1. Introduction
The rapid and reliable detection of multiple biomarkers simultaneously in small sample volumes is increasingly requested in current clinical practice and represents a fundamental step towards personalized medicine [
1]. Nowadays, most of the available diagnostic devices are solid-state based analytical assays, where a functionalized surface works as the active element for biorecognition. Site-specific immobilization of multiple active elements on the same surface to probe low biosample volumes requires miniaturization and is generally technologically demanding, since it entails successive steps specifically tailored to each biomarker probe.
One promising strategy for multiplexing probe immobilization is DDI (DNA-directed immobilization) [
2,
3,
4,
5], where different DNA-conjugated antibodies targeting different biomarkers are immobilized via Watson-Crick base pairing on surface-tethered complementary DNA sequences. However the synthesis of DNA-protein conjugates is quite challenging, requires click-chemistry kits and careful optimization in order to assure a final construct that is both suitable for immobilization and preserves the original affinity.
Nucleic acid aptamers can offer a valid alternative to antibodies. Aptamers are oligosequences selected in vitro to bind a target with high affinity. In particular, they show highly specific binding activity since aptamer-target interaction is based on three-dimensional folding patterns, resulting in dissociation constants in the picomolar range. In addition, aptamers, if compared to standard proteins, show higher stability, and ease of chemical modification. Aptamer production is fully automated, highly reproducible, and low-cost. Moreover, aptamers are naturally integrable in the context of DDI: DNA-aptamer constructs are produced simply by adding a surface binding sequence to the aptamer during oligo-synthesis.
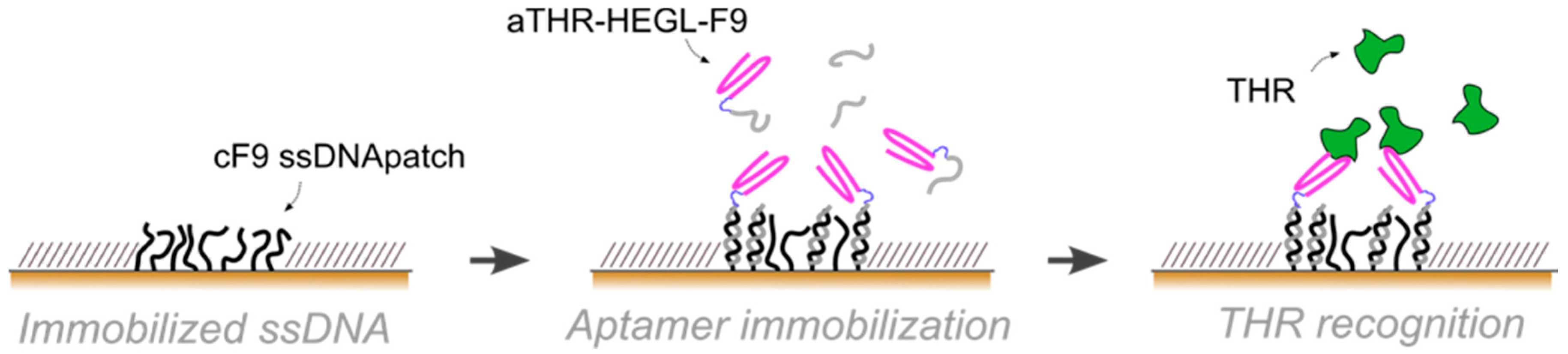
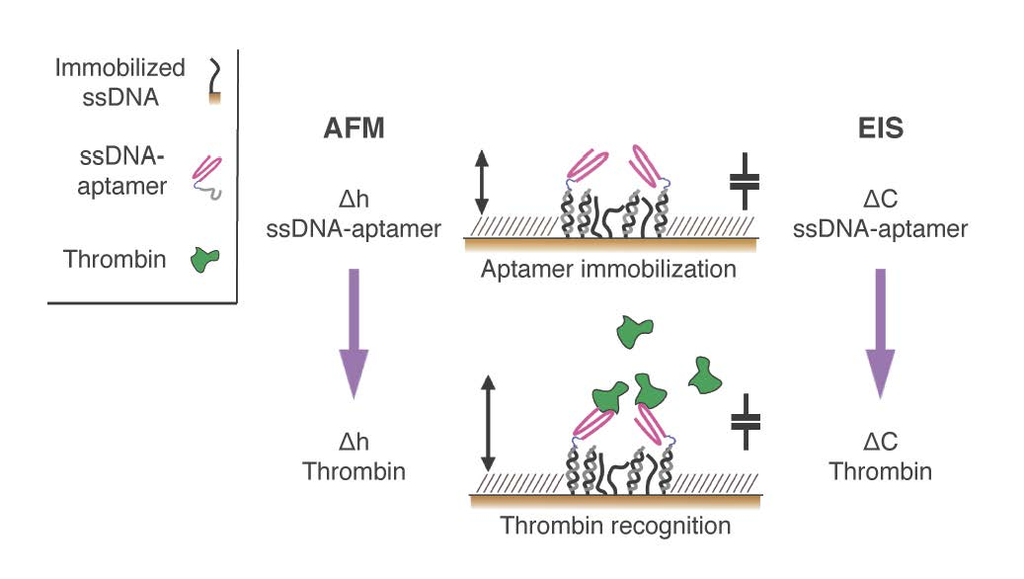
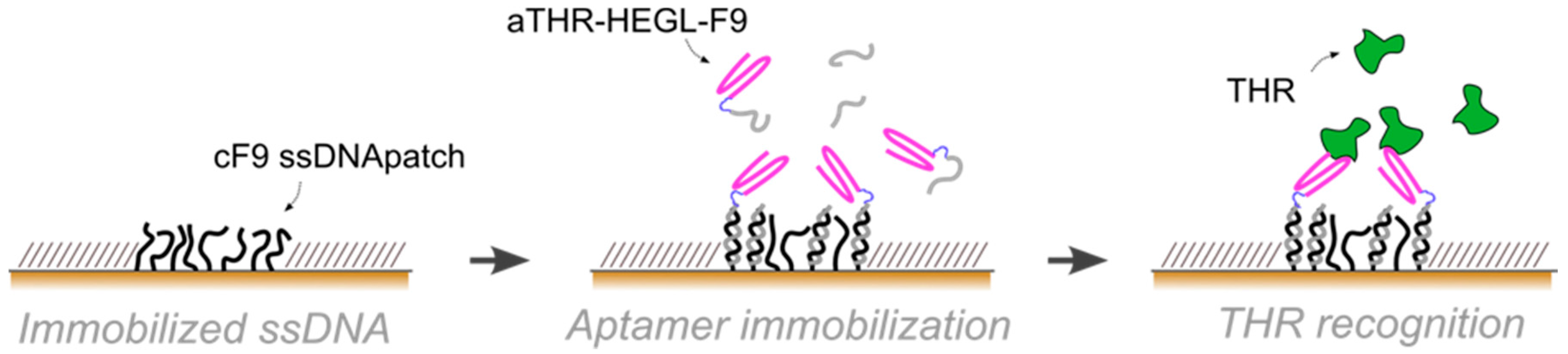
In this paper, we exploited aptamers as the active recognition elements of miniaturized DNA-based biosensors. We modified aptamers with a DNA tag (cDNA), meant to hybridize to micron-sized surface-grafted complementary DNA monolayers, to create the desired functional areas (
Figure 1). In particular, we carefully optimized the immobilization strategy to implement DNA-modified aptamers onto two innovative, DNA-based miniaturized sensor platforms: (i) atomic force microscopy (AFM)-based nanoarrays, and (ii) electrochemical impedance spectroscopy (EIS)-based microelectrodes. Both of these platforms have been designed and developed to allow for the sensitive detection of biomolecules in small sample volumes.
