Developing a Quantitative Profiling Method for Detecting Free Fatty Acids in Crude Lanolin Based on Analytical Quality by Design


Abstract
1. Introduction
2. Reagents
3. Methods
3.1. Chromatographic Analysis Conditions
3.2. Solution Preparation
3.2.1. Preparation of the Mixed Standard Solution
3.2.2. Preparation of the Test Solution
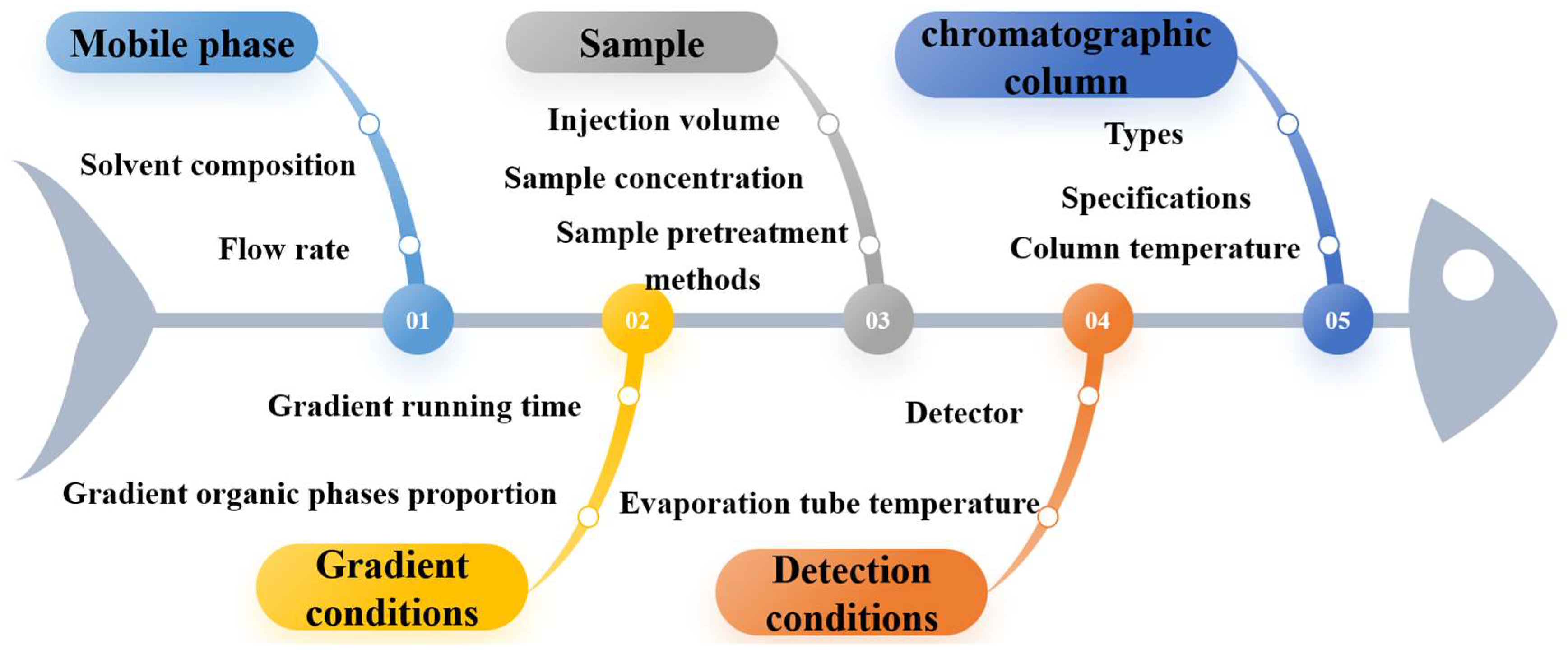
3.3. Preliminary Experiments and Identification of Analytical Method Parameters
3.4. Experimental Design
3.5. Method Validation
