
Estimation of Enantiomeric Excess Based on Rapid Host–Guest Exchange

 ,
,  ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
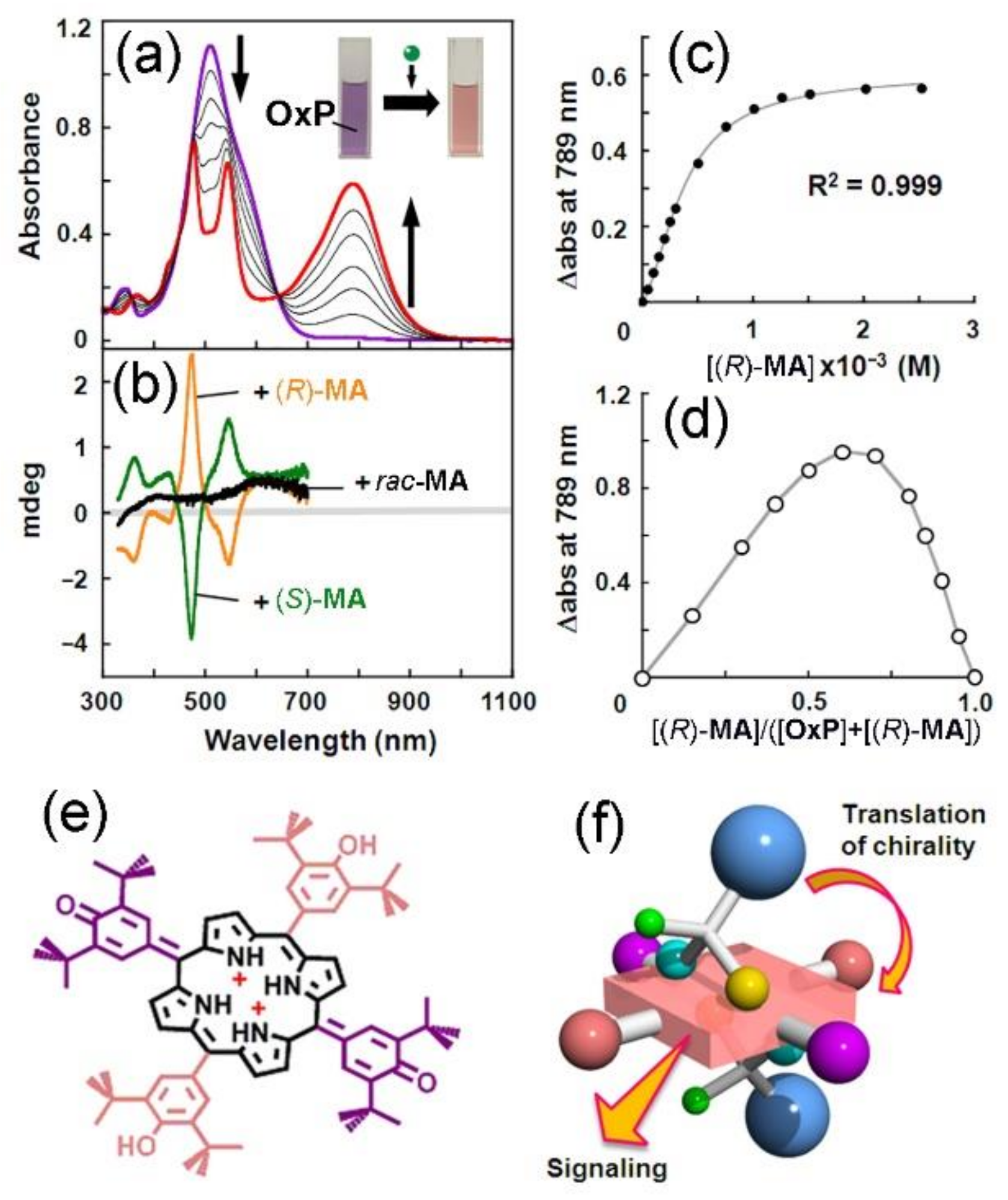
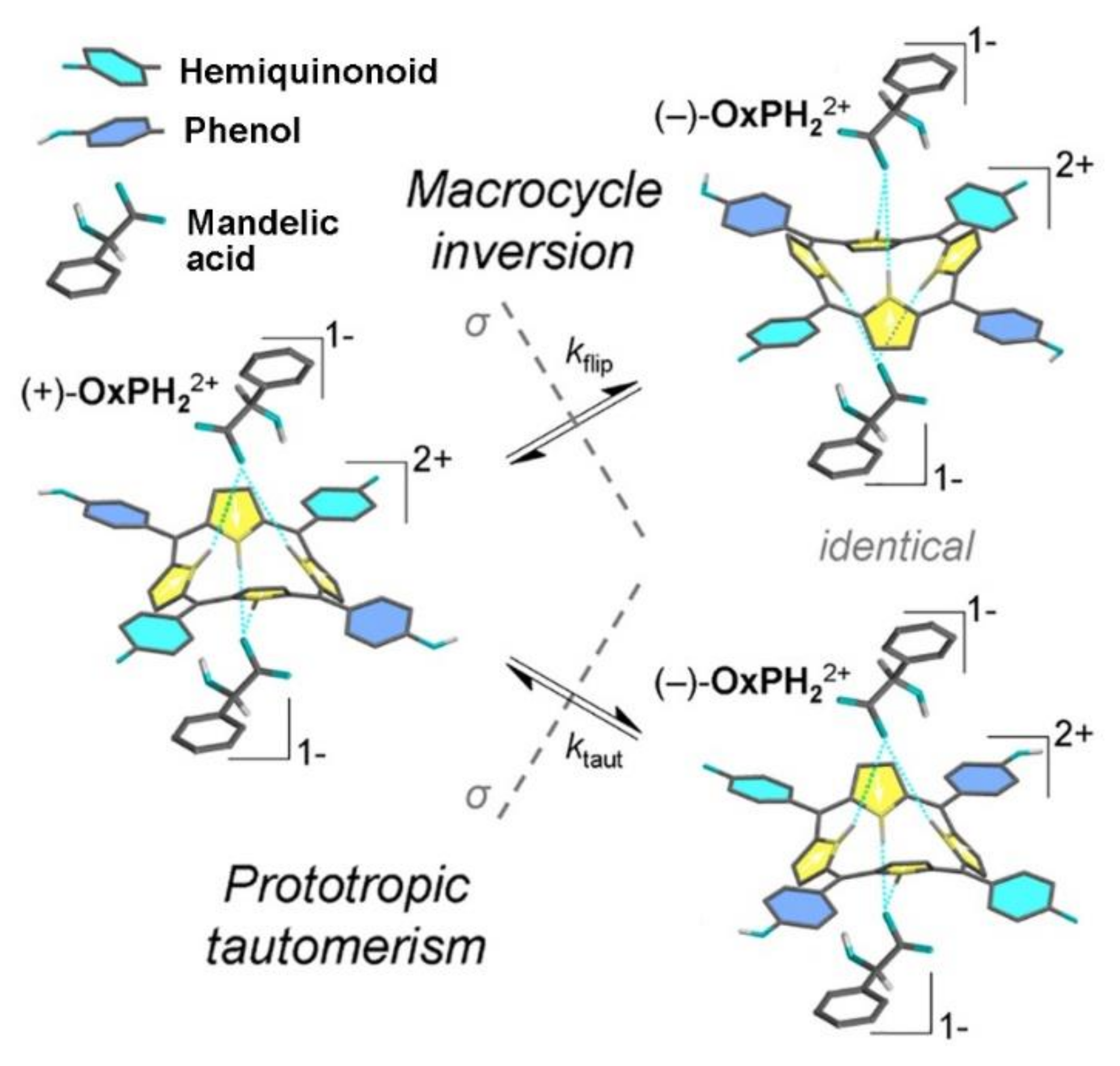
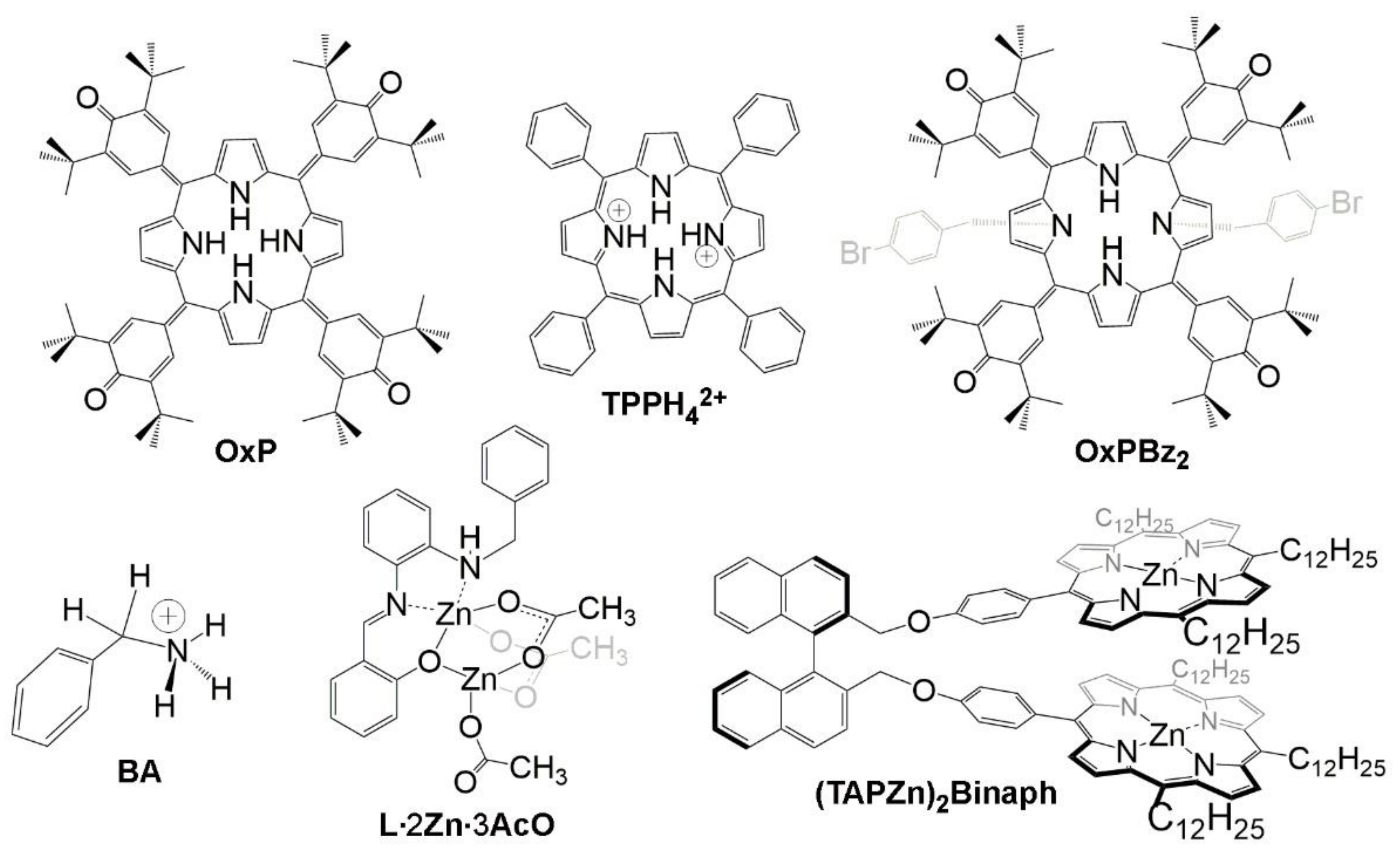
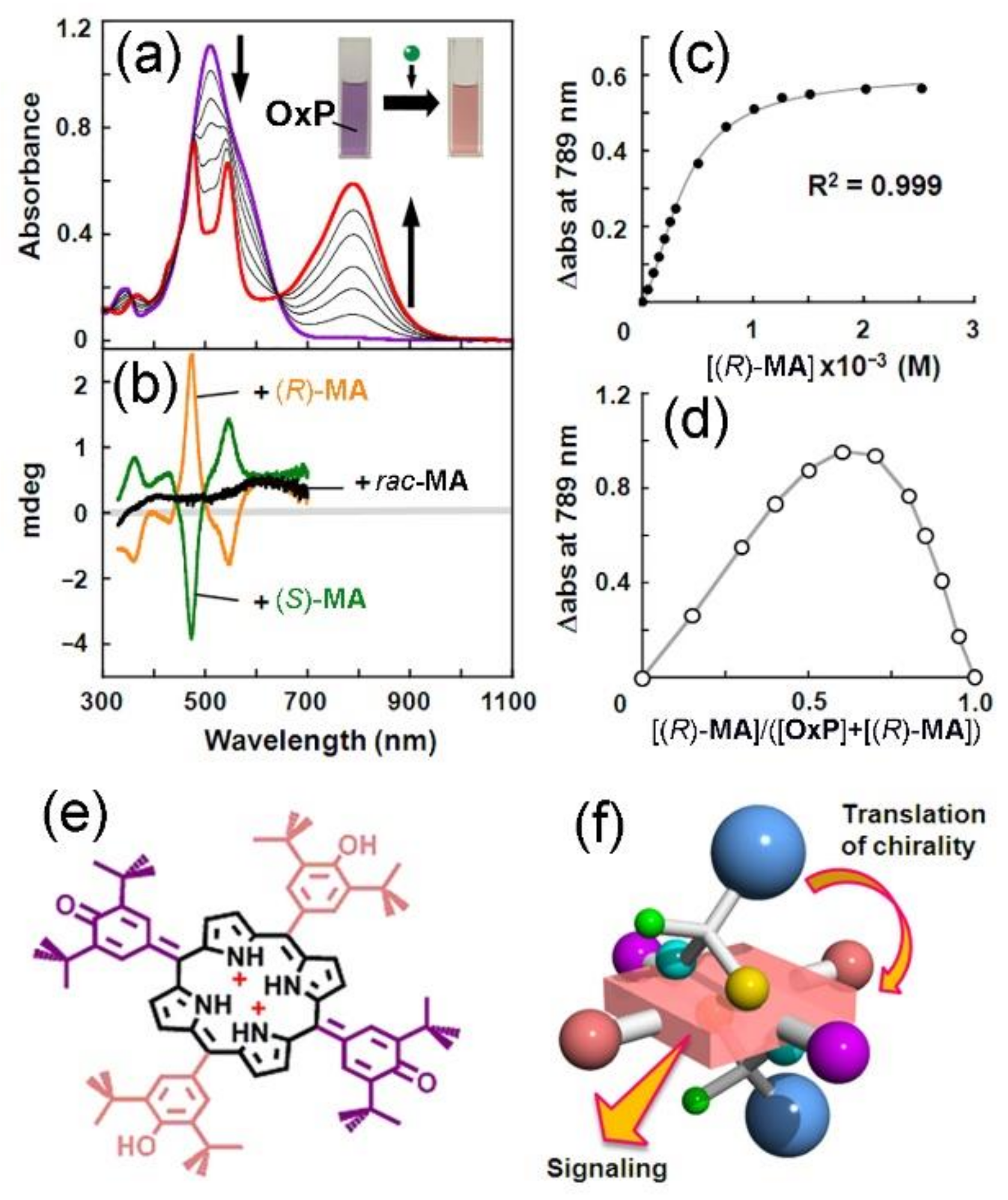
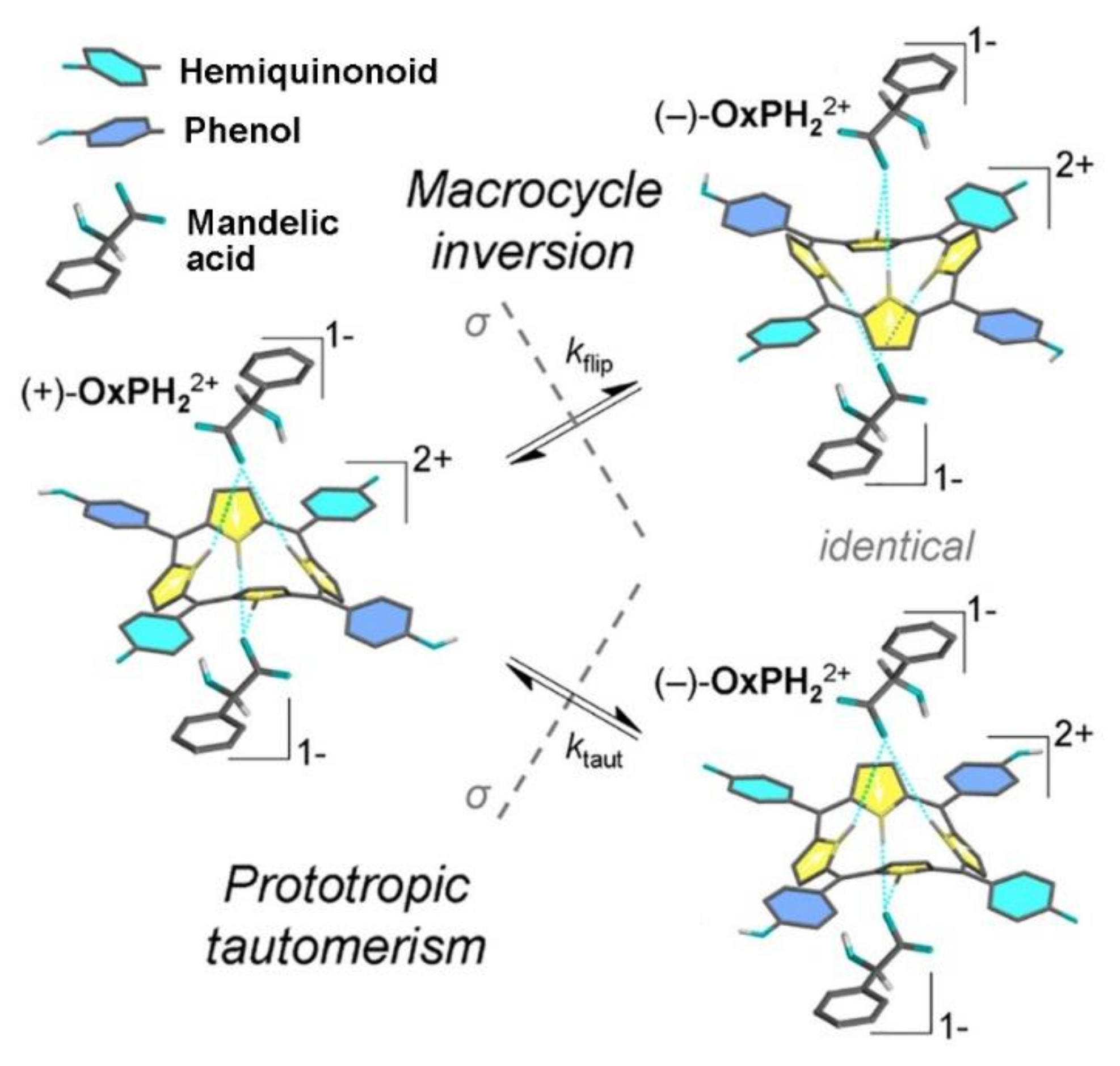
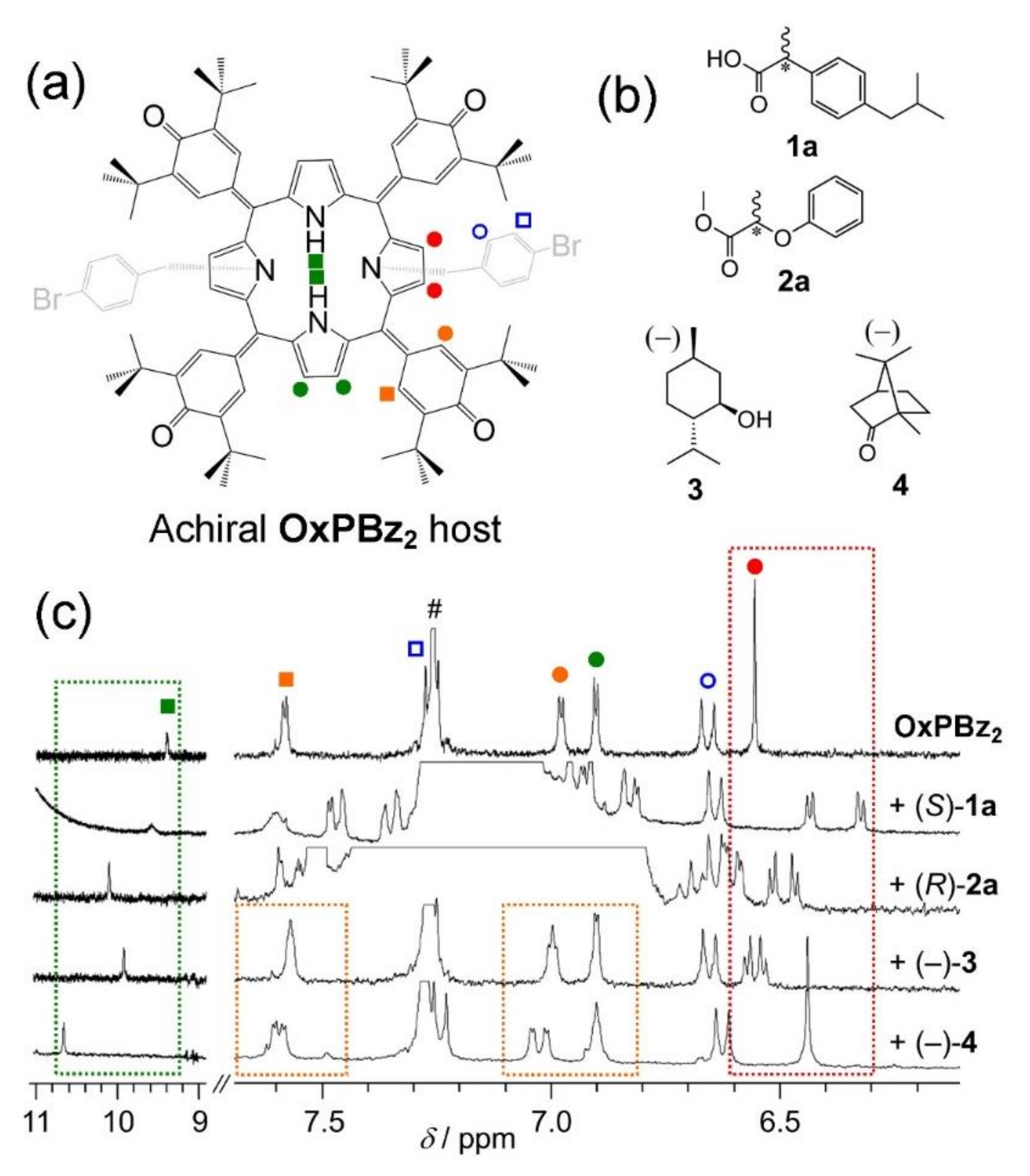
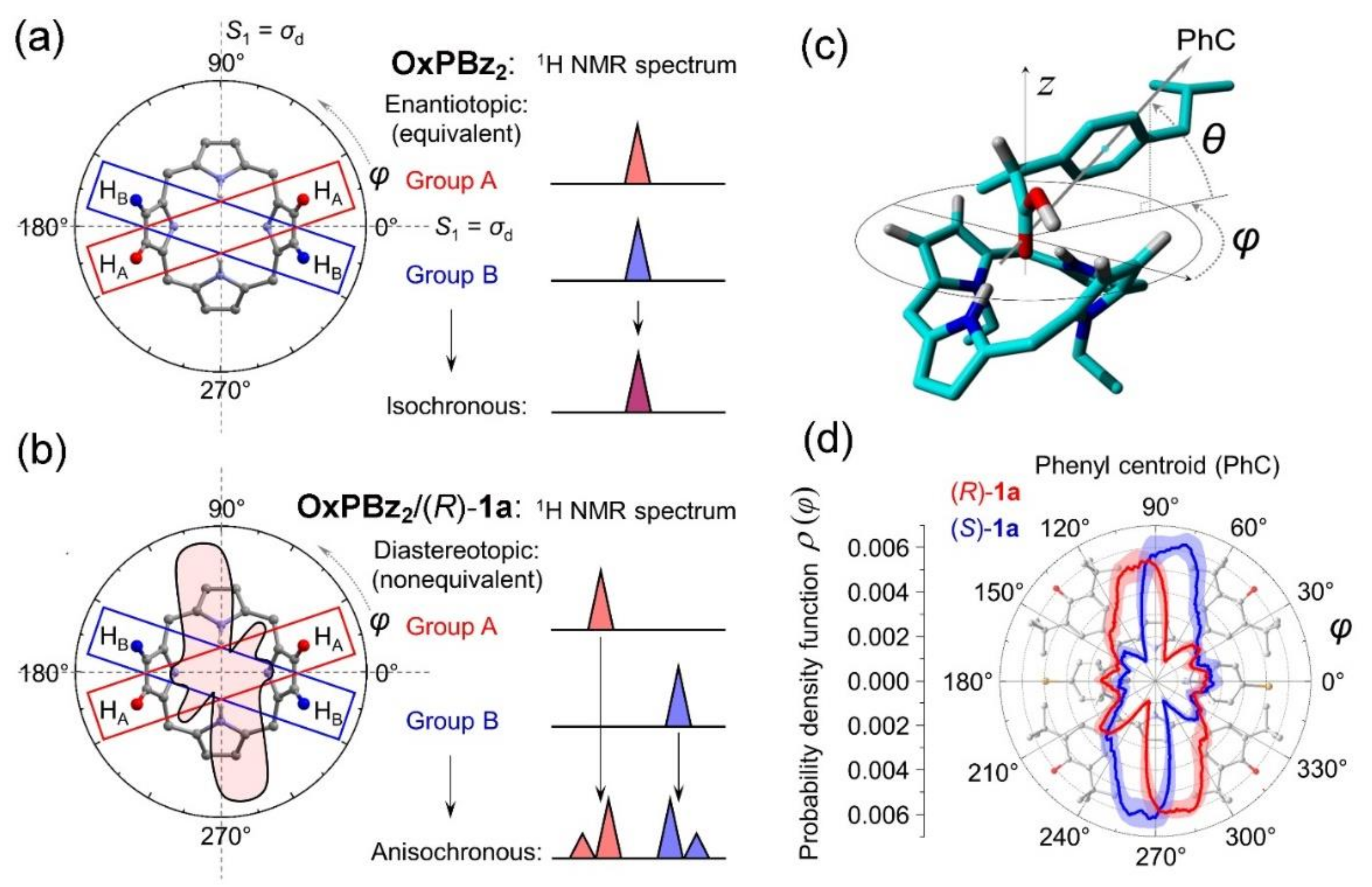
2. Oxoporphyrinogens and Porphyrin Dications
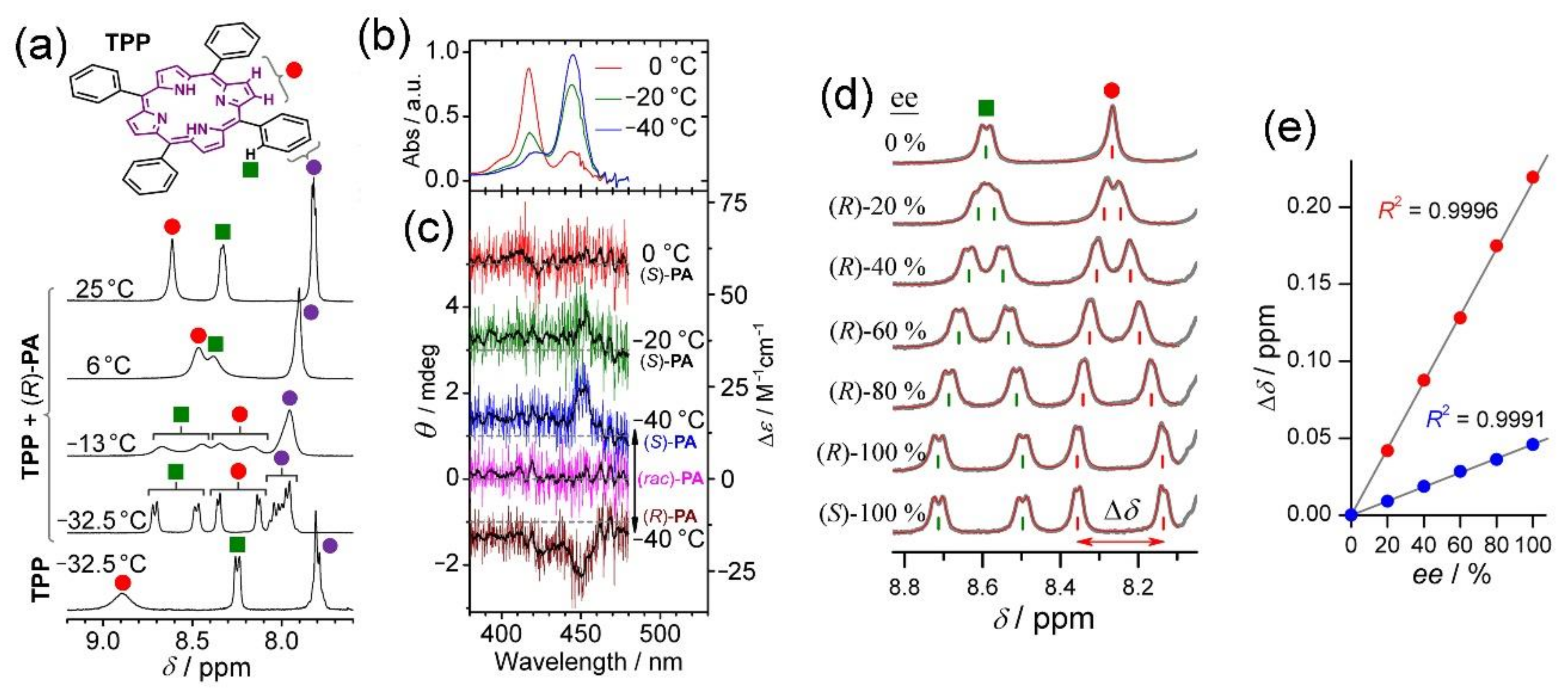
3. Extension to Small Molecules—The Case of Benzylamine
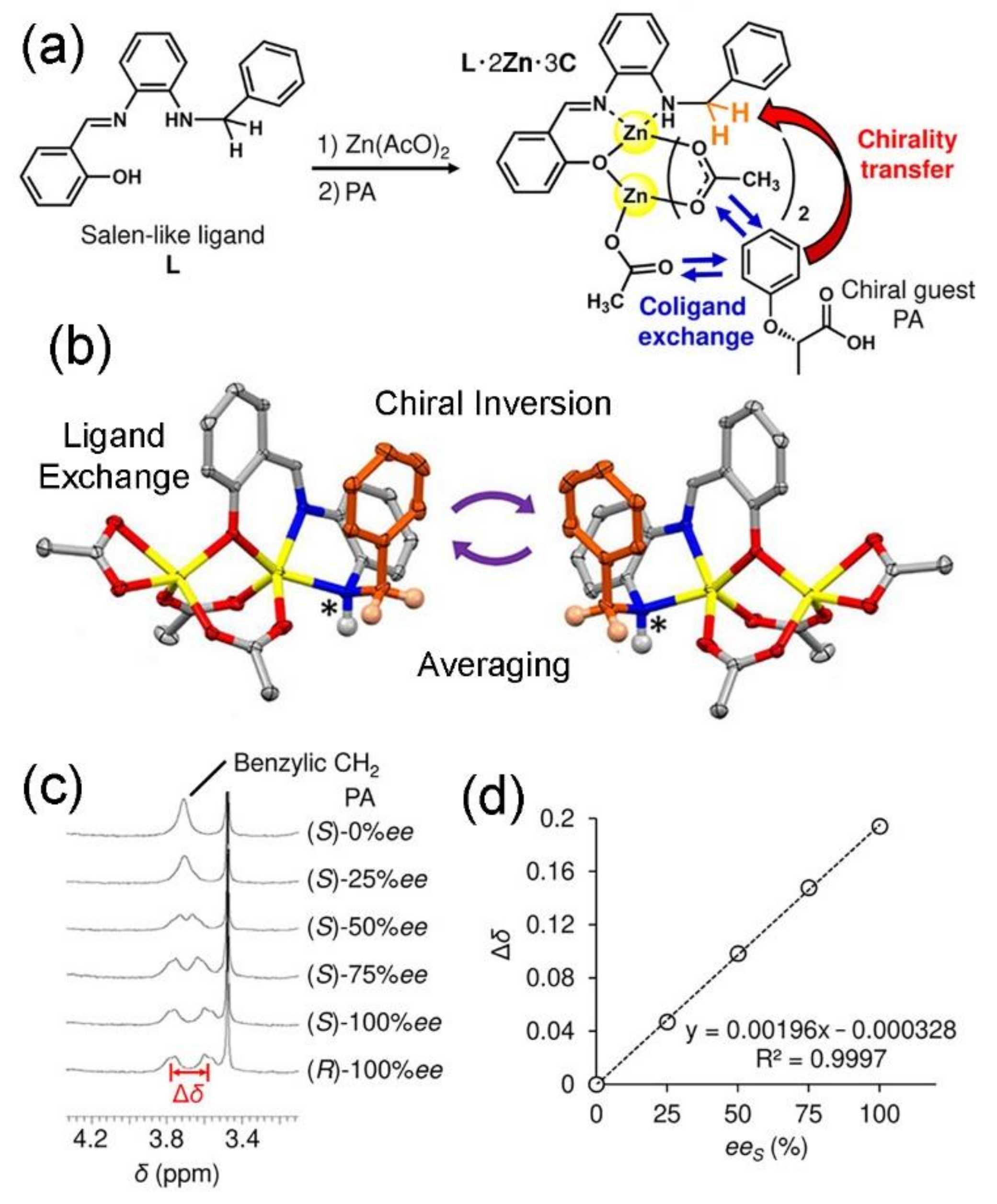
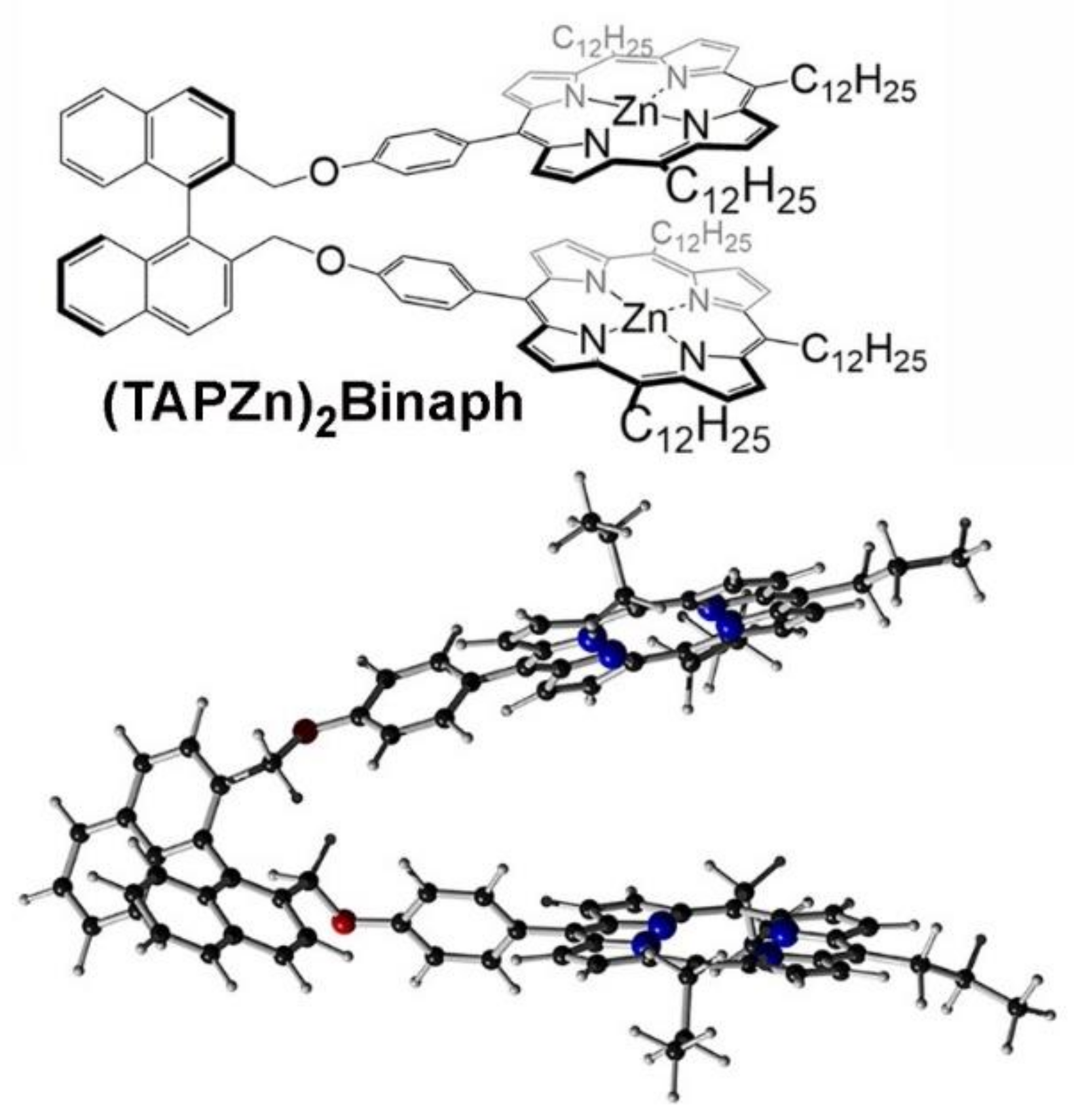
4. Coordination Complexes
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hutt, A.J. Drug Chirality and Its Pharmacological Consequences. In Introduction to the Principles of Drug Design and Action; Smith, H.J., Williams, H., Eds.; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Jacobsen, E.N.; Pfaltz, A.; Yamamoto, H. Comprehensive Organic Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: New York, NY, USA, 1999. [Google Scholar]
- Heitbaum, M.; Glorius, F.; Escher, I. Asymmetric heterogeneous catalysis. Angew. Chem. Int. Ed. 2006, 45, 4732–4762. [Google Scholar] [CrossRef]
- Yoon, T.P.; Jacobsen, E.N. Privileged chiral catalysts. Science 2003, 299, 1691–1693. [Google Scholar] [CrossRef]
- Maeda, K.; Morino, K.; Okamoto, Y.; Sato, T.; Yashima, E. Mechanism of helix induction on a stereoregular poly((4-carboxyphenyl)acetylene) with chiral amines and memory of the macromolecular helicity assisted by interaction with achiral amines. J. Am. Chem. Soc. 2004, 126, 4329–4342. [Google Scholar] [CrossRef] [PubMed]
- Ishi-i, T.; Crego-Calama, M.; Timmerman, P.; Reinhoudt, D.N.; Shinkai, S. Enantioselective formation of a dynamic hydrogen-bonded assembly based on the chiral memory concept. J. Am. Chem. Soc. 2002, 124, 14631–14641. [Google Scholar] [CrossRef]
- Kondo, T.; Oyama, K.; Yoshida, K. Chiral molecular recognition on formation of a metalloanthocyanin: A supramolecular metal complex pigment from blue flowers of Salvia patens. Angew. Chem. Int. Ed. 2001, 40, 894–897. [Google Scholar] [CrossRef]
- Hill, J.P.; Shrestha, L.K.; Ishihara, S.; Ji, Q.; Ariga, K. Self-assembly: From amphiphiles to chromophores and beyond. Molecules 2014, 19, 8589–8609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, S.; Labuta, J.; van Rossom, W.; Ishikawa, D.; Minami, K.; Hill, J.P.; Ariga, K. Porphyrin-based sensor nanoarchitectonics in diverse physical detection modes. Phys. Chem. Chem. Phys. 2014, 16, 9713–9746. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ishihara, S.; Ji, Q.; Akada, M.; Hill, J.P.; Ariga, K. Paradigm shift from self-assembly to commanded assembly of functional materials: Recent examples in porphyrin/fullerene supramolecular systems. Sci. Tech. Adv. Mater. 2012, 13, 053001. [Google Scholar] [CrossRef]