In the case of nanoarrays, we capitalize here on the exploitation of AFM-based lithography, nanografting [
6,
7,
8], and AFM topographic imaging in physiological environment successfully demonstrated in the work of Bano et al. for the detection of multiple proteins in standardized human serum [
9], and successively by Bosco and Ganau et al., for the ultimate integration with a device for cell sorting, to measure the secretome of few selected cells [
10]. By AFM nanografting we confined nanospots of thiol-modified single-stranded DNA monolayers inside a bio-repellent, self-assembled monolayer (SAM) of oligoethyleneglycol-terminated alkanethiols. The density of the DNA-confined SAM can be tuned via nanografting, and adjusted to the steric requirements of the biorecognition elements. On such active DNA nanospots, the aptamer-cDNA conjugate is then loaded through Watson-Crick base-pairing, in a process known as DNA-directed immobilization [
9]. On the other side, an electrochemical impedance-based biosensor, with a three-electrode design developed in our group for the recognition of nucleic acids [
11,
12], are tested towards protein detection through modified aptamers. The working micro-electrode is fully covered by a functional, thiolated, single-stranded DNA SAM, on which the aptamer-cDNA conjugate is then loaded via DDI.
As a proof of principle, in this study we used an extensively-investigated aptamer for thrombin (THR) [
13] that shows a well-characterized structure and binding properties (K
D = 50 nM), confirmed by several studies [
14,
15]. Two constructs were proposed in this regard: a simple design consisting of the aptamer region on the 5′ side, extending with the immobilization region complementary to the DNA grafted monolayer on the 3′ side (
Supplementary Materials, Figure S1a); a second design similar to the first one, but containing a hexaethilenglycol-spacer (HEGL) between the two regions to improve the functionality of aptamer binding site (
Supplementary Materials, Figure S1b). As stated in several works, the use of polyethyleneglycol groups can significantly improve biorecognition sensitivity by reducing nonspecific interactions and steric hindrance effects [
16,
17]. In both cases the surface linking oligo sequence (with an alkanethiol linker to bind to the gold surface), and the complementary one on the aptamer side, were carefully selected to have minimal influence on the protein binding—aptamer region in order to avoid possible interferences during aptamer immobilization and then on the aptamer binding interaction with THR.
2. Materials and Methods
2.1. Preliminary Affinity Characterization of the DNA-Aptamer Constructs
Affinity characterization of the DNA-aptamer constructs was first carried out in silico using the web service UNAFold, developed by Zucker and his coworkers [
18]. The software application [
19] for hybridization of two different oligonucleotide strands was used to generate temperature-dependent concentration plots of the tested sequence pairs in buffer, considering an ionic composition of 150 mM Na
+, 2 mM Mg
2+, similar to the binding buffer used in further wet experiments. The calculated concentrations from the plot data for hybridized and free sequences at 25 °C were further used to determine the theoretical dissociation constants (K
D). In this regard theoretical K
D values were calculated and compared for the thiolated linker sequence cF9 (5′-CTTCACGATTGCCACTTTCCAC-3′) vs. the protein binding aptamer sequence (5′-GGTTGGTGTGGTTGG-3′) and the complementary hybridization region F9 (5′-GTGGAAAGTGGCAATCGTGAAG-3′) used in the aptamer constructs. The designed aptamer constructs were further tested in bulk conditions for their functionality, testing the binding affinity for human thrombin (HT) in a qualitative electrophoretic mobility shift assay (EMSA) (
Supplementary Materials). Two different aptamer constructs were prepared: the one in which the F9 sequence is directly extending from the aptamer sequence (5′-GGTTGGTGTGGTTGGGTGGAAAGTGGCAATCGTGAAG-3′), named F9aTHR and with hexaethyleneglycol (HEGL) linker in between, (5′-GGTTGGTGTGGTTGG-HEGL-GTGGAAAGTGGCAATCGTGAAG-3′), named F9-HEGL-aTHR. All the reagents in the binding experiments including buffers, human thrombin and materials used for synthesis of aptamer constructs F9aTHR and F9-HEGL-aTHR, were ordered from Sigma-Aldrich Corp. (Saint Louis, MO, USA).
