scholar] [CrossRef] [PubMed]
- Lang, J.E.; Wecsler, J.S.; Press, M.F.; Tripathy, D. Molecular Markers for Breast Cancer Diagnosis, Prognosis and Targeted Therapy. J. Surg Oncol. 2015, 111, 81–90. [Google Scholar] [CrossRef]
- Fallahpour, S.; Navaneelan, T.; De, P.; Borgo, A. Breast Cancer Survival by Molecular Subtype: A Population-Based Analysis of Cancer Registry Data. CMAJ Open 2017, 5, E734–E739. [Google Scholar] [CrossRef]
- Howlader, N.; Cronin, K.A.; Kurian, A.W.; Andridge, R. Cancer Surveillance Research Differences in Breast Cancer Survival by Molecular Subtypes in the United States. Cancer Epidemiol. Biomark. Prev. 2018, 27, 619–626. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.; Dent, R.; Mackey, J.R.; Laing, K.; Mcleod, D.; Verma, S. Targeting Triple-Negative Breast Cancer: Optimising Therapeutic Outcomes. Ann. Oncol. 2012, 23, 2223–2234. [Google Scholar] [CrossRef] [PubMed]
- Salomon, D.S.; Brandta, R.; Ciardiellob, F.; Normannoc, N. Epidermal Growth Factor-Related Peptides and Their Receptors in Human Malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Lurje, G.; Lenz, H.-J. EGFR Signaling and Drug Discovery. Oncology 2009, 77, 400–410. [Google Scholar] [CrossRef]
- Martinazzi, M.; Crivelli, F.; Zampatti, C.; Martinazzi, S. Epidermal Growth Factor Receptor Immunohistochemistry in Different Histological Types of Infiltrating Breast Carcinoma. J. Clin. Pathol. 1993, 46, 1009–1010. [Google Scholar] [CrossRef][Green Version]
- Richard, J.; Sainsbury, C.; Needham, G.; Farndon, J.; Malcolm, A.; Harris, A. Epidermal-growth-factor receptor status as predictor of early recurrence of and death from breast cancer. Lancet 1987, 329, 1398–1402. [Google Scholar] [CrossRef]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Burness, M.L.; Grushko, T.A.; Olopade, O.I. Epidermal Growth Factor Receptor in Triple-Negative and Basal-Like Breast Cancer. Cancer J. 2010, 16, 23–32. [Google Scholar] [CrossRef]
- Guérin, M.; Gabillot, M.; Mathieu, M.C.; Travagli, J.P.; Spielmann, M.; Andrieu, N.; Riou, G. Structure and Expression of C-ErbB-2 and EGF Receptor Genes in Inflammatory and Non-Inflammatory Breast Cancer: Prognostic Significance. Int. J. Cancer 1989, 43, 201–208. [Google Scholar] [CrossRef]
- Schechter, A.; Hung, M.; Vaidyanathan, L.; Weinberg, R.; Yang-Feng, T.; Francke, U.; Ullrich, A.; Coussens, L. The Neu Gene: An ErbB-Homologous Gene Distinct from and Unlinked to the Gene Encoding the EGF Receptor. Science 1985, 229, 976–978. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic Kinase Signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Burgess, A.W. EGFR Family: Structure Physiology Signalling and Therapeutic Targets. Growth Factors 2008, 26, 263–274. [Google Scholar] [CrossRef]
- Riese, D.J.; Bermingham, Y.; van Raaij, T.M.; Buckley, S.; Plowman, G.D.; Stern, D.F. Betacellulin Activates the Epidermal Growth Factor Receptor and ErbB-4, and Induces Cellular Response Patterns Distinct from Those Stimulated by Epidermal Growth Factor or Neuregulin-Beta. Oncogene 1996, 12, 345–353. [Google Scholar] [PubMed]
- Cappuzzo, F.; Ciuleanu, T.; Stelmakh, L.; Cicenas, S.; Szczésna, A.; Juhász, E.; Esteban, E.; Molinier, O.; Brugger, W.; Melezínek, I.; et al. Erlotinib as Maintenance Treatment in Advanced Non-Small-Cell Lung Cancer: A Multicentre, Randomised, Placebo-Controlled Phase 3 Study. Lancet Oncol. 2010, 11, 521–529. [Google Scholar] [CrossRef]