- Siegel, J.S. (Ed.) Supramolecular Stereochemistry; Springer Netherlands: Dordrecht, The Netherlands, 1995. [Google Scholar]
- Karnik, A.V.; Hasan, M. Stereochemistry: A Three-Dimensional Insight; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Schurig, V. Chiral separations using gas chromatography. TrAC Trends Anal. Chem. 2002, 21, 647–661. [Google Scholar] [CrossRef]
- Okamoto, Y.; Ikai, T. Chiral HPLC for efficient resolution of enantiomers. Chem. Soc. Rev. 2008, 37, 2593–2608. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, J.; Tiritan, M.E.; Pinto, M.M.M.; Fernandes, C. Chiral stationary phases for liquid chromatography: Recent developments. Molecules 2019, 24, 865. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.W. Optical isomer separation by liquid chromatography. Anal. Chem. 1987, 59, 84A–91A. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M.; Borkowski, M.; Strzelecka, D.; Kielar, K.; Kicińska, W.; Karevych, S.; Jasiński, R.; Demchuk, O.M. A general phenomenon of spontaneous amplification of optical purity under achiral chromatographic conditions. Symmetry 2019, 11, 680. [Google Scholar] [CrossRef] [Green Version]
- Pirkle, W.H.; Sikkenga, D.L.; Pavlin, M.S. Nuclear magnetic resonance determination of enantiomeric composition and absolute configuration of γ-lactones using chiral 2,2,2,-trifluoro-1-(9-anthryl)ethanol. J. Org. Chem. 1977, 42, 384–387. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Świerczynska, W.; Pietrusiewicz, K.M.; Woźnica, M.; Wójcik, D.; Frelek, J. A convenient application of the NMR and CD methodologies for the determination of enantiomeric ratio and absolute configuration of chiral atropoisomeric phosphine oxides. Tetrahedron Asymmetry 2008, 19, 2339–2345. [Google Scholar] [CrossRef]
- Pakulski, Z.; Demchuk, O.M.; Kwiatosz, R.; Osiński, P.W.; Świerczynska, W.; Pietrusiewicz, K.M. The classical Kagan’s amides are still practical NMR chiral shift reagents: Determination of enantiomeric purity of P-chirogenic phospholene oxides. Tetrahedron Asymmetry 2003, 14, 1459–1462. [Google Scholar] [CrossRef]
- Mizuno, Y.; Aida, T.; Yamaguchi, K. Chirality-memory molecule: Crystallographic and spectroscopic studies on dynamic molecular recognition events by fully substituted chiral porphyrins. J. Am. Chem. Soc. 2000, 122, 5278–5285. [Google Scholar] [CrossRef]
