Abstract
The influence of the experimental conditions (glutathione concentration and incubation time and temperature) concerning the covalent immobilization of glutathione via carbodiimide coupling on the behavior of a glutathione modified screen-printed carbon electrode obtained by electrografting is evaluated. The optimized parameters fasten the modification process and improve the performance of the sensor as compared to the usual procedure. This suggests the convenience of a tailored preparation of metal sensors based on metal-binding biomolecules such as glutathione.
1. Introduction
Environmental contamination by heavy metals is a serious problem enhanced by the large quantity and variety of natural and anthropogenic sources of these species, their accumulation in organisms and their negative effects on human health [1]. Against such contamination, organisms have developed biochemical mechanisms of defense, based on the synthesis of molecules with high affinity for heavy metal ions than can chemically bind them to decrease their toxicity and to favor their elimination. The basis of such biochemical defense includes high concentrations of small molecules, mainly aminothiols like cysteine (Cys), cysteine-glycine (Cys-Gly) or glutathione (GSH), and low concentrations of larger molecules which are enzymatically synthesized from the previous ones as a response to heavy metal contamination. In the case of plants, algae and fungi, such larger molecules are phytochelatins, of general structure (γ-Glu-Cys)nGly with n usually ranging from 2 to 5 [2,3,4]. In the case of mammals metallothioneins, a more complex combination of aminoacids with a high proportion of cysteine, play a similar role as phytochelatins [2].
While organisms reinforce their mechanisms to minimize the effects of heavy metal contamination, analytical chemists try to develop more effective methods to determine and monitor the concentration of heavy metals in environmental and biological samples. Although techniques like atomic absorption or ICP are especially powerful in the analysis of metals [5,6], they should be complemented by portable, low-cost techniques for these applications where simplicity, ease of use, economy and possibility of measuring on-line or in-situ are required. Among these techniques, voltammetry is a sensitive, cheap and versatile choice, especially when it includes analyte preconcentration by reduction (anodic stripping voltammetry, ASV) or by adsorption (adsorptive stripping voltammetry, AdSV) [7]. For a long time, stripping voltammetric measurements were mostly carried out with mercury drop or mercury film electrodes [8]. However, in the last years, the toxicity of mercury has encouraged its replacement by alternative materials such as bismuth and antimony films on carbon supports or conventional solid electrodes chemically modified with nanoparticles and molecules having some affinity for heavy metals [9,10,11]. More recently, the popularization of screen-printed electrodes (SPE) and their commercial availability in a wide variety of materials has largely expanded the field of application of modified electrodes as substitutes for mercury [12]. Commercial screen-printed electrodes are an excellent substrate for modification. They are cheap and reproducible and their disposable character avoids tedious and time-consuming polishing and cleaning steps which are mandatory in the modification of conventional solid electrodes.
Among the different strategies proposed for the stripping analysis of heavy metals beyond the use of mercury, some efforts have been devoted to mimic the defense mechanisms of plants and preconcentrate the target metals by binding them to the electrode with the same molecules used for metal bioaccumulation. Thus, the molecule immobilization procedure is a key aspect in the design of these electrodes [13]. In this regard, the literature describes different strategies including the formation of alkanethiol self-assembled monolayers (SAMs) and the molecule immobilization based on aryl diazonium salt anchored on the electrode surface, which is a more viable approach that can overcome the major limitations of SAMs and has proven its usefulness in the development of metal ion sensors [14,15,16]. For example, the development of modified sensors in which glutathione (GSH) and its fragments Cys-Gly and γ-Glu-Cys were immobilized through aryl diazonium electrochemical grafting onto the surface of graphite-epoxy composite electrodes (GEC), was used for the simultaneous determination of Cd(II), Pb(II) and Zn(II) [17]. More recently, the procedure was successfully adapted to the special characteristics of screen-printed electrodes to develop a glutathione-modified carbon nanofiber screen-printed electrode [18].
