

SARS-CoV-2 Variability in Patients and Wastewaters—Potential Immuno-Modulation during the Shift from Delta to Omicron
 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Wastewater Samples
3. Results
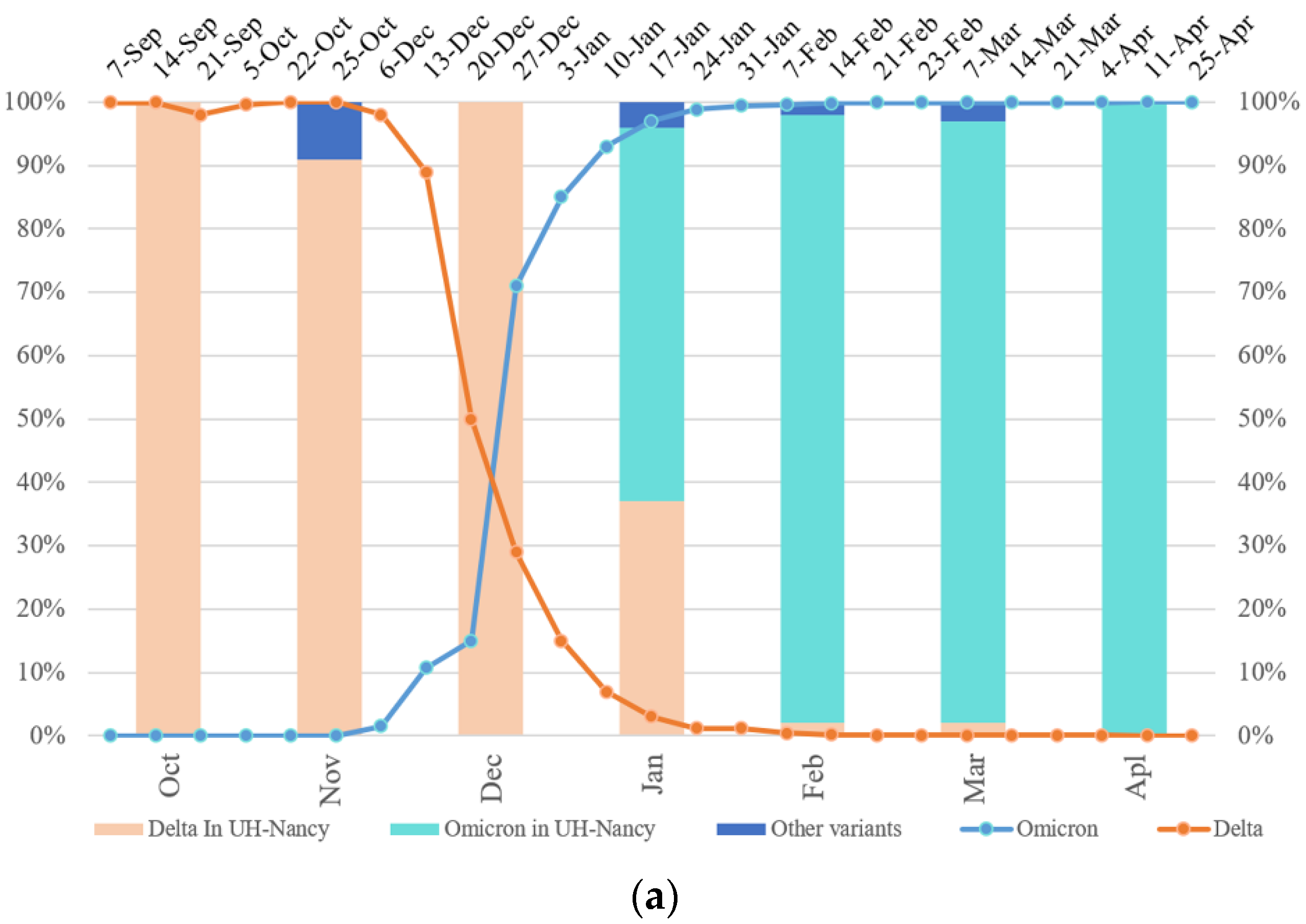
3.1. Temporal Variability and Mixtures of Variants in Nasopharyngeal Samples
3.2. Detection of SARS-CoV-2 Genetic Variability in Raw Wastewater Samples
3.3. Mapping of Variants Signatures
3.4. Humoral Immunogenicity and MHC-I Binding Affinity Prediction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domingo, E.; García-Crespo, C.; Lobo-Vega, R.; Perales, C. Mutation Rates, Mutation Frequencies, and Proofreading-Repair Activities in RNA Virus Genetics. Viruses 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Jetzt, A.E.; Yu, H.; Klarmann, G.J.; Ron, Y.; Preston, B.D.; Dougherty, J.P. High Rate of Recombination throughout the Human Immunodeficiency Virus Type 1 Genome. J. Virol. 2000, 74, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ma, W.; Dang, S.; Chen, L.; Zhang, R.; Mei, S.; Wang, J. Possible recombination between two variants of concern in a COVID-19 patient. Emerg. Microbes Infect. 2022, 11, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses 2022, 14, 78. [Google Scholar] [CrossRef] [PubMed]
- Combes, P.; Bisseux, M.; Bal, A.; Marin, P.; Latour, J.; Archimbaud, C.; Mirand, A. Evidence of co-infections during Delta and Omicron SARS-CoV-2 variants co-circulation through prospective screening and sequencing. Clin. Microbiol. Infect. 2022, in press. [Google Scholar] [CrossRef] [PubMed]
- Nabel, K.G.; Clark, S.A.; Shankar, S.; Pan, J.; Clark, L.E.; Yang, P.; Abraham, J. Structural basis for continued antibody evasion by the SARS-CoV-2 receptor binding domain. Science 2021, 375, eabl6251. [Google Scholar] [CrossRef] [PubMed]
- Hartard, C.; Chaqroun, A.; Settembre, N.; Gauchotte, G.; Lefevre, B.; Marchand, E.; Schvoerer, E. Multiorgan and Vascular Tropism of SARS-CoV-2. Viruses 2022, 14, 515. [Google Scholar] [CrossRef]
- Alhama, J.; Maestre, J.P.; Martín, M.A.; Michán, C. Monitoring COVID-19 through SARS-CoV-2 quantification in wastewater: Progress, challenges and prospects. Microb. Biotechnol. 2022, 15, 1719–1728. [Google Scholar] [CrossRef]
- Etievant, S.; Bal, A.; Escuret, V.; Brengel-Pesce, K.; Bouscambert, M.; Cheynet, V.; Gaymard, A. Performance Assessment of SARS-CoV-2 PCR Assays Developed by WHO Referral Laboratories. J. Clin. Med. 2020, 9, 1871. [Google Scholar] [CrossRef]
- Quick, J. nCoV-2019 Sequencing Protocol v3 (LoCost) V.3. 2020. Available online: https://protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye (accessed on 25 August 2020).