- Nieto, S.; Lynch, V.M.; Anslyn, E.V.; Kim, H.; Chin, J. High-throughput screening of identity, enantiomeric excess, and concentration using MLCT transitions in CD spectroscopy. J. Am. Chem. Soc. 2008, 130, 9232–9233. [Google Scholar] [CrossRef] [PubMed]
- Sato, H. A new horizon for vibrational circular dichroism spectroscopy: A challenge for supramolecular chirality. Phys. Chem. Chem. Phys. 2020, 22, 7671–7679. [Google Scholar] [CrossRef] [PubMed]
- Kubo, Y.; Maeda, S.; Tokita, S.; Kubo, M. Colorimetric chiral recognition by a molecular sensor. Nature 1996, 382, 522–524. [Google Scholar] [CrossRef]
- Zhu, L.; Anslyn, E.V. Facile quantification of enantiomeric excess and concentration with indicator-displacement assays: An example in the analyses of α-hydroxyacids. J. Am. Chem. Soc. 2004, 126, 3676–3677. [Google Scholar] [CrossRef]
- Tsubaki, K.; Nuruzzaman, M.; Kusumoto, T.; Hayashi, N.; Wang, B.-G.; Fuji, K. Visual enantiomeric recognition using chiral phenolphthalein derivatives. Org. Lett. 2001, 3, 4071–4073. [Google Scholar] [CrossRef]
- James, T.D.; Sandanayake, K.R.A.S.; Shinkai, S. Chiral discrimination of monosaccharides using a fluorescent molecular sensor. Nature 1995, 374, 345–347. [Google Scholar] [CrossRef]
- Matsushita, M.; Yoshida, K.; Yamamoto, N.; Wirsching, P.; Lerner, R.A.; Janda, K.D. High-throughput screening by using a blue-fluorescent antibody sensor. Angew. Chem. Int. Ed. 2003, 42, 5984–5987. [Google Scholar] [CrossRef] [PubMed]
- Mei, X.; Wolf, C. Enantioselective sensing of chiral carboxylic acids. J. Am. Chem. Soc. 2004, 126, 14736–14737. [Google Scholar] [CrossRef]
- Yu, S.; Pu, L. Pseudoenantiomeric fluorescent sensors in a chiral assay. J. Am. Chem. Soc. 2010, 132, 17698–17700. [Google Scholar] [CrossRef] [PubMed]
- Mirri, G.; Bull, S.D.; Horton, P.N.; James, T.D.; Male, L.; Tucker, J.H.R. Electrochemical method for the determination of enantiomeric excess of binol using redox-active boronic acids as chiral sensors. J. Am. Chem. Soc. 2010, 132, 8903–8905. [Google Scholar] [CrossRef]
- Rekharsky, M.V.; Yamamura, H.; Inoue, C.; Kawai, M.; Osaka, I.; Arakawa, R.; Shiba, K.; Sato, A.; Young, H.K.; Selvapalam, N.; et al. Chiral recognition in cucurbituril cavities. J. Am. Chem. Soc. 2006, 128, 14871–14880. [Google Scholar] [CrossRef] [PubMed]
- Borovkov, V.V.; Lintuluoto, J.M.; Inoue, Y. Supramolecular chirogenesis in zinc porphyrins: Mechanism, role of guest structure, and application for the absolute configuration determination. J. Am. Chem. Soc. 2001, 123, 2979–2989. [Google Scholar] [CrossRef]