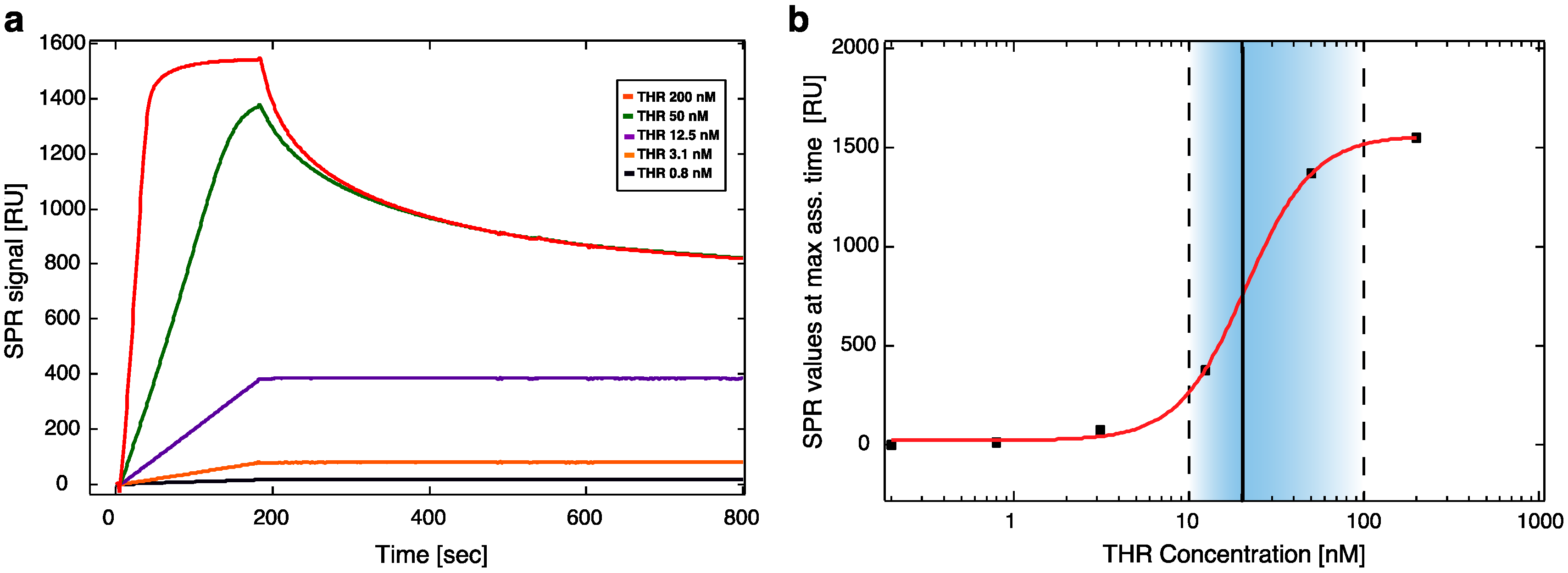
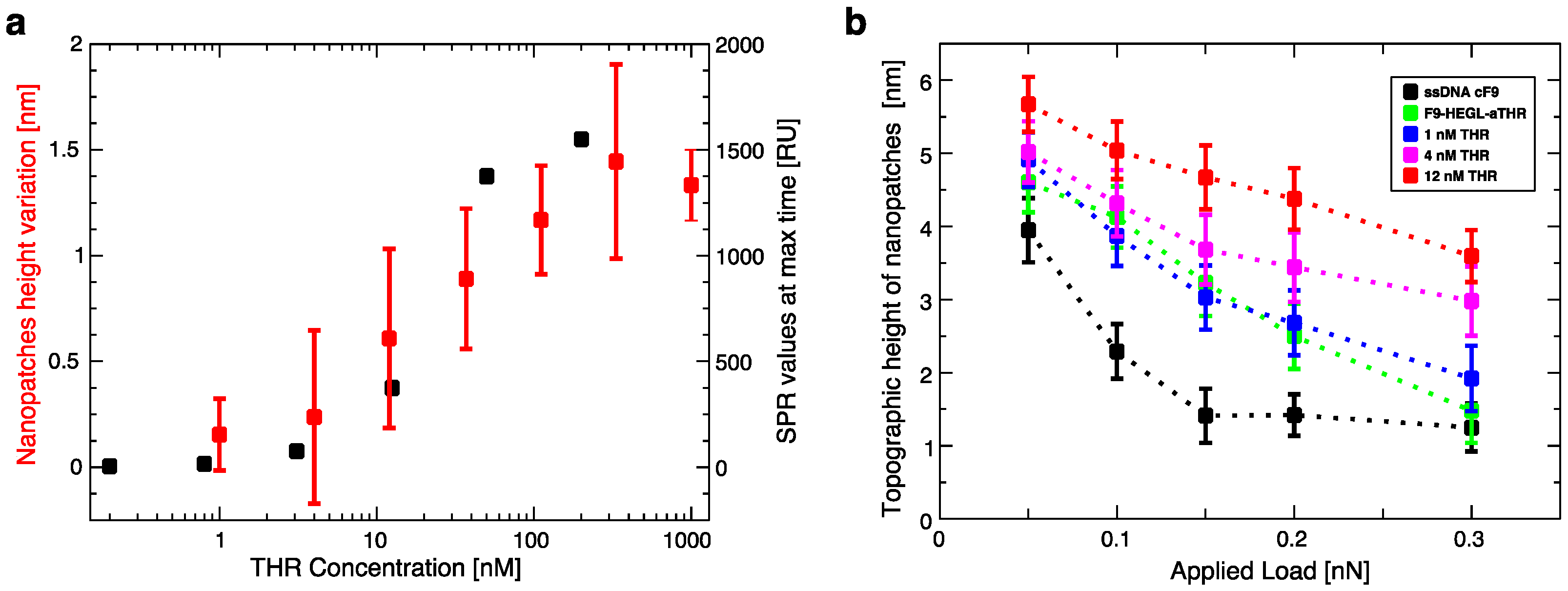
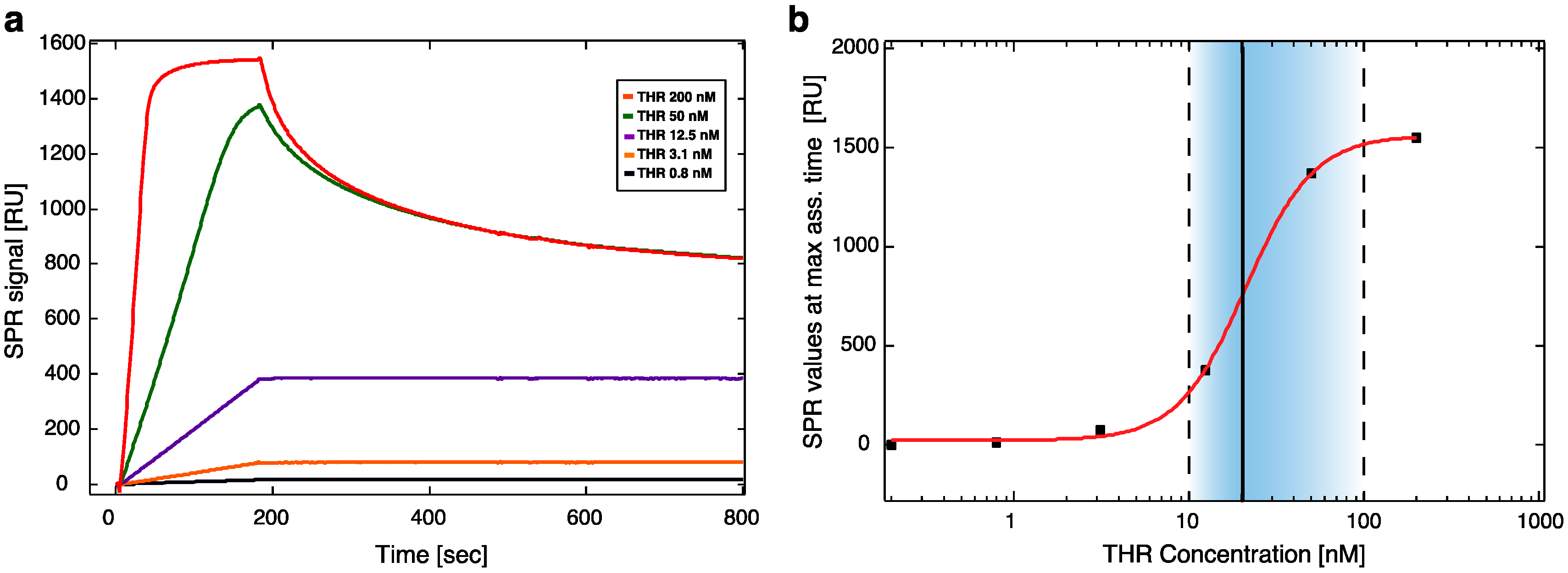
To test aptamer affinity on surfaces, a Biacore X100 Surface Plasmon Resonance (GE Healthcare, Little Chalfont, Buckinghamshire, UK) instrument was used at a constant temperature of 25 °C. A continuous flow (5 μL/min) of PBS buffer (running buffer) was maintained during all the experiments. First, a biotinilated cF9 sequence (cF9-biotin, 2 μM in PBS buffer) was immobilized over the Biacore SA gold chip surface. The immobilization through streptavidin-biotin binding was stopped after reaching a binding level of ~1200 RU, corresponding to an amount of ssDNA on the surface that ensure an efficient attachment of the molecules in the following steps of the experiment; then the surface was rinsed with two 1 min pulses of 50 mM NaOH solution, in order to remove unbound cF9-biotin [
20]. The hybridization was carried out by incubation with F9-HEGL-aTHR at 30 μM in TE buffer with 1 M NaCl until reaching a binding level of ~500 RU, to form an active layer suitable to detect a binding signal also with the lowest concentrations of analyte. The immobilization procedure was followed by a flow of running buffer for 2 h in order to remove aptamers non-specifically bound to the surface and to stabilize the baseline. After this procedure the signal remained constant without any baseline drifting. Binding affinity tests were performed injecting different dilutions of thrombin (0, 0.2, 0.8, 3.1, 12.5, 50, and 200 nM) in running buffer at a flow rate of 30 μL/min for 3 min (association phase) and afterwards flushing with running buffer for 5 min (dissociation phase). For the regeneration of the surface 1 min pulses of a 50 mM NaOH solution were used, followed by a stabilization time of 5 min [