In the present work we revisit the glutathione-modified electrode to gain a deeper insight on the main factors concerning the immobilization of the peptide by electrografting. In this way, we try not only to improve the analytical performance of this sensor, but also to deduce some practical guidelines for the immobilization of more complex metal-binding biomolecules.
2. Materials and Methods
2.1. Chemicals
Potassium ferrocyanide K4[Fe(CN)6]·3H2O, 2-(N-morpholino)-ethanesulfonic acid (MES), sodium acetate, acetic acid, hydrochloric acid and glutathione (GSH), in the reduced form, with purity greater than 99% were supplied from Merck (Darmstadt, Germany). N-hydroxysulfosuccinimide (sulfo-NHS), 4-aminobenzoic acid (ABA), perchloric acid, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), potassium dihydrogen phosphate, sodium monophosphate, sodium nitrite and methanol were provided by Sigma-Aldrich (St. Louis, MO, USA). Potassium ferricyanide K3[Fe(CN)6] was purchased from Panreac (Barcelona, Spain). All reagents were of analytical grade. Cd(II) and Pb(II) stock solutions 10−2 mol·L−1 were prepared from Cd(NO3)2·4H2O and Pb(NO3)2·4H2O respectively and standardized complexometrically [19]. Ultrapure water (Milli-Q plus 185 system, Millipore) was used in all experiments.
2.2. Instrumentation
An Autolab System PGSTAT12 (EcoChemie, Utrecht, The Netherlands) attached to a Metrohm 663 VA Stand (Metrohm, Herisau, Switzerland) and using GPES 4.9 software package (EcoChemie) was used for differential pulse anodic stripping voltammetric (DPASV) measurements.
For experiments, the working electrode was a modified glutathione electrode prepared from carbon nanofibers modified screen-printed electrode (GSH-SPCNFE) of 4 mm diameter and supplied by Dropsens (Oviedo, Spain) (ref. 110CNF, DS SPCE). A flexible cable (ref. CAC, DropSens) was used to connect screen-printed electrodes to the Autolab System. The auxiliary electrode was a Pt wire (Metrohm, Switzerland) and the reference electrode was Ag|AgCl|KCl (3 mol·L−1).
A Crison micro pH 2000 pH-meter was used for pH measurements, and all measurements were performed in a glass cell at room temperature (20 °C) without oxygen removal.
2.3. Preparation of the Glutathione Modified Screen-Printed Carbon Nanofiber Electrode (GSH-SPCNFE)
GSH-SPCNFEs were prepared based on a two-step procedure previously reported [18] and briefly described below. Factors concerning the immobilization of glutathione onto the functionalized SPE such as concentration of glutathione, and incubation time and temperature for the binding of the activated carboxyl groups and glutathione were considered for optimization.
2.3.1. Diazonium Salt Electrografting
The aryl diazonium salt was generated in-situ by adding 2 mmol·L−1 of sodium nitrite to a cooled solution of 73 mmol·L−1 of ABA in 1 mol·L−1 aqueous HCl. This solution was mixed in an ice bath for 30 min before the electrochemical grafting process [20] was conducted. For this purpose, the SPCNFE was immersed in the diazonium salt solution and 15 cyclic voltammetry (CV) cycles were applied from 0 V to −1 V at 0.2 V·s−1. The functionalized electrodes were thoroughly rinsed with Mili-Q water and methanol in order to remove any physisorbed compounds.
2.3.2. Covalent Immobilization of Glutathione via Carbodiimide Coupling
The carboxylic groups were activated by dropping 10 µL of a 35 mmol·L−1 sulfo-NHS and 26 mmol·L−1 EDC solution in 100 mmol·L−1 MES buffer (pH 4.5) onto the electrode surface and leaving it for 1 h. Then the electrodes were rinsed with Mili-Q water. The activated carboxyl groups reacted 30 min, 2 h or 24 h at 4 °C or 25 °C with the amine terminal groups of glutathione by placing 10 µL of a glutathione solution of a concentration of 1.45, 2.90 or 5.80 mg/100 μL in 0.1 mol·L−1 MES buffer (pH 4.5).