3.6. Data Processing and Analysis
3.7. High-Resolution Mass Spectrometry Analysis
4. Results and Discussions
4.1. Data Analysis of DSD Experiments
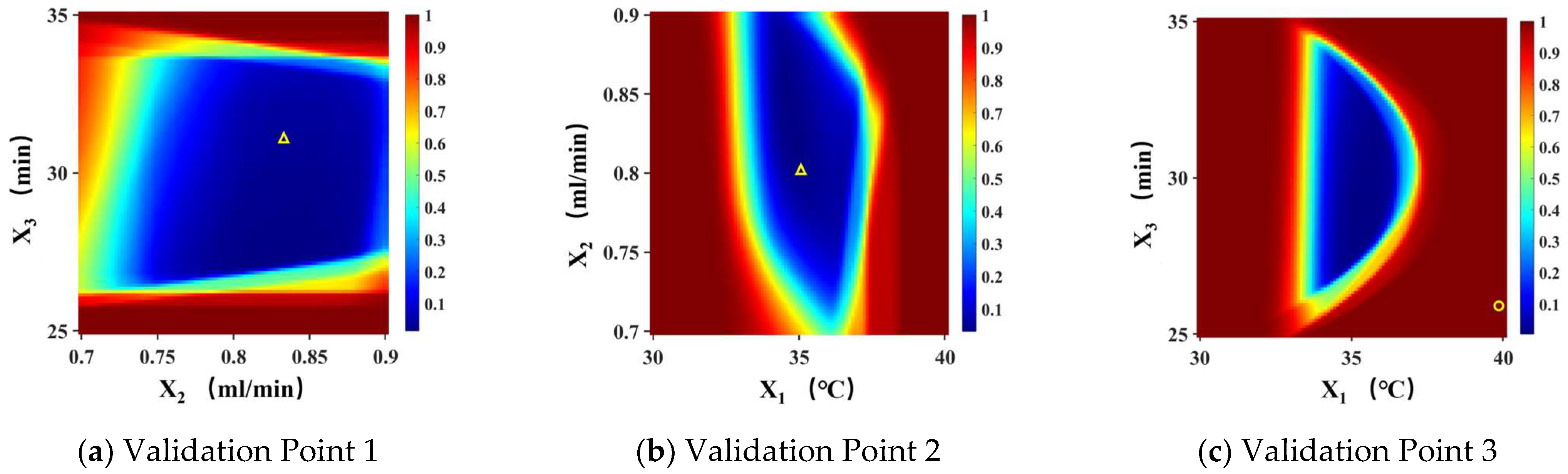
4.2. Establishment and Verification of the Design Space