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of Globally Shared SARS-CoV-2 Data to Identify and Characterize New Variants. Bioinformatics 2021, 38, 1735–1737. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosa, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 26, vev003. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. J. Virol. 2005, 21, 98–102. [Google Scholar] [CrossRef]
- Maynard Smith, J. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved algorithmic complexity for the 3SEQ recombination detection algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef]
- The Immune Epitope Database (IEDB). Continuously Updated. Available online: https://www.iedb.org/ (accessed on 1 March 2022).
- Barbé, L.; Schaeffer, J.; Besnard, A.; Jousse, S.; Wurtzer, S.; Moulin, L.; Obepine Consortium. SARS-CoV-2 Whole-Genome Sequencing Using Oxford Nanopore Technology for Variant Monitoring in Wastewaters. Front. Microbiol. 2022, 13, 889811. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Wang, X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Li, Y.; Lai, D.Y.; Zhang, H.N.; Jiang, H.W.; Tian, X.; Ma, M.L.; Tao, S.C. Linear epitopes of SARS-CoV-2 spike protein elicit neutralizing antibodies in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 1095–1097. [Google Scholar] [CrossRef]
- Burki, T.K. Omicron variant and booster COVID-19 vaccines. Lancet Respir. Med. 2022, 10, e17. [Google Scholar] [CrossRef]
- Wurtzer, S.; Levert, M.; Dhenain, E.; Accrombessi, H.; Manco, S.; Fagour, N.; Moulin, L. Monitoring of SARS-CoV-2 variant dynamics in wastewater by digital RT-PCR: From Alpha to Omicron BA.2 VOC. medRxiv 2022. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Sato, K. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. 2021, 29, 1124–1136.e11. [Google Scholar] [CrossRef]
- Vellas, C.; Del Bello, A.; Debard, A.; Steinmeyer, Z.; Tribaudeau, L.; Ranger, N.; Izopet, J. Influence of treatment with neutralizing monoclonal antibodies on the SARS-CoV-2 nasopharyngeal load and quasispecies. Clin. Microbiol. Infect. 2022, 28, 139.e5–139.e8. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Sample | Sampling Date GenBank Number | NexClade/Pangolin Main SARS-CoV-2 Assignation for Each Sample | Other SARS-CoV-2 Variants | Other Variants Features: Signature Mutations | Other Variants: Mutations Percentage (%) |
|---|---|---|---|---|---|
| E1 | Nov-2021(1) ON832655 | 21J(Delta)/AY.43 | BA.1/BA.2 BA.4 | ORF1a:E91del S:D796Y ORF7a:S44del ORF1a:S142del | 5.6 6.9 5.2 5.1 |
| E2 | Nov-2021(2) ON832659 | 21J(Delta)/AY.122 | BA.4 BA.1/BA.2 | ORF1a:S142del S:D796Y | 7.6 5.2 |