The electrochemical characterisation of each GSH-SPCNFE was carried out using 2 mmol·L−1 ferrocyanide/ferricyanide as redox probe in 100 mmol·L−1 phosphate buffer (pH 7.4) at each functionalization step by CV leading voltammograms that confirm the modifications taking place on the electrode surface (figure not shown).
2.4. Voltammetric Measurements
The linear calibration plots for the simultaneous determination of Pb(II) and Cd(II) on GSH-SPCNFE were obtained from DPASV measurements, performed at a deposition potential (Ed) of −1.4 V during a deposition time (td) of 120 s with stirring and followed by a rest period (tr) of 5 s, by increasing metal ion concentrations in 0.1 mol·L−1 acetate buffer (pH 4.5) solution. Determinations were done by scanning the potential from −1.4 to −0.3 V using pulse times of 50 ms, a step potential of 5 mV and pulse amplitudes of 50 mV.
3. Results and Discussion
The preparation of the GSH-SPCNFE by electrografting methodology is based on two main steps. The first one, the diazonium salt electrografting, is mainly dependent on both the substrate and the support of the working electrode. There is not a clear agreement about the number of CV cycles (ranging from 1 to 200) that should be performed during the electrografting process [18]. Thus, the number of CV cycles as well as their scan rate should be optimized according to the working electrode. In our case, for a SPCNFE this step was previously optimized to 15 cycles at 200 mV·s−1 [18]. The second step, the covalent immobilization of the ligand via carbodiimide coupling, is crucial in the modification of working electrodes with different ligands through the aryl diazonium salt electrografting. In the case of electrodes modified with glutathione, slightly different conditions have been reported in the literature for this step, with concentrations of glutathione ranging from 2.5 to 5 mg/100 µL MES buffer pH 4.5, either at 4 °C or without temperature control and usually overnight [17,18,21,22,23,24]. The influence of these three parameters (glutathione concentration, incubation temperature and incubation time) in the performance of the resulting sensor was studied taking the conditions used in [18] (2.9 mg/100 µL MES, 4 °C and overnight) as starting point and using the determination of Cd(II) and Pb(II) as a model of application.
First of all, three different glutathione concentrations, 1.45, 2.90 and 5.80 mg/100 µL MES, were tested maintaining the incubation time and temperature at 24 h and 4 °C respectively. Figure 1 shows the obtained voltammetric responses with each resulting sensor for a solution containing 77 µg·L−1 of both Cd(II) and Pb(II). As it can be observed, well-shaped peaks were obtained using 1.45 and 2.90 mg GSH/100 µL MES whereas 5.80 mg GSH/100 µL MES resulted in less defined and smaller peaks. This fact could be due to an excess of glutathione blocking the active sites. This blocking also affects the interaction of Pb(II) and Cd(II) with the surface of the electrodes, resulting in a slight shift in the potential, which is also highlighted by the worse baseline. Also, the performance of the resulting sensors was evaluated in terms of sensitivity, limit of detection (LOD) and linearity range for both Cd(II) and Pb(II) (Table 1). Looking for a linear proportionality between metal concentration and analytical response, the use of peak intensity or peak area as analytical response was evaluated during the experimental research of this work. The second approach showed a better linearity, particularly for Pb(II) ions, than the peak intensity. Therefore the peak area approach was used as analytical response in this study. Sensitivities were calculated as the slope of the calibration curve and LODs were calculated as 3 times the standard deviation of the intercept over the slope of the calibration curve. In all cases, linear calibration curves were obtained up to 125.2 and 150.1 µg·L−1 for Cd(II) and Pb(II) respectively. As shown in Table 1, the highest sensitivity for Pb(II) and Cd(II) were obtained using 1.45 and 5.80 mg GSH/100 µL MES respectively whereas the lowest LODs were obtained using 2.90 and 5.80 mg GSH/100 µL MES respectively. Taking into account the better defined peaks achieved using either 1.45 or 2.90 mg GSH/100 µL MES compared to those obtained using 5.80 mg GSH/100 µL MES and the lower LODs provided by 2.90 in comparison to 1.45 mg GSH/100 µL MES, 2.90 mg GSH/100 µL MES was selected as the optimal glutathione concentration. It should be pointed out though, that if a more expensive peptide was to be used as a ligand, a concentration of 1.45 mg/100 µL MES would result in a less expensive sensor without highly affecting its analytical performance.