- Kubo, Y.; Ohno, T.; Yamanaka, J.-I.; Tokita, S.; Iida, T.; Ishimaru, Y. Chirality-transfer control using a heterotopic zinc(II) porphyrin dimer. J. Am. Chem. Soc. 2001, 123, 12700–12701. [Google Scholar] [CrossRef]
- Li, X.; Tanasova, M.; Vasileiou, C.; Borhan, B. Fluorinated porphyrin tweezer: A powerful reporter of absolute configuration for erythro and threo diols, amino alcohols, and diamines. J. Am. Chem. Soc. 2008, 130, 1885–1893. [Google Scholar] [CrossRef]
- Huang, X.F.; Fujioka, N.; Pescitelli, G.; Koehn, F.E.; Williamson, R.T.; Nakanishi, K.; Berova, N. Absolute configurational assignments of secondary amines by CD-sensitive dimeric zinc porphyrin host. J. Am. Chem. Soc. 2002, 124, 10320–10335. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, M.d.C.; Gallo, M.A.; Espinosa, A.; Campos, J.M. Rapid Development of Chiral Drugs in the Pharmaceutical Industry. In New Developments in Medicinal Chemistry; Taft, C.A., Ed.; Bentham Science Publisher: Sharjah, United Arab Emirates, 2010; Volume 1, pp. 95–113. [Google Scholar]
- Halpern, J.; Trost, B. Asymmetric catalysis (highlighting PNAS special issue). Proc. Nat. Acad. Sci. USA 2004, 101, 5347. [Google Scholar] [CrossRef] [Green Version]
- Dale, J.A.; Dull, D.L.; Mosher, H.S. alpha.-Methoxy-.alpha.-trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric composition of alcohols and amines. J. Org. Chem. 1969, 34, 2543–2549. [Google Scholar] [CrossRef]
- Parker, D. NMR determination of enantiomeric purity. Chem. Rev. 1991, 91, 1441–1457. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Wilcox, J.D. Chiral reagents for the determination of enantiomeric excess and absolute configuration using NMR spectroscopy. Chirality 2003, 15, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Shundo, A.; Labuta, J.; Hill, J.P.; Ishihara, S.; Ariga, K. Nuclear magnetic resonance signaling of molecular chiral information using an achiral reagent. J. Am. Chem. Soc. 2009, 131, 9494–9495. [Google Scholar] [CrossRef]
- Labuta, J.; Ishihara, S.; Šikorský, T.; Futera, Z.; Shundo, A.; Hanyková, L.; Burda, J.V.; Ariga, K.; Hill, J.P. NMR spectroscopic detection of chirality and enantiopurity in referenced systems without formation of diastereomers. Nat. Commun. 2013, 4, 2188. [Google Scholar] [CrossRef]
- Labuta, J.; Futera, Z.; Ishihara, S.; Kouřilová, H.; Tateyama, Y.; Ariga, K.; Hill, J.P. Chiral guest binding as a probe of macrocycle dynamics and tautomerism in a conjugated tetrapyrrole. J. Am. Chem. Soc. 2014, 136, 2112–2118. [Google Scholar] [CrossRef]
- Ishihara, S.; Labuta, J.; Futera, Z.; Mori, S.; Sato, H.; Ariga, K.; Hill, J.P. NMR spectroscopic determination of enantiomeric excess using small prochiral molecules. J. Phys. Chem. B 2018, 122, 5114–5120. [Google Scholar] [CrossRef]