- Corkery, B.; Crown, J.; Clynes, M.; O’Donovan, N. Epidermal Growth Factor Receptor as a Potential Therapeutic Target in Triple-Negative Breast Cancer. Ann. Oncol. 2009, 20, 862–867. [Google Scholar] [CrossRef]
- Dickler, M.N.; Rugo, H.S.; Eberle, C.A.; Brogi, E.; Caravelli, J.F.; Panageas, K.S.; Boyd, J.; Yeh, B.; Lake, D.E.; Dang, C.T.; et al. A Phase II Trial of Erlotinib in Combination with Bevacizumab in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2008, 14, 7878–7883. [Google Scholar] [CrossRef]
- Dickler, M.N.; Cobleigh, M.A.; Miller, K.D.; Klein, P.M.; Winer, E.P. Efficacy and Safety of Erlotinib in Patients with Locally Advanced or Metastatic Breast Cancer. Breast Cancer Res. Treat 2009, 115, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wu, Y.L.; Chen, G.; Feng, J.; Liu, X.Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus Chemotherapy as First-Line Treatment for Patients with Advanced EGFR Mutation-Positive Non-Small-Cell Lung Cancer (OPTIMAL, CTONG-0802): A Multicentre, Open-Label, Randomised, Phase 3 Study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Raab, G.; Klagsbrun, M. Heparin-Binding EGF-like Growth Factor. Biochim. Biophys. Acta 1997, 1333, F179–F199. [Google Scholar] [CrossRef]
- Sharma, P.; Khan, Q.; Kimler, B.; Klemp, J.; Connor, C.; McGinness, M.; Mammen, J.; Tawfik, O.; Fan, F.; Fabian, C.J. Abstract P1-11-07: Results of a Phase II Study of Neoadjuvant Platinum/Taxane Based Chemotherapy and Erlotinib for Triple Negative Breast Cancer. Cancer Res. 2010, 70, P1–P11. [Google Scholar] [CrossRef]
- Maurer, M.A.; Kalinsky, K.; Crew, K.; Jayasena, R.; Forman, J.; Lau, Y.-K.; Parsons, R.; Hershman, D. Abstract 28: Phase I Trial of Combined Temsirolimus, Erlotinib, and Cisplatin in Advanced Solid Tumors. Cancer Res. 2013, 73, 28. [Google Scholar] [CrossRef]
- Tan, A.R.; Yang, X.; Hewitt, S.M.; Berman, A.; Lepper, E.R.; Sparreboom, A.; Parr, A.L.; Figg, W.D.; Chow, C.; Steinberg, S.M.; et al. Evaluation of Biologic End Points and Pharmacokinetics in Patients with Metastatic Breast Cancer after Treatment with Erlotinib, an Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. J. Clin. Oncol. 2004, 22, 3080–3090. [Google Scholar] [CrossRef]
- Xiang, Q.; Chen, W.; Ren, M.; Wang, J.; Zhang, H.; Deng, D.Y.B.; Zhang, L.; Shang, C.; Chen, Y. Cabozantinib Suppresses Tumor Growth and Metastasis in Hepatocellular Carcinoma by a Dual Blockade of VEGFR2 and MET. Clin. Cancer Res. 2014, 20, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a Novel MET and VEGFR2 Inhibitor, Simultaneously Suppresses Metastasis, Angiogenesis, and Tumor Growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Sameni, M.; Tovar, E.A.; Essenburg, C.J.; Chalasani, A.; Linklater, E.S.; Borgman, A.; Cherba, D.M.; Anbalagan, A.; Winn, M.E.; Graveel, C.R.; et al. Cabozantinib (XL184) Inhibits Growth and Invasion of Preclinical TNBC Models. Clin. Cancer Res. 2016, 22, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Gordon, M.; Smith, D.C.; Kurzrock, R.; Daud, A.; Vogelzang, N.J.; Lee, Y.; Scheffold, C.; Shapiro, G.I. Phase II Randomised Discontinuation Trial of Cabozantinib in Patients with Advanced Solid Tumours. Eur. J. Cancer 2017, 86, 296–304. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Nechushtan, H.; Ron, I.-G.; Schöffski, P.; Awada, A.; Yasenchak, C.A.; Laird, A.D.; O’Keeffe, B.; Shapiro, G.I.; Winer, E.P. Cabozantinib for Metastatic Breast Carcinoma: Results of a Phase II Placebo-Controlled Randomized Discontinuation Study. Breast Cancer Res. Treat 2016, 160, 305–312. [Google Scholar] [CrossRef]