| E3 | Nov-2021(3) ON832662 | 21J(Delta)/AY.125 | BA.1/BA.2 | S:T95I, K417N | 99 95.1 |
| E4 | Dec-2021(1) ON832661 | 21J(Delta)/B1.617.2 | BA.1/BA.2 | S:G484A | 99.4 |
| E5 | Dec-2021(2) ON832692 | 21J(Delta)/AY.124 | BA.1/BA.2 | S:T95I G339D | 97.5 10 |
| E6 | Dec-2021(3) ON832691 | 21J(Delta)/AY.43 | BA.4 BA.1/BA.2 | ORF1a:S142del S:D796Y | 6.1 16.3 |
| E7 | Dec-2021(4) ON832695 | 21J(Delta)/AY.122 | BA.1/BA.2 | S:A67V | 99.3 |
| E8 | Dec-2021(5) ON832711 | 21J(Delta)/AY.43 | BA.1/BA.2 | S:A67V | 99.4 |
| E9 | Feb-2022 ON832717 | 21K(Omicron)/BA.x | BA.4 | L452R | 72 |
| E10 | Apr-2022 ON832714 | 21L(Omicron)/BA.2.11 | BA.4 | ORF1a:S142del S:L452R | 6.1 93.2 |
| WW1 | Jan-2022 ON832058 | 21K(Omicron)/BA.1 | 21J(Delta)/BA.2 | 21J(Delta)-S:L452R,P681R; ORF1a:P2046L BA.2-ORF1a:S135R,T842I | |
| WW2 | Feb-2022 ON832065 | 21M(Omicron)/n.a. | 21J(Delta)/BA.2 | 21J(Delta)-ORF1b:L829I BA.2-S:S371F,T376A,D405N,R408S; ORF1a-G1307S;ORF1b:R1315C,T2163I | |
| WW3 | Mar-2022 ON832097 | 21L(Omicron)/n.a. | BA.1/BA.2 | BA.1-ORF1a:K856R BA.2-ORF1a:S2083del,L2048I,A2710T | |
| WW4 | Apr-2022 ON832102 | 21L(Omicron)/BA.2 | 21J(Delta) | 21J(Delta)-N:G215C | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaqroun, A.; Hartard, C.; Josse, T.; Taverniers, A.; Jeulin, H.; Gantzer, C.; Murray, J.M.; Obepine Consortium; Bertrand, I.; Schvoerer, E. SARS-CoV-2 Variability in Patients and Wastewaters—Potential Immuno-Modulation during the Shift from Delta to Omicron. Biomedicines 2022, 10, 2080. https://doi.org/10.3390/biomedicines10092080
Chaqroun A, Hartard C, Josse T, Taverniers A, Jeulin H, Gantzer C, Murray JM, Obepine Consortium, Bertrand I, Schvoerer E. SARS-CoV-2 Variability in Patients and Wastewaters—Potential Immuno-Modulation during the Shift from Delta to Omicron. Biomedicines. 2022; 10(9):2080. https://doi.org/10.3390/biomedicines10092080
Chicago/Turabian StyleChaqroun, Ahlam, Cédric Hartard, Thomas Josse, Audrey Taverniers, Hélène Jeulin, Christophe Gantzer, John M. Murray, Obepine Consortium, Isabelle Bertrand, and Evelyne Schvoerer. 2022. "SARS-CoV-2 Variability in Patients and Wastewaters—Potential Immuno-Modulation during the Shift from Delta to Omicron" Biomedicines 10, no. 9: 2080. https://doi.org/10.3390/biomedicines10092080
APA StyleChaqroun, A., Hartard, C., Josse, T., Taverniers, A., Jeulin, H., Gantzer, C., Murray, J. M., Obepine Consortium, Bertrand, I., & Schvoerer, E. (2022). SARS-CoV-2 Variability in Patients and Wastewaters—Potential Immuno-Modulation during the Shift from Delta to Omicron. Biomedicines, 10(9), 2080. https://doi.org/10.3390/biomedicines10092080