- Payne, D.T.; Chahal, M.K.; Březina, V.; Webre, W.A.; Ariga, K.; D’Souza, F.; Labuta, J.; Hill, J.P. Diporphyrin tweezer for multichannel spectroscopic analysis of enantiomeric excess. Front. Chem. Sci. Eng. 2020, 14, 28–40. [Google Scholar] [CrossRef]
- Takimoto, K.; Ishihara, S.; Labuta, J.; Březina, V.; Hill, J.P.; Ariga, K.; Sumita, M.; Mori, S.; Sato, H. Enantiomeric excess dependent splitting of NMR signal through co-ligand dynamic exchange in a coordination complex. J. Phys. Chem. Lett. 2020, 11, 8164–8169. [Google Scholar] [CrossRef]
- Milgrom, L.R. The facile aerial oxidation of a porphyrin. Tetrahedron 1983, 39, 3895–3898. [Google Scholar] [CrossRef]
- Hill, J.P.; Hewitt, I.J.; Anson, C.E.; Powell, A.K.; McCarthy, A.L.; Karr, P.A.; Zandler, M.E.; D’Souza, F. Highly non-planar, electron deficient, N-substituted tetraoxocyclohexadienylidene porphyrinogens: Structural, computational, and electrochemical investigations. J. Org. Chem. 2004, 69, 5861–5869. [Google Scholar] [CrossRef]
- Golder, A.J.; Milgrom, L.R.; Nolan, K.B.; Povey, D.C. 5,10,15,20-Meso-tetrakis(3,5-di-t-butyl-4-quinomethide) porphyrinogen: A highly puckered tetrapyrrolic macrocycle from the facile aerial oxidation of a phenolic porphyrin. J. Chem. Soc. Chem. Commun. 1989, 22, 1751–1753. [Google Scholar] [CrossRef]
- Dong, S.K.; Sessler, J.L. Calix[4]pyrroles: Versatile molecular containers with ion transport, recognition, and molecular switching functions. Chem. Soc. Rev. 2015, 44, 532–546. [Google Scholar]
- Hill, J.P.; Schumacher, A.L.; D’Souza, F.; Labuta, J.; Redshaw, C.; Elsegood, M.R.J.; Aoyagi, M.; Nakanishi, T.; Ariga, K. A chromogenic indicator for anion reporting based on an N-substituted oxoporphyrinogen. Inorg. Chem. 2006, 45, 8288–8296. [Google Scholar] [CrossRef] [PubMed]
- Chahal, M.K.; Payne, D.T.; Labuta, J.; Karr, P.A.; D’Souza, F.; Ariga, K.; Hill, J.P. Selective phase transfer reagents (OxP-crowns) for chromogenic detection of nitrates especially ammonium nitrate. Chem. Eur. J. 2020, 26, 13177–13183. [Google Scholar] [CrossRef]
- Ishihara, S.; Labuta, J.; Šikorský, T.; Burda, J.V.; Okamoto, N.; Abe, H.; Ariga, K.; Hill, J.P. Colorimetric detection of trace water in tetrahydrofuran using N,N’-substituted oxoporphyrinogens. Chem. Commun. 2012, 48, 3933–3935. [Google Scholar] [CrossRef]
- Ishihara, S.; Iyi, N.; Labuta, J.; Deguchi, K.; Ohki, S.; Tansho, M.; Shimizu, T.; Yamauchi, Y.; Naito, M.; Abe, H.; et al. Naked-eye discrimination of methanol and ethanol using composite film of oxoporphyrinogen and layered double hydroxide. ACS Appl. Mater. Interfaces 2013, 5, 5927–5930. [Google Scholar] [CrossRef]