- Miller, S.M.; Goulet, D.R.; Johnson, G.L. Targeting the Breast Cancer Kinome. J. Cell Physiol. 2017, 232, 53–60. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR Mutation and Resistance of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Traxl, A.; Wanek, T.; Mairinger, S.; Stanek, J.; Filip, T.; Sauberer, M.; Muller, M.; Kuntner, C.; Langer, O. Breast Cancer Resistance Protein and P-Glycoprotein Influence In Vivo Disposition of 11C-Erlotinib. J. Nucl. Med. 2015, 56, 1930–1936. [Google Scholar] [CrossRef] [PubMed]
- Suda, K.; Tomizawa, K.; Fujii, M.; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Epithelial to Mesenchymal Transition in an Epidermal Growth Factor Receptor-Mutant Lung Cancer Cell Line with Acquired Resistance to Erlotinib. J. Thorac. Oncol. 2011, 6, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.-Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET Amplification Occurs with or without T790M Mutations in EGFR Mutant Lung Tumors with Acquired Resistance to Gefitinib or Erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 Amplification: A Potential Mechanism of Acquired Resistance to Egfr Inhibition in EGFR -Mutant Lung Cancers That Lack the Second-Site EGFR T790M Mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef]
- Lin, L.; Bivona, T.G. Mechanisms of Resistance to Epidermal Growth Factor Receptor Inhibitors and Novel Therapeutic Strategies to Overcome Resistance in NSCLC Patients. Chemother. Res. Pract. 2012, 2012, 817297. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Jubb, A.M.; Lyle, K.; Xiao, Q.; Ong, C.C.; Desai, R.; Fu, L.; Gnad, F.; Song, Q.; Haverty, P.M.; et al. PAK1 Mediates Pancreatic Cancer Cell Migration and Resistance to MET Inhibition. J. Pathol. 2014, 234, 502–513. [Google Scholar] [CrossRef]
- Corso, S.; Ghiso, E.; Cepero, V.; Sierra, J.R.; Migliore, C.; Bertotti, A.; Trusolino, L.; Comoglio, P.M.; Giordano, S. Activation of HER Family Members in Gastric Carcinoma Cells Mediates Resistance to MET Inhibition. Mol. Cancer 2010, 9, 121. [Google Scholar] [CrossRef]
- Ji, F.; Liu, X.; Wu, Y.; Fang, X.; Huang, G. Overexpression of PI3K P110α Contributes to Acquired Resistance to MET Inhibitor, in MET-Amplified SNU-5 Gastric Xenografts. Drug Des. Dev. Ther. 2015, 9, 5697. [Google Scholar] [CrossRef] [PubMed]
- Hanker, A.B.; Pfefferle, A.D.; Balko, J.M.; Kuba, M.G.; Young, C.D.; Sanchez, V.; Sutton, C.R.; Cheng, H.; Perou, C.M.; Zhao, J.J.; et al. Mutant PIK3CA Accelerates HER2-Driven Transgenic Mammary Tumors and Induces Resistance to Combinations of Anti-HER2 Therapies. Proc. Natl. Acad. Sci. USA 2013, 110, 14372–14377. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.; Eustace, A.J.; Busschots, S.; Breen, L.; Crown, J.; Clynes, M.; Stordal, B.; Mini, E.; Michiels, C.; O’Donovan, N.; et al. In Vitro Development of Chemotherapy and Targeted Therapy Drug-Resistant Cancer Cell Lines: A Practical Guide with Case Studies. Front. Oncol. 2014, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Allan, A.L. Anti-Proliferative and Anti-Migratory Effects of EGFR and c-Met Tyrosine Kinase Inhibitors in Triple Negative Breast Cancer Cells. Precis. Cancer Med. 2021, 4, 2. [Google Scholar] [CrossRef]