21]. Binding affinity parameters were calculated using the BIAevaluation 3.1 software (Biacore GE Healthcare, Little Chalfont, Buckinghamshire, UK).
2.2. AFM-Based Nanoarrays
All AFM experiments were carried out on a XE-100 Park Instrument (Park System Corp., KANC 4F, Suwon, Korea) with a customized liquid cell. A tip-assisted AFM-based nanolithography technique has been used to fabricate DNA nanoarrays with high surface density (1–2 × 10
13 molecules/cm
2): using Si cantilevers (NSC36B Mikromasch (Mikromasch, Innovative Solutions Bulgaria Ltd., Sofia, Bulgaria), spring constant: 0.6 N/m) multiple nanografting assembled monolayers (NAM) of thiol-modified single-stranded DNA (ssDNA), named cF9, were prepared by serial AFM-based nanografting inside a self-assembled monolayer (SAM) of a top oligo ethylene glycol terminated alkanethiol, TOEG ((1-mercaptoundec-11-yl)hexa(ethyleneglycol), HS-(CH2)11-(OCH2CH2)6-OH from Sigma Aldrich) on ultraflat gold surfaces [
22] following standard protocols reported earlier [
9,
23]. The DNA patches were obtained promoting the replacement of the TOEG molecules with the oligonucleotides by the AFM tip scanning an area of 1 μm × 1 μm or less at high force (about 100 nN) in the presence of a solution of thiolated ssDNA sequences (5 μM in TE buffer 1 M NaCl) at a scan rate of 2 Hz. Several patches (6–8 for each experiment, to guarantee good statistics) of cF9 ssDNA were produced.
Aptamer immobilization was performed via DDI, incubating the ssDNA SAM with 1:1 mix of F9-HEGL-aTHR and F9 at 2 μM in TE buffer with 1 M NaCl in order to avoid steric hindrance by reducing the aptamer surface density, hence, preserving the activity.