- Chahal, M.K.; Labuta, J.; Březina, V.; Karr, P.A.; Matsushita, Y.; Webre, W.A.; Payne, D.T.; Ariga, K.; D’Souza, F.; Hill, J.P. Knock-on synthesis of tritopic calix[4]pyrrole host for enhanced anion interactions. Dalton Trans. 2019, 48, 15563–15596. [Google Scholar] [CrossRef]
- Shundo, A.; Ishihara, S.; Labuta, J.; Onuma, Y.; Sakai, H.; Abe, M.; Ariga, K.; Hill, J.P. Colorimetric visualization of acid-base equilibria in non-polar solvent. Chem. Commun. 2013, 49, 6870–6872. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, F.; Subbaiyan, N.K.; Xie, Y.; Hill, J.P.; Ariga, K.; Ohkubo, K.; Fukuzumi, S. Anion-complexation-induced stabilization of charge separation. J. Am. Chem. Soc. 2009, 131, 16138–16146. [Google Scholar] [CrossRef] [PubMed]
- Chahal, M.K.; Liyanage, A.; Alsaleh, A.Z.; Karr, P.A.; Hill, J.P.; D’Souza, F. Anion-enhanced excited state charge separation in a spiro-locked N-heterocycle-fused push-pull zinc porphyrin. Chem. Sci. 2021, 12, 4925–4930. [Google Scholar] [CrossRef] [PubMed]
- Chahal, M.K.; Liyanage, A.; Gobeze, H.B.; Payne, D.T.; Ariga, K.; Hill, J.P.; D’Souza, F. Supramolecular ultrafast energy and electron transfer in a directly Linked BODIPY-oxoporphyrinogen dyad upon fluoride ion binding. Chem. Commun. 2020, 56, 3855–3858. [Google Scholar] [CrossRef] [PubMed]
- Chahal, M.K.; Gobese, H.B.; Webre, W.A.; Karr, P.A.; Payne, D.T.; Ariga, K.; D’Souza, F.; Hill, J.P. Electron and energy transfer in a porphyrin-oxoporphyrinogen-fullerene triad, ZnP-OxP-C60. Phys. Chem. Chem. Phys. 2020, 22, 14356–14363. [Google Scholar] [CrossRef] [PubMed]
- Chahal, M.K.; Velychkivska, N.; Webre, W.A.; Labuta, J.; Ishihara, S.; Ariga, K.; D’Souza, F.; Hill, J.P. Increasing the complexity of oxoporphyrinogen colorimetric sensing chromophores: N-alkylation and β-substitution. J. Porphyrins Phthalocyanines 2019, 23, 1184–1194. [Google Scholar] [CrossRef] [Green Version]
- Chahal, M.K.; Payne, D.T.; Matsushita, Y.; Labuta, J.; Ariga, K.; Hill, J.P. Molecular Engineering of β-Substituted Oxoporphyrinogens for Hydrogen-Bond Donor Catalysis. Eur. J. Org. Chem. 2020, 2020, 82–90. [Google Scholar] [CrossRef]
- Milgrom, L.R. The Colors of Life; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Scheer, H. Chlorophylls; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Lewis, D.F.W. Guide to Cytochromes P450; Taylor and Francis: London, UK, 2002. [Google Scholar]
- Proni, G.; Pescitelli, G.; Huang, X.F.; Nakanishi, K.; Berova, N. Magnesium tetraarylporphyrin tweezer: A CD-sensitive host for absolute configurational assignments of α-chiral carboxylic acids. J. Am. Chem. Soc. 2003, 125, 12914–12927. [Google Scholar] [CrossRef]