- Bendall, S.C.; Hughes, C.; Stewart, M.H.; Doble, B.; Bhatia, M.; Lajoie, G.A. Prevention of Amino Acid Conversion in SILAC Experiments with Embryonic Stem Cells. Mol. Cell. Proteom. 2008, 7, 1587–1597. [Google Scholar] [CrossRef]
- Marcilla, M.; Alpizar, A.; Paradela, A.; Albar, J.P. A Systematic Approach to Assess Amino Acid Conversions in SILAC Experiments. Talanta 2011, 84, 430–436. [Google Scholar] [CrossRef]
- Cox, J.; Matic, I.; Hilger, M.; Nagaraj, N.; Selbach, M.; Olsen, J.V.; Mann, M. A Practical Guide to the MaxQuant Computational Platform for SILAC-Based Quantitative Proteomics. Nat. Protoc. 2009, 4, 698–705. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant Computational Platform for Mass Spectrometry-Based Shotgun Proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus Computational Platform for Comprehensive Analysis of (Prote)Omics Data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Casado, P.; Rodriguez-Prados, J.-C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-Substrate Enrichment Analysis Provides Insights into the Heterogeneity of Signaling Pathway Activation in Leukemia Cells. Sci. Sign. 2013, 6, rs6. [Google Scholar] [CrossRef]
- Wiredja, D.D.; Koyutürk, M.; Chance, M.R. The KSEA App: A Web-Based Tool for Kinase Activity Inference from Quantitative Phosphoproteomics. Bioinformatics 2017, 33, 3489–3491. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wang, W.; Aldahdooh, J.; Malyutina, A.; Shadbahr, T.; Pessia, A.; Tang, J. SynergyFinder Plus: Towards a Better Interpretation and Annotation of Drug Combination Screening Datasets. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Ianevski, A.; Giri, A.; Aittokallio, T. SynergyFinder 2.0: Visual Analytics of Multi-Drug Combination Synergies. Nucleic Acids Res. 2020, 48, W488–W493. [Google Scholar] [CrossRef]
- Hidalgo, M.; Bloedow, D. Pharmacokinetics and Pharmacodynamics: Maximizing the Clinical Potential of Erlotinib (Tarceva). Semin. Oncol. 2003, 30, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Rivas, S.; Marín, A.; Samtani, S.; González-Feliú, E.; Armisén, R. MET Signaling Pathways, Resistance Mechanisms, and Opportunities for Target Therapies. Int. J. Mol. Sci. 2022, 23, 13898. [Google Scholar] [CrossRef]
- Lacy, S.A.; Miles, D.R.; Nguyen, L.T. Clinical Pharmacokinetics and Pharmacodynamics of Cabozantinib. Clin. Pharmacokinet. 2017, 56, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wennerberg, K.; Aittokallio, T. What Is Synergy? The Saariselkä Agreement Revisited. Front. Pharmacol. 2015, 6, 181. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose–Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef]
- Bixby, D.; Talpaz, M. Mechanisms of Resistance to Tyrosine Kinase Inhibitors in Chronic Myeloid Leukemia and Recent Therapeutic Strategies to Overcome Resistance. Hematology 2009, 2009, 461–476. [Google Scholar] [CrossRef]