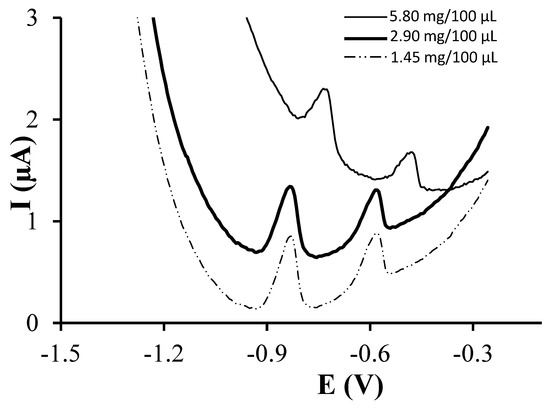
Figure 1.
Differential pulse anodic stripping voltammetric (DPASV) measurements of 77 µg·L−1 Pb(II) and Cd(II) on GSH-SPCNFE applying a Ed of −1.4 V during 120 s at 0.1 mol·L−1 acetate buffer (pH 4.5). GSH-SPCNFE modified with a glutathione solution of a concentration 1.45, 2.90 or 5.80 mg/100 μL in 0.1 mol·L−1 MES buffer (pH 4.5) during 24 h at 4 °C.

Table 1.
Calibration data for the simultaneous determination of Pb(II) and Cd(II) on GSH-SPCNFE at Ed of −1.4 V, td of 120 s and pH 4.5 at different GSH immobilization conditions.
Once the glutathione concentration was selected, the effect of the incubation time was studied. Figure 2 shows the voltammograms obtained for a solution containing 77 µg·L−1 of both Cd(II) and Pb(II) using incubation times of 30 min, 2 h and 24 h. Well-defined peaks were obtained in all cases. Again, linear calibration curves were obtained from 1 and up to 125.2 and 150.1 µg·L−1 for Cd(II) and Pb(II) respectively. Sensitivities associated to Cd(II) increased using longer incubation times whereas Pb(II) sensitivity increased when the incubation time was varied from 30 min to 2 h but decreased when it was extended to 24 h. On the other hand, the lowest LODs were obtained using incubation times of 30 min and 2 h for Pb(II) and Cd(II) respectively. Therefore, these results suggest that 30 min is not enough time to reach the full analytical performance of the modified sensor whereas an incubation time of 24 h considerably increases the manufacturing process without providing a much better analytical performance. Thus, an incubation time of 2 h was selected.
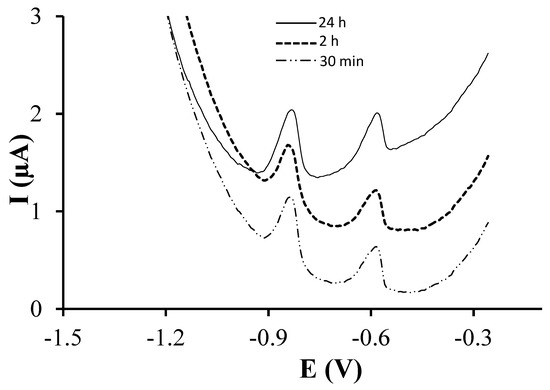
Figure 2.
DPASV measurements of 77 µg·L−1 Pb(II) and Cd(II) on GSH-SPCNFE applying a Ed of −1.4 V during 120 s at 0.1 mol·L−1 acetate buffer (pH 4.5). GSH-SPCNFE modified with a glutathione solution of a concentration 2.90 mg/100 μL in 0.1 mol·L−1 MES buffer (pH 4.5) during 30 min, 2 h or 24 h at 4 °C.