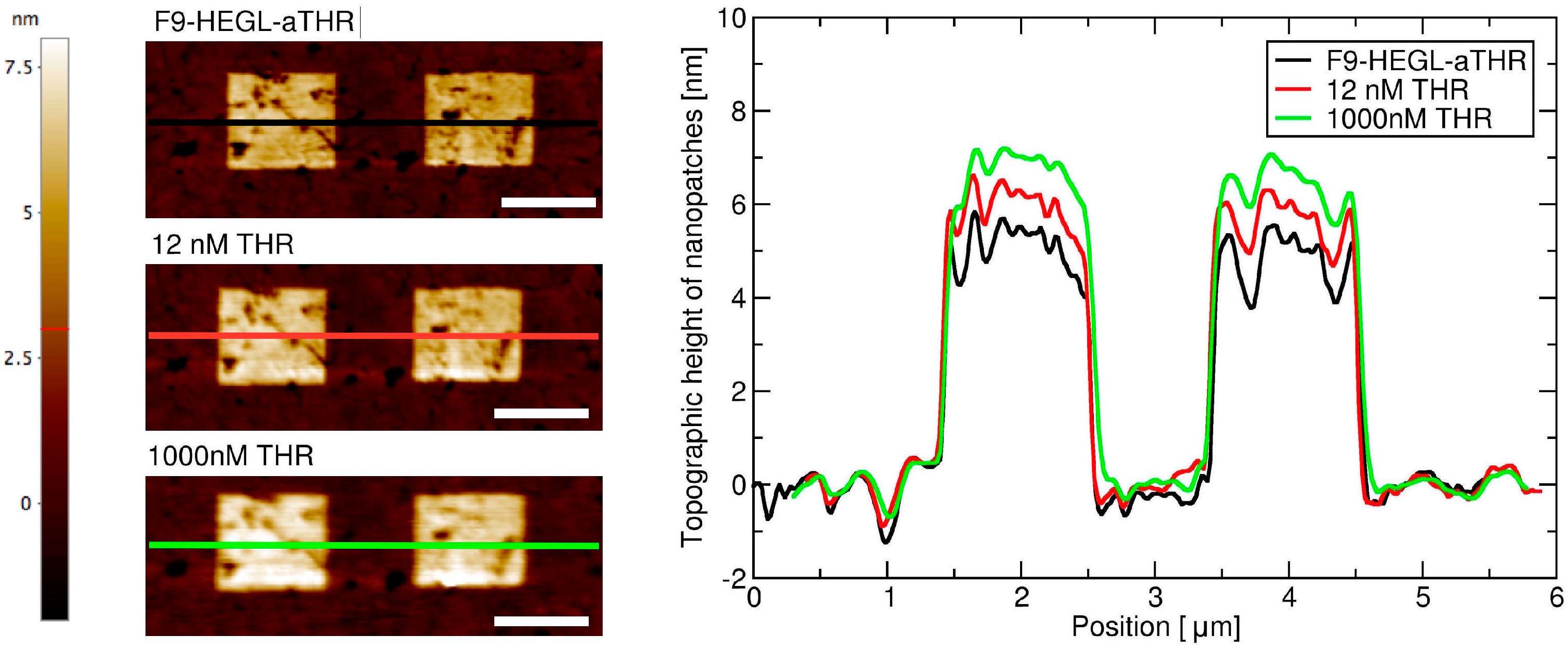
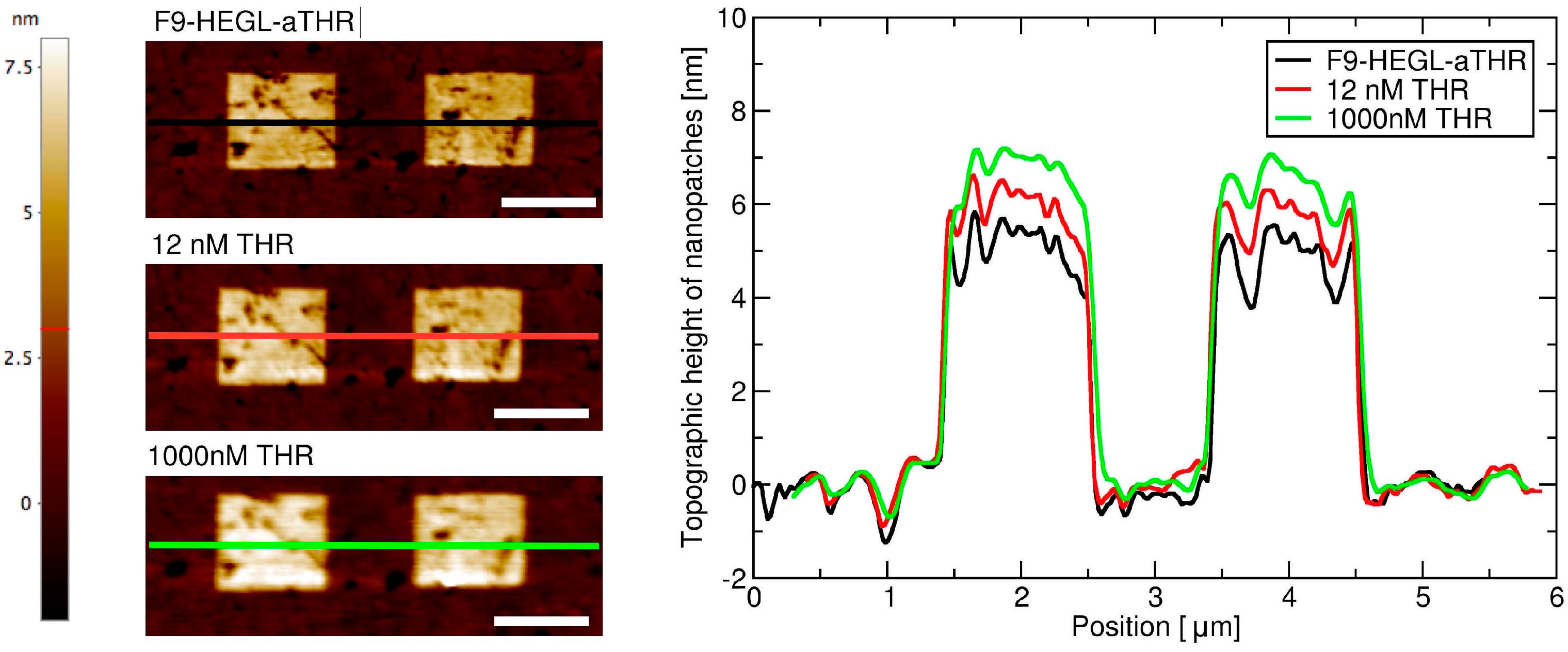
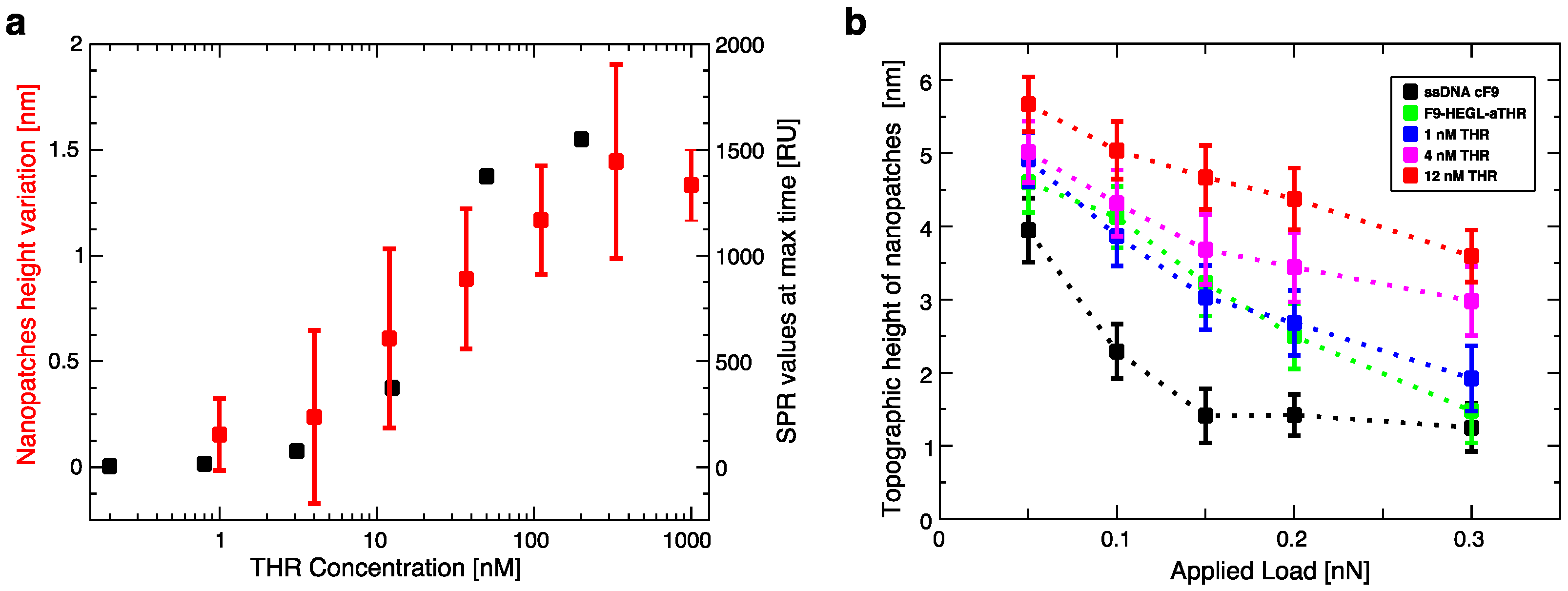
Aptamer-thrombin binding was promoted through the incubation of aptamer nanopatches for one hour with a solution (volume of about 100 μL) containing thrombin at different concentrations (THR buffer: 20 mM Tris pH 7.4, 140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1 mM CaCl2); topographic height variations over the NAMs were measured with AFM in gentle contact with standard silicon cantilevers (CSC38 Mikromasch, spring constant: 0.06 N/m) at a 1 Hz scan rate, applying a force of 0.1 nN to detect and quantify binding affinity.