- Jorge, S.E.D.C.E.D.C.; Kobayashi, S.S.S.; Costa, D.B.B. Epidermal Growth Factor Receptor (EGFR) Mutations in Lung Cancer: Preclinical and Clinical Data. Braz. J. Med. Biol. Res. 2014, 47, 929–939. [Google Scholar] [CrossRef]
- Minuti, G.; Cappuzzo, F.; Duchnowska, R.; Jassem, J.; Fabi, A.; O’Brien, T.; Mendoza, A.D.; Landi, L.; Biernat, W.; Czartoryska-Arłukowicz, B.; et al. Increased MET and HGF Gene Copy Numbers Are Associated with Trastuzumab Failure in HER2-Positive Metastatic Breast Cancer. Br. J. Cancer 2012, 107, bjc2012335. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Ziehr, D.R.; Guo, H.; Ng, M.R.; Barry, W.T.; Higgins, M.J.; Isakoff, S.J.; Brock, J.E.; Ivanova, E.V.; Paweletz, C.P.; et al. Phase II and Biomarker Study of Cabozantinib in Metastatic Triple-Negative Breast Cancer Patients. Oncologist 2017, 22, 25–32. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating Viruses and Cellular Organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Zemp, I.; Wild, T.; O’Donohue, M.; Wandrey, F.; Widmann, B.; Gleizes, P.E.; Kutay, U. Distinct Cytoplasmic Maturation Steps of 40S Ribosomal Subunit Precursors Require HRio2. J. Cell Biol. 2009, 185, 1167–1180. [Google Scholar] [CrossRef]
- Huang, D.-Y.; Chen, W.-Y.; Chen, C.-L.; Wu, N.-L.; Lin, W.-W. Synergistic Anti-Tumour Effect of Syk Inhibitor and Olaparib in Squamous Cell Carcinoma: Roles of Syk in EGFR Signalling and PARP1 Activation. Cancers 2020, 12, 489. [Google Scholar] [CrossRef]
- Yu, Y.; Rahmanto, Y.S.; Shen, Y.-A.; Ardighieri, L.; Davidson, B.; Gaillard, S.; Ayhan, A.; Shi, X.; Xuan, J.; Wang, T.-L.; et al. Spleen Tyrosine Kinase Activity Regulates Epidermal Growth Factor Receptor Signaling Pathway in Ovarian Cancer. EBioMedicine 2019, 47, 184–194. [Google Scholar] [CrossRef]
- Lamb, D.J.; Rust, A.; Rudisch, A.; Glüxam, T.; Harrer, N.; Machat, H.; Christ, I.; Colbatzky, F.; Wernitznig, A.; Osswald, A.; et al. Inhibition of SYK Kinase Does Not Confer a Pro-Proliferative or pro-Invasive Phenotype in Breast Epithelium or Breast Cancer Cells. Oncotarget 2020, 11, 1257–1272. [Google Scholar] [CrossRef] [PubMed]
- Coopman, P.; Do, M.; Barth, M.; Bowden, E.; Hayes, A.J.; Basyuk, E.; Blancato, J.K.; Vezza, P.R.; McLeskey, S.W.; Mangeat, P.H.; et al. The Syk Tyrosine Kinase Suppresses Malignant Growth of Human Breast Cancer Cells. Nature 2000, 406, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Krisenko, M.O.; Geahlen, R.L. Calling in SYK: SYK’s Dual Role as a Tumor Promoter and Tumor Suppressor in Cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 254–263. [Google Scholar] [CrossRef]
- Chorner, P.M.; Moorehead, R.A. A-674563, a Putative AKT1 Inhibitor That Also Suppresses CDK2 Activity, Inhibits Human NSCLC Cell Growth More Effectively than the Pan-AKT Inhibitor, MK-2206. PLoS ONE 2018, 13, e0193344. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.Y.; Hou, Y.J.; Yin, Y.B.; Wang, F.Z.; Yang, M.F.; Zhang, Y.Y.; Fan, C.D.; Sun, B. liang CCT128930 Induces G1-Phase Arrest and Apoptosis and Synergistically Enhances the Anticancer Efficiency of VS5584 in Human Osteosarcoma Cells. Biomed. Pharmacother. 2020, 130, 110544. [Google Scholar] [CrossRef]