Finally, the effect of the incubation temperature was also evaluated. Figure 3 shows the voltammograms obtained for a solution containing 77 µg·L−1 of both Cd(II) and Pb(II) using incubation temperatures of 4 °C and 25 °C. As it can be observed, an incubation temperature of 25 °C resulted in more intense peaks for both Cd(II) and Pb(II) ions. Also, as it can be observed in Table 1, higher sensitivities were achieved for both Cd(II) and Pb(II) using an incubation temperature of 25 °C whereas lower LODs were obtained using 4 and 25 °C for Cd(II) and Pb(II) respectively. Therefore, 25 °C was selected as the optimal incubation temperature. It should be pointed out that, out of the three parameters studied, the incubation temperature is the one that mostly affects the target ions sensitivities. However, although increasing the temperature from 4 to 25 °C improves the sensitivity, it should be taken into account that higher temperatures could affect the integrity of glutathione.
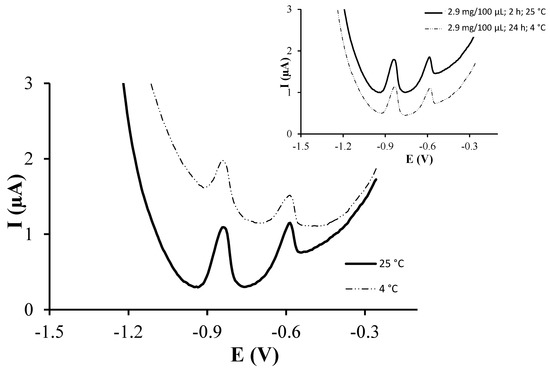
Figure 3.
DPASV measurements of 77 µg·L−1 Pb(II) and Cd(II) on GSH-SPCNFE applying a Ed of −1.4 V during 120 s at 0.1 mol·L−1 acetate buffer (pH 4.5). GSH-SPCNFE modified with a glutathione solution of a concentration 2.90 mg/100 μL in 0.1 mol·L−1 MES buffer (pH 4.5) during 2 h at 4 or 25 °C. Inset: comparison between the response of the GSH-SPCNFE under ideal and base conditions.
Therefore, these reported results suggest that the better conditions for the covalent immobilization of glutathione during the preparation of GSH-SPCNFE are a concentration of 2.9 mg GSH/100 µL, an incubation time of 2 h and an incubation temperature of 25 °C. As it can be observed in Figure 4, these optimized conditions result in a sensor that provides well-defined peaks that increase linearly with concentration for both Cd(II) and Pb(II) ions. Also, the optimized sensor provides more intensive peaks compared to that modified using the starting conditions (inset in Figure 3) as well as higher sensitivities and lower LODs for both Cd(II) and Pb(II) ions (Table 1). It should be pointed out though, that the optimal incubation time and glutathione concentration could slightly vary depending on the type and the area of the carbon surface where the modification is performed.
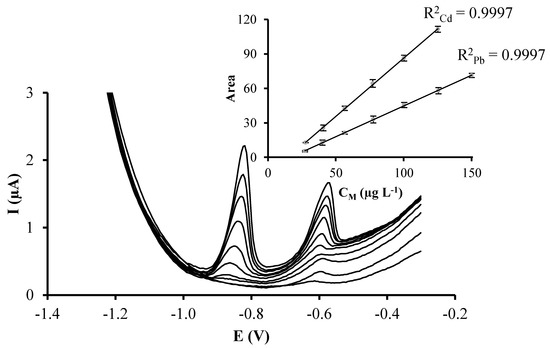
Figure 4.
DPASV measurements of increasing concentrations of Pb(II) and Cd(II) simultaneously recorded on GSH-SPCNFE applying a Ed of −1.4 V during 120 s at 0.1 mol·L−1 acetate buffer (pH 4.5). GSH-SPCNFE modified with a glutathione solution of a concentration 2.90 mg/100 μL in 0.1 mol·L−1 MES buffer (pH 4.5) during 2 h at 25 °C. Inset: Pb(II) and Cd(II) calibration plots.