- Balaz, M.; De Napoli, M.; Holmes, A.E.; Mammana, A.; Nakanishi, K.; Berova, N.; Purrello, R. A cationic zinc porphyrin as a chiroptical probe for Z-DNA. Angew. Chem. Int. Ed. 2005, 44, 4006–4009. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Fujii, I.; Muranaka, A.; Hembury, G.A.; Tanaka, T.; Ceulemans, A.; Kobayashi, N.; Inoue, Y. Rationalization of supramolecular chirality in a bisporphyrin system. Angew. Chem. Int. Ed. 2004, 43, 5481–5485. [Google Scholar] [CrossRef]
- D’Souza, F.; Schumacher, A.L.; Hill, J.P.; Karr, P.A.; Zandler, M.E.; Xie, Y.; Ariga, K.; Sandanayaka, A.S.D.; Araki, Y.; Ito, O. Oxoporphyrinogens: From redox and spectroscopic probe for anion sensing to a platform for construction of supramolecular donor-acceptor conjugates. Electrochem. Soc. Trans. 2008, 13, 127–136. [Google Scholar] [CrossRef]
- Hembury, G.A.; Borovkov, V.V.; Inoue, Y. Chirality-sensing supramolecular systems. Chem. Rev. 2008, 108, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Furusho, Y.; Kimura, T.; Mizuno, Y.; Aida, T. Chirality-memory molecule: A D2-symmetric fully substituted porphyrin as a conceptually new chiral sensor. J. Am. Chem. Soc. 1997, 119, 5267–5268. [Google Scholar] [CrossRef]
- Labuta, J.; Ishihara, S.; Shundo, A.; Arai, S.; Takeoka, S.; Ariga, K.; Hill, J.P. Chirality sensing by nonchiral porphines. Chem. Eur. J. 2011, 17, 3558–3561. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.; Fleischer, E.B. The molecular and crystal structure of porphyrin diacids. J. Am. Chem. Soc. 1968, 90, 2735–2748. [Google Scholar] [CrossRef]
- Labuta, J.; Hill, J.P.; Elsegood, M.R.J.; Ariga, K. Synthesis of stable pseudotetrahedral supermolecules based on an oxoporphyrinogen. Tetrahedron Lett. 2010, 51, 2935–2938. [Google Scholar] [CrossRef]
- Labuta, J.; Ishihara, S.; Ariga, K.; Hill, J.P. Dynamic processes in prochiral solvating agents (pro-CSAs) studied by NMR spectroscopy. Symmetry 2014, 6, 345–367. [Google Scholar] [CrossRef] [Green Version]
- Soai, K.; Shibata, T.; Morioka, H.; Choji, K. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 1995, 378, 767–768. [Google Scholar] [CrossRef]
- Lawrance, G.A. Introduction to Coordination Chemistry; John Wiley & Sons Ltd: Chichester, UK, 2009. [Google Scholar]
- Zhao, Y.; Swager, T.M. Simultaneous chirality sensing of multiple amines by 19F NMR. J. Am. Chem. Soc. 2015, 137, 3221–3224. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xia, X.; Bian, G.; Song, L. A chiral sensor for recognition of varied amines based on 19F NMR signals of newly designed rhodium complexes. Chem. Comm. 2019, 55, 6098–6101. [Google Scholar] [CrossRef]