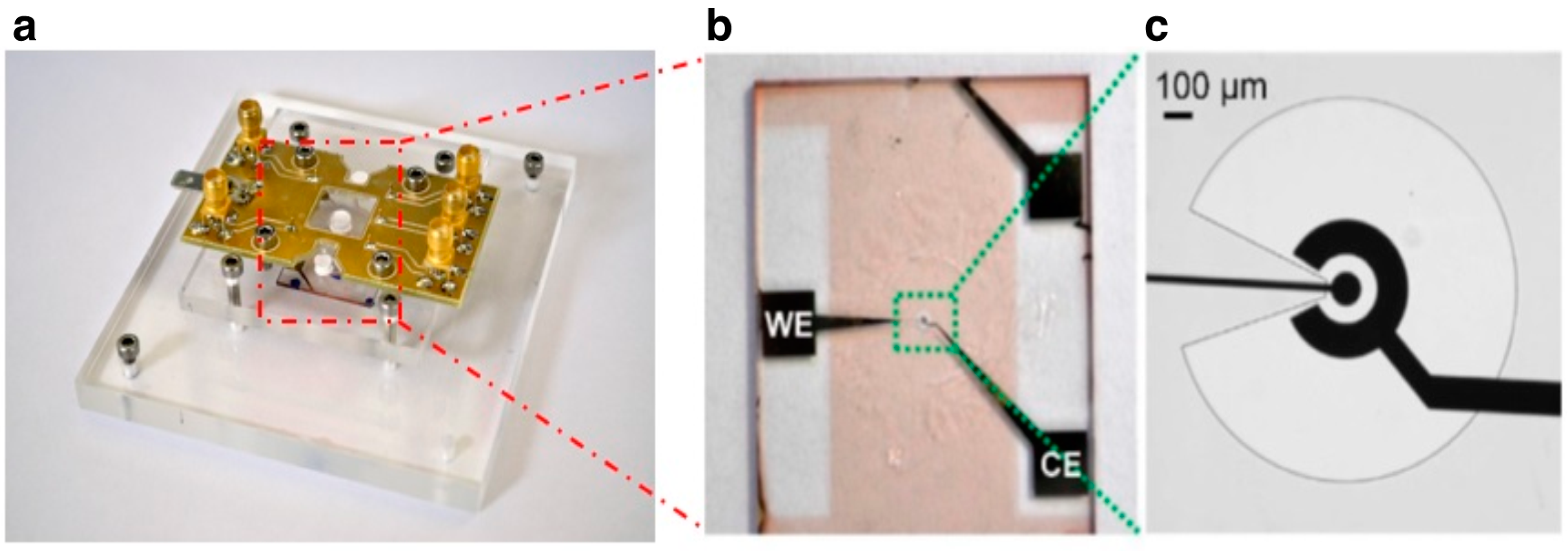
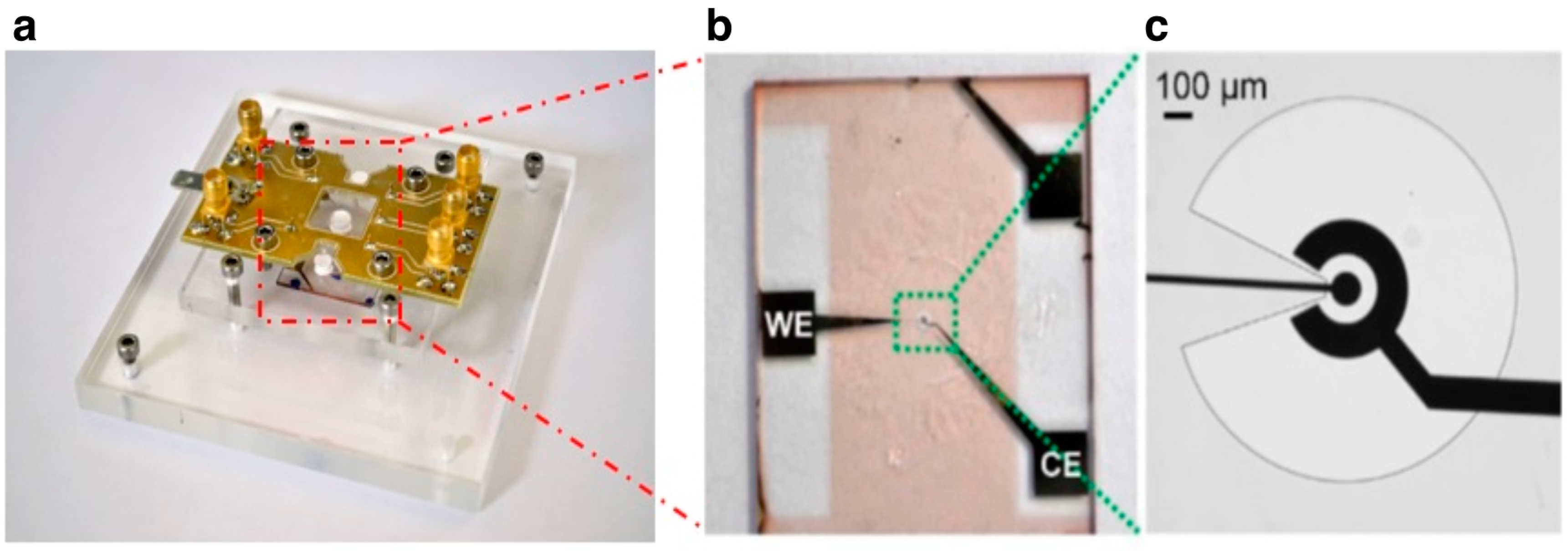
2.3. EIS-Based Devices
Electrochemical impedance spectroscopy (EIS)-based devices were fabricated using optical lithography techniques. They consist of a three-electrode electrochemical cell with microfabricated working (WE) and the counter (CE) gold electrodes and a classical mm-sized Ag/AgCl reference electrode (RE). As in standard electrochemical setup, the potential (AC, 10 mV rms) is applied across WE and RE, whereas the current is measured across WE and CE. The differential capacitance (C
d) defines the charge density (σ
M) change at the metal surface for a small variation of the applied potential (φ):
is obtained by fitting the current response of the device upon application of the AC voltage at four frequencies: 100 Hz, 200 Hz, 250 Hz, and 400 Hz, by using the HEKA PG340 USB bipotentiostat (HEKA Elektronik, Dr. Schulze, GmbH, Lambrecht, Germany). As already explained in [
11,
12], at each frequency we collect 200 complete periods from which we compute the root mean squared value of the measured current, I
rms = 2πfV
rmsC
d, and the relative uncertainties using error propagation analysis. We also proved that the device is reusable up to several times and the data are reproducible on different sensors with a standard deviation of only a few percent.