4. Conclusions
The results described so far suggest that, beyond the standard receipts for electrode modification by electrografting and taking into account that the first step should be optimized according to the working electrode, the systematic optimization of the experimental conditions concerning the covalent immobilization of a particular ligand via carbodiimide coupling step can substantially improve the performance of the sensor.
In the case of glutathione immobilization, the incubation temperature has evolved as a relevant parameter. A typical room temperature of 25 °C produces better results than the usual refrigerator temperature of 4 °C, possibly due to faster kinetics. As for the incubation time, it can be notoriously reduced from 24 to 2 h keeping an optimal sensing performance. Finally, the usual concentration of ligand (2.9 mg/100 µL) has shown to be very close to the optimum value. A decrease of such concentration produces a slight increase of detection and quantification limits. Anyway the relatively low impact of reducing the amount of ligand could justify this option in the case of rare or expensive substances. As for the increase of the ligand concentration until 5.8 mg/100 µL, it causes a dramatic enhancement of the baseline and its curvature, which deteriorates sensor performance. This suggests that the concentration of the ligand should be carefully optimized depending on the nature of both subtract and modifier. In this sense, mol·L−1 units would be more convenient than mg/100 µL for a better comparison among ligands with different molecular weight (e.g., glutathione and phytochelatins of different chain length).
Thus, in general, we can conclude that the use of tailored modification procedures which adapt experimental conditions such as the incubation temperature, the incubation time and the concentration of the ligand can considerably improve the performance of modified sensors based on the electrografting methodology, especially in studies where different members of a family of compounds are compared. This is a key point to take into account in further investigation on the bioinspired application of glutathione and related molecules as ligands in the development of heavy metal sensors.
Acknowledgments
This work is supported by the University of Barcelona and by the Generalitat of Catalonia (Project 2014SGR269). Clara Pérez-Ràfols acknowledges the Spanish Ministry of Economy and Competitiveness for a Ph.D. grant.
Author Contributions
C. Pérez-Ràfols, N. Serrano and J.M. Díaz-Cruz conceived and designed the experiments and made a first draft of the paper, C. Pérez-Ràfols performed the experiments and the data treatment, C. Pérez-Ràfols, N. Serrano, J.M. Díaz-Cruz, C. Ariño and M. Esteban discussed the results and revised the writing of the paper from the first draft to the definitive version.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lawrence, E.; Jackson, A.R.W.; Jackson, J.M. Longman Dictionary of Environmental Science; Addison Wesley Longman: Essex, UK, 1998. [Google Scholar]
- Cobbett, C.S.; Goldsbrough, P. Phytochelatins and metallothioneins: Roles in heavy metal detoxification and homeostasis. Annu. Rev. Plant Biol. 2002, 53, 159–182. [Google Scholar] [CrossRef] [PubMed]
- Grill, E.; Loffler, S.; Winnacker, E.L.; Zenk, M.H. Phytochelatins, the heavy-metal binding peptides of plants, are synthesized from glutathione by a specific γ-glutamylcysteine dipeptidyl transpeptidase (phytochelatin synthase). Proc. Natl. Acad. Sci. USA 1989, 86, 6838–6842. [Google Scholar] [CrossRef] [PubMed]
- Serrano, N.; Díaz-Cruz, J.M.; Ariño, C.; Esteban, M. Recent contributions to the study of phytochelatins with an analytical approach. Trends Anal. Chem. 2015, 73, 129–145. [Google Scholar] [CrossRef]