4.3. High-Resolution Mass Spectrometry Analysis
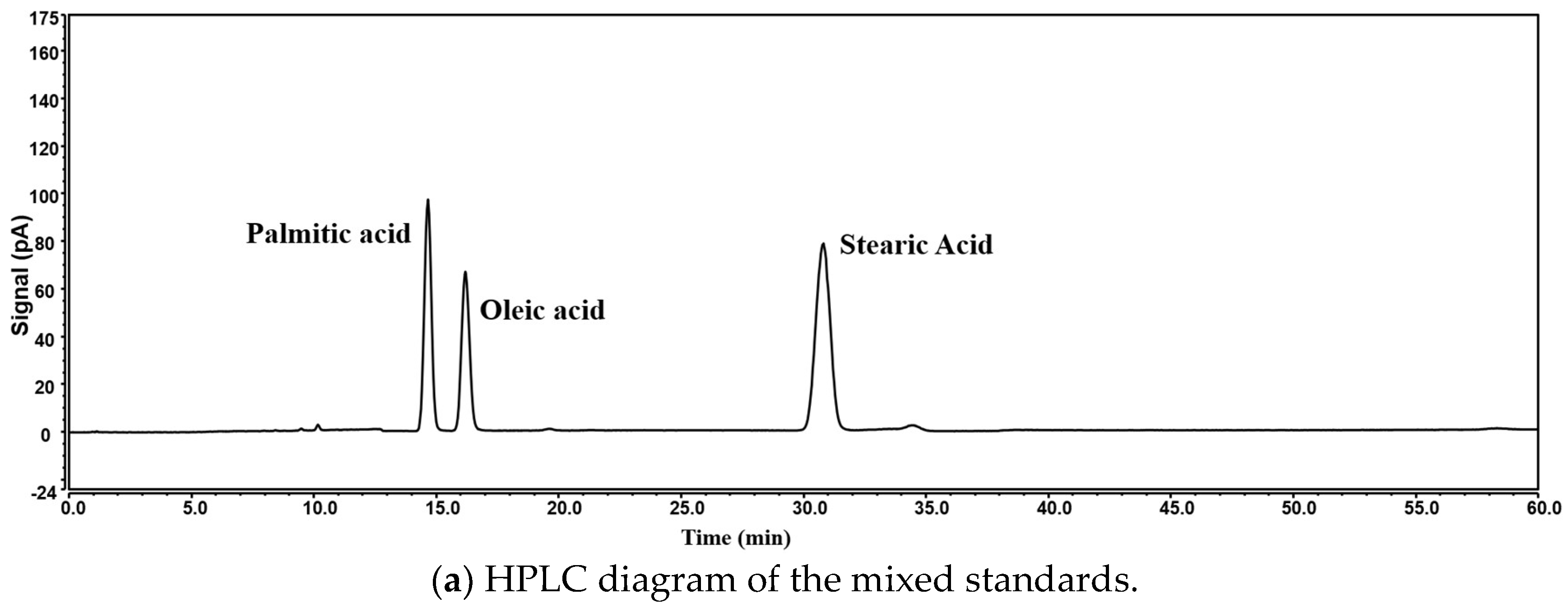
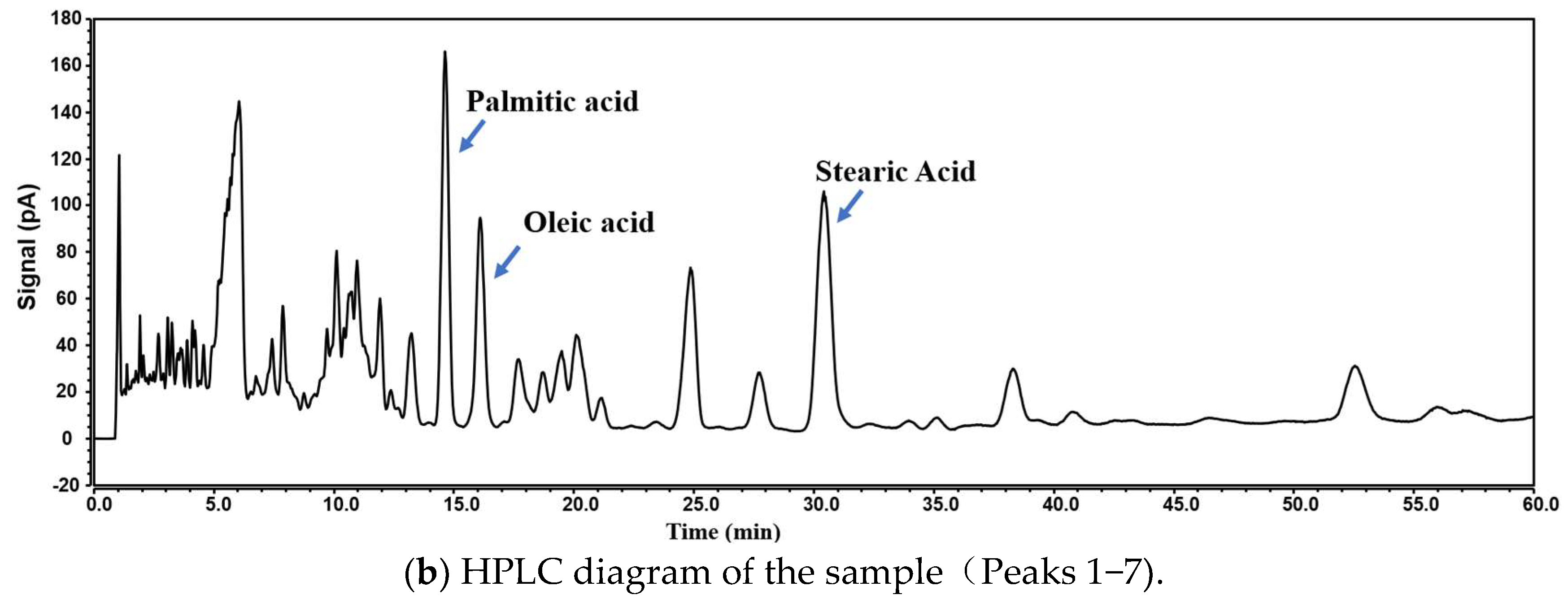
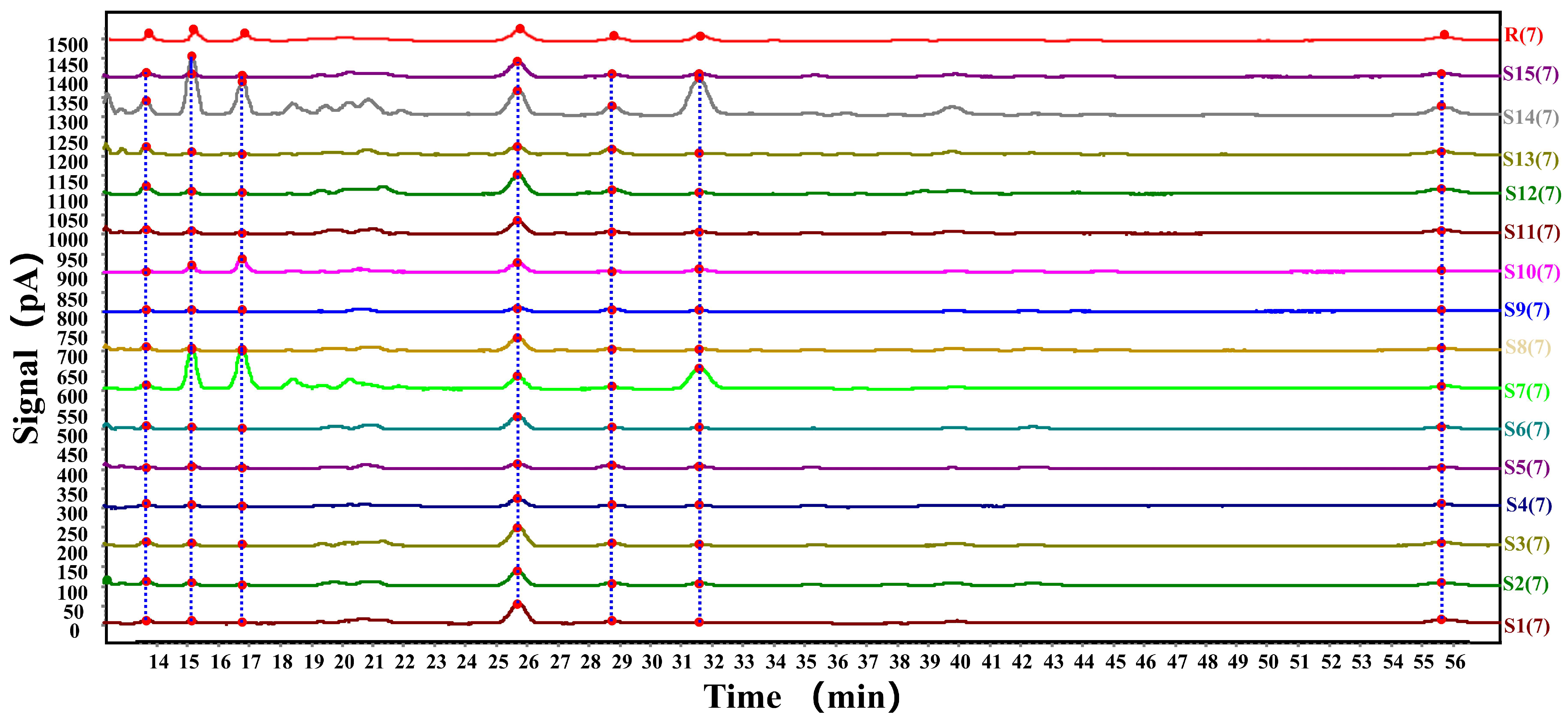
4.4. Fingerprinting Method Validation
4.5. Method Validation of Content Determination