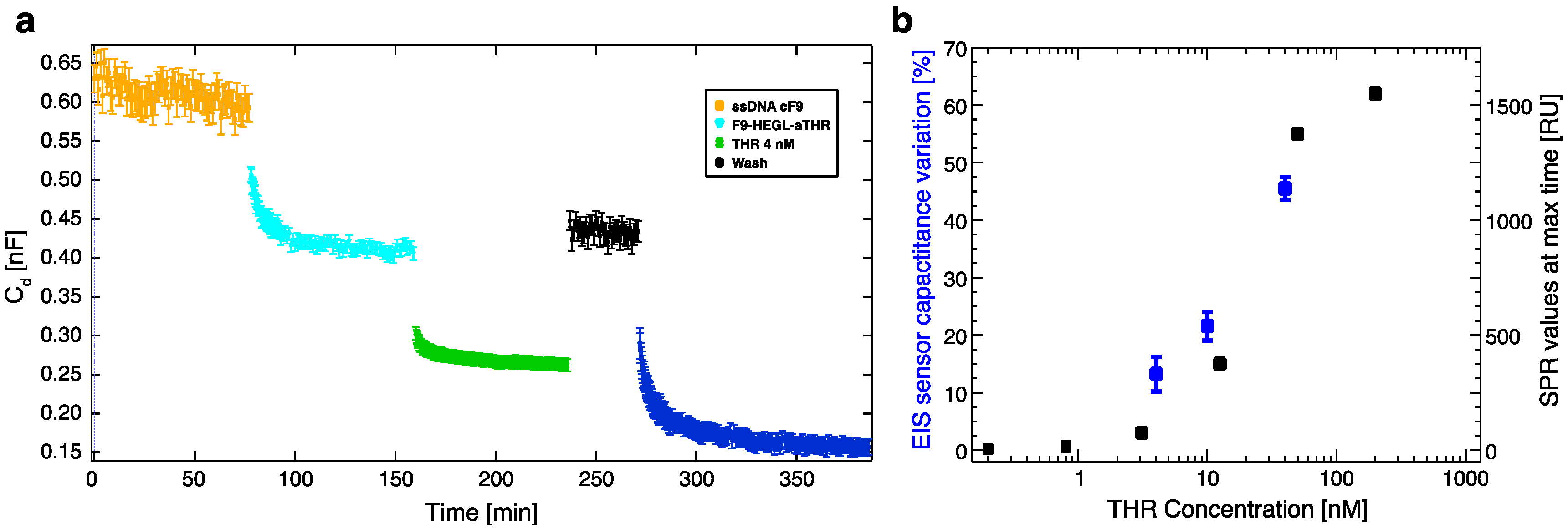
Cd variations, recorded upon exposing the electrochemical cell to aptamer-DNA conjugate/analyte containing solutions, were quantitatively connected to the number of biorecognition events. In particular, Cd is mostly affected by molecular layer height changes, the replacement of water molecules in the biological layer, and by electrical charge redistribution upon biorecognition.
Electrodes were first patterned on clean microscope slides using MEGAPOSIT
TM SPR
TM 220 1.2 (Series Photo-Resist) (The Dow Chemical Company, Michigan, USA) as the optical positive resist. The slides were then metalized in an e-beam evaporator, with a 20-nm Ti layer, in order to promote the adhesion, followed by 80-nm Au layer deposition and then kept in an acetone bath overnight in order to perform the lift-off process. After lift-off, the electrodes were coated again with an insulating layer of NANO
TMSU8-2002 (MicroChem Corp., Westborough, MA, USA), shaped to expose only the circular part of WE and CE. SU8 was used as insulating material to reduce the active surface area for biorecognition. The circular WE exposed to the solution, the arc of the CE and the profile of the SU8 layer, which covers the rest of the metal electrodes, are visible in
Figure 2.
The insulating SU8-layer had a thickness of about 1.5 μm, measured using a 3D surface profilometer. Gold electrode functionalization with ssDNA molecules was carried out using the well-established procedures for creating DNA SAMs on gold [
24,
25,
26]. Initially the electrodes were wetted for 15 min with a drop of a high-ionic-strength buffer, TE NaCl 1 M, containing cF9-SH to create a low density DNA SAM (2.1 × 10
12 molecules/cm
2) [
12]. Low density monolayers were chosen to avoid steric hindrance limitations to the hybridization efficiency. In this regime DNA hybridization follows Langmuir-like kinetics [
12,
27], while electrostatic charges are largely screened allowing for fast hybridization kinetics and reducing the limit of detection of the device. After each functionalization step the devices were rinsed with the proper DNA/protein buffer solution, prior to capacitance measurements. SAM hybridization was performed with 1 μM DNA-aptamer construct in about 100 μL solution (same volume as for the nanoarray measurements) containing 100 mM KCl. To follow hybridization kinetics, we initially measured at a rate of four measurements/min for 15 min, then we slowed down to one measurement/min until the C
d differential variations between successive points were lower than 6%, which we considered to be the “steady-state” of our measure.
4. Conclusions
Within this work we proved that short oligo-labelled aptamers can be used for the detection of biomolecules in miniaturized, surface-based devices, as nanoarrays and electrochemical microfabricated cells, in a scheme which allows for multiplexing analysis. First, we demonstrated in bulk that by linking a short oligo via a polyethileneglycol linker to an aptamer designed and optimized for thrombin recognition, the binding affinity was preserved. Then, by immobilizing the DNA-aptamer construct on a SPR chips via DNA-directed immobilization and performing binding affinity measurements via SPR, we obtained a value of KD in the range of 10–100 nM, in good agreement with the available literature for in bulk studies, demonstrating that the aptamer functionality was retained upon surface immobilization. Successful thrombin detection was achieved by tuning aptamer surface density on the active area to low values in order to allow aptamers conformational rearrangement necessary to bind the ligands. Additionally, we demonstrated that carefully optimized miniaturized devices could be used in a quantitative manner to determine thrombin concentration in solution, with the given, ultimate advantage with respect to other assays to be used with low volume biosamples. We used two miniaturized platforms, one based on AFM lithography and topography readout, and one on electrochemical impedance spectroscopy measurements. In both cases the measured binding affinity curves overlapped with SPR data impressively well. In particular, although these measurements are preliminary, we observed that at low thrombin concentrations (<5 nM), the EIS sensor is more sensitive than SPR, and that could be pushed to reach lower detection limits (we expect, in the pM range). On the other hand, DNA nanoarrays also allowed for an estimation of the binding affinity which resulted in good agreement with SPR data. Here, the change of conformation of aptamer upon THR binding towards a more compact structure worked reducing the expected height variation due to immobilization of THR on the device. Although still detectable, the error associated to the measurements makes this device less reliable than SPR chips to measure binding affinity. However, mechanical compressibility measurements performed on the nanopatches helped to validate the result. The height change measurements on the nanoarrays, moreover, can be combined with the monitoring of roughness variations on the patch and outside it to infer the occurrence of specific binding only on the patch, and exclude the presence of aspecific binding outside it, even without the use of (expensive) sandwich schemes.
We put forward the idea to complement our data with AFM-based force spectroscopy measurements to investigate the variation of the mechanical properties of the DNA-aptamer conjugate structures upon ligand binding. This additional strategy might be particularly profitable given the peculiar properties of aptamers, which undergo a global conformational change upon binding. Furthermore, combining the output of these two techniques, a structural-mechanical model for the interface may be derived to be, in turn, used to describe the details of capacitance changes at the gold electrode.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}