- Holland, W.S.; Chinn, D.C.; Lara, P.N., Jr.; Gandara, D.R.; Mack, P.C. Effects of AKT Inhibition on HGF-Mediated Erlotinib Resistance in Non-Small Cell Lung Cancer Cell Lines. J. Cancer Res. Clin. Oncol. 2015, 141, 615. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C. Targeting the PI3K/Akt/MTOR Pathway in Non-Small Cell Lung Cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef]
- Rabalski, A.J.; Gyenis, L.; Litchfield, D.W. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a Potential Target to Inhibit Survival and DNA Damage Response and Repair Pathways in Cancer Cells. Clin. Cancer Res. 2016, 22, 2840–2847. [Google Scholar] [CrossRef]
- Izadi, S.; Nikkhoo, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Mohammadi, H.; Yousefi, M.; Jadidi-Niaragh, F. CDK1 in Breast Cancer: Implications for Theranostic Potential. Anticancer Agents Med. Chem. 2020, 20, 758–767. [Google Scholar] [CrossRef]
- McDermott, M.; Sharko, A.; Munie, J.; Kassler, S.; Melendez, T.; Lim, C.; Broude, E. CDK7 Inhibition Is Effective in All the Subtypes of Breast Cancer: Determinants of Response and Synergy with EGFR Inhibition. Cells 2020, 9, 638. [Google Scholar] [CrossRef]
- Lee, D.-F.; Hung, M.-C. Advances in Targeting IKK and IKK-Related Kinases for Cancer Therapy. Clin. Cancer Res. 2008, 14, 5656. [Google Scholar] [CrossRef] [PubMed]
- Prunier, C.; Prudent, R.; Kapur, R.; Sadoul, K.; Lafanechère, L. LIM Kinases: Cofilin and Beyond. Oncotarget 2017, 8, 41749. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective Small-Molecule Inhibitor Reveals Critical Mitotic Functions of Human CDK1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef] [PubMed]
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 Inhibitors as Anticancer Drugs. Cancer Metastasis Rev. 2020, 39, 805. [Google Scholar] [CrossRef] [PubMed]
- Laface, C.; Maselli, F.M.; Santoro, A.N.; Iaia, M.L.; Ambrogio, F.; Laterza, M.; Guarini, C.; De Santis, P.; Perrone, M.; Fedelem, P. The Resistance to EGFR-TKIs in Non-Small Cell Lung Cancer: From Molecular Mechanisms to Clinical Application of New Therapeutic Strategies. Pharmaceutics 2023, 15, 1604. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefebvre, C.; Pellizzari, S.; Bhat, V.; Jurcic, K.; Litchfield, D.W.; Allan, A.L. Involvement of the AKT Pathway in Resistance to Erlotinib and Cabozantinib in Triple-Negative Breast Cancer Cell Lines. Biomedicines 2023, 11, 2406. https://doi.org/10.3390/biomedicines11092406
Lefebvre C, Pellizzari S, Bhat V, Jurcic K, Litchfield DW, Allan AL. Involvement of the AKT Pathway in Resistance to Erlotinib and Cabozantinib in Triple-Negative Breast Cancer Cell Lines. Biomedicines. 2023; 11(9):2406. https://doi.org/10.3390/biomedicines11092406
Chicago/Turabian StyleLefebvre, Cory, Sierra Pellizzari, Vasudeva Bhat, Kristina Jurcic, David W. Litchfield, and Alison L. Allan. 2023. "Involvement of the AKT Pathway in Resistance to Erlotinib and Cabozantinib in Triple-Negative Breast Cancer Cell Lines" Biomedicines 11, no. 9: 2406. https://doi.org/10.3390/biomedicines11092406
APA StyleLefebvre, C., Pellizzari, S., Bhat, V., Jurcic, K., Litchfield, D. W., & Allan, A. L. (2023). Involvement of the AKT Pathway in Resistance to Erlotinib and Cabozantinib in Triple-Negative Breast Cancer Cell Lines. Biomedicines, 11(9), 2406. https://doi.org/10.3390/biomedicines11092406