- Li, L.P.; Peng, H.L.; Ye, B.H. Chiral sensor for enantiomeric purity of amines, amino alcohols and amino esters based on bis-cyclometalated Ir(III) complex using 1H NMR spectroscopy. Inorg. Chim. Acta 2018, 482, 691–697. [Google Scholar] [CrossRef]
- Labuta, J.; Hill, J.; Ishihara, S.; Hanyková, L.; Ariga, K. Chiral sensing by nonchiral tetrapyrroles. Acc. Chem. Res. 2015, 48, 521–529. [Google Scholar] [CrossRef]
- Labuta, J.; Ishihara, S.; Hill, J.P. Meso–tetraphenylporphine as prochiral solvating agent (pro–CSA): A physicochemical study. J. Porphyrins Phthalocyanines 2020, 24, 320–329. [Google Scholar] [CrossRef]
- Burgess, J. Inorganic Reaction Mechanism; Royal Society of Chemistry: London, UK, 1971; Volume 2. [Google Scholar]
- Cozzi, P.G. Metal-salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Shinoda, T.; Onaka, M.; Izumi, Y. The reason why K10 is an effective promoter for meso-tetraalkylporphyrin synthesis. Chem. Lett. 1995, 24, 493–494. [Google Scholar] [CrossRef]
- Plamont, R.; Kikkawa, Y.; Takahashi, M.; Kanesato, M.; Giorgi, M.; Shun, A.C.K.; Roussel, C.; Balaban, T.S. Nanoscopic imaging of meso-tetraalkylporphyrins prepared in high yields enabled by Montmorillonite K10 and 3 Å molecular sieves. Chem. Eur. J. 2013, 19, 11293–11300. [Google Scholar] [CrossRef]
- Lu, W.; Yang, H.; Li, X.; Wang, C.; Zhan, X.; Qi, D.; Bian, Y.; Jiang, J. Chiral discrimination of diamines by a binaphthylene-bridged porphyrin dimer. Inorg. Chem. 2017, 56, 8223–8231. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labuta, J.; Ishihara, S.; Payne, D.T.; Takimoto, K.; Sato, H.; Hanyková, L.; Ariga, K.; Hill, J.P. Estimation of Enantiomeric Excess Based on Rapid Host–Guest Exchange. Chemosensors 2021, 9, 259. https://doi.org/10.3390/chemosensors9090259
Labuta J, Ishihara S, Payne DT, Takimoto K, Sato H, Hanyková L, Ariga K, Hill JP. Estimation of Enantiomeric Excess Based on Rapid Host–Guest Exchange. Chemosensors. 2021; 9(9):259. https://doi.org/10.3390/chemosensors9090259
Chicago/Turabian StyleLabuta, Jan, Shinsuke Ishihara, Daniel T. Payne, Kazuyoshi Takimoto, Hisako Sato, Lenka Hanyková, Katsuhiko Ariga, and Jonathan P. Hill. 2021. "Estimation of Enantiomeric Excess Based on Rapid Host–Guest Exchange" Chemosensors 9, no. 9: 259. https://doi.org/10.3390/chemosensors9090259
APA StyleLabuta, J., Ishihara, S., Payne, D. T., Takimoto, K., Sato, H., Hanyková, L., Ariga, K., & Hill, J. P. (2021). Estimation of Enantiomeric Excess Based on Rapid Host–Guest Exchange. Chemosensors, 9(9), 259. https://doi.org/10.3390/chemosensors9090259