- Soodan, R.K.; Pakade, Y.B.; Nagpal, A.; Katnoria, J.K. Analytical techniques for estimation of heavy metals in soil ecosystem: A tabulated review. Talanta 2014, 125, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Bings, N.H.; Bogaerts, A.; Broekaert, J.A.C. Atomic Spectroscopy. Anal. Chem. 2013, 85, 670–704. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Stripping Analysis: Principles, Instrumentation and Applications; VCH: New York, NY, USA, 1985. [Google Scholar]
- Barek, J.; Fogg, A.G.; Muck, A.; Zima, J. Polarography and voltammetry at mercury electrodes. Crit. Rev. Anal. Chem. 2001, 31, 291–309. [Google Scholar] [CrossRef]
- Serrano, N.; Alberich, A.; Díaz-Cruz, J.M.; Ariño, C.; Esteban, M. Coating methods, modifiers and applications of bismuth screen-printed electrodes. Trends Anal. Chem. 2013, 46, 15–29. [Google Scholar] [CrossRef]
- Serrano, N.; Díaz-Cruz, J.M.; Ariño, C.; Esteban, M. Antimony-based electrodes for analytical determinations. Trends Anal. Chem. 2016, 77, 203–213. [Google Scholar] [CrossRef]
- March, G.; Nguyen, T.D.; Piro, B. Modified electrodes used for electrochemical detection of metal ions in environmental analysis. Biosensors 2015, 5, 241–275. [Google Scholar] [CrossRef] [PubMed]
- Barton, J.; González-García, M.B.; Hernández-Santos, D.; Fanjul-Bolado, P.; Ribotti, A.; McCaul, M.; Diamond, D.; Magni, P. Screen-printed electrodes for environmental monitoring of heavy metal ions: A review. Microchim. Acta 2016, 183, 503–517. [Google Scholar] [CrossRef]
- Wawrzyniak, U.E.; Ciosek, P.; Zaborowski, M.; Liu, G.; Gooding, J.J. Gly-Gly-His Immobilized on Monolayer Modified Back-Side Contact Miniaturized Sensors for Complexation of Copper Ions. Electroanalysis 2013, 25, 1461–1471. [Google Scholar] [CrossRef]
- Gooding, J.J. Advances in interfacial design for electrochemical biosensors and sensors: Aryl diazonium salts for modifying carbon and metal electrodes. Electroanalysis 2008, 20, 573–582. [Google Scholar] [CrossRef]
- Delamar, M.; Hitmi, R.; Pinson, J.; Savbnt, J.M. Covalent modification of carbon surfaces by grafting of functionalized aryl radicals produced from electrochemical reduction of diazonium salts. J. Am. Chem. Soc. 1992, 114, 5883–5884. [Google Scholar] [CrossRef]
- Baranton, S.; Bélanger, D. Electrochemical derivatization of carbon surface by reduction of in situ generated diazonium cations. J. Phys. Chem. B 2005, 109, 24401–24410. [Google Scholar] [CrossRef] [PubMed]
- Serrano, N.; Prieto-Simón, B.; Cetó, X.; del Valle, M. Array of peptide-modified electrodes for the simultaneous determination of Pb(II), Cd(II) and Zn(II). Talanta 2014, 125, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ràfols, C.; Serrano, N.; Díaz-Cruz, J.M.; Ariño, C.; Esteban, M. Glutathione modified screen-printed carbon nanofiber electrode for the voltammetric determination of metal ions in natural samples. Talanta 2016, 155, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.I. Textbook of Quantitative Chemical Analysis, 5th ed.; Pearson Education Limited: Harlow, UK, 1989. [Google Scholar]
- Bélanger, D.; Pinson, J. Electrografting: A powerful method for surface modification. Chem. Soc. Rev. 2011, 40, 3995–4048. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Böcking, T.; Gooding, J.J. Diazonium salts: Stable monolayers on gold electrodes for sensing applications. J. Electroanal. Chem. 2007, 600, 335–344. [Google Scholar] [CrossRef]
- Chow, E.; Ebrahimi, D.; Gooding, J.J.; Hibbert, D.B. Application of N-PLS calibration to the simultaneous determination of Cu2+, Cd2+ and Pb2+ using peptide modified electrochemical sensors. Analyst 2006, 131, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, D.; Chow, E.; Gooding, J.J.; Hibbert, D.B. Multi-analyte sensing: A chemometrics approach to understanding the merits of electrode arrays versus single electrodes. Analyst 2008, 133, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.; Hibbert, D.B.; Gooding, J.J. Voltammetric detection of cadmium ions at glutathione-modified gold electrodes. Analyst 2005, 130, 831–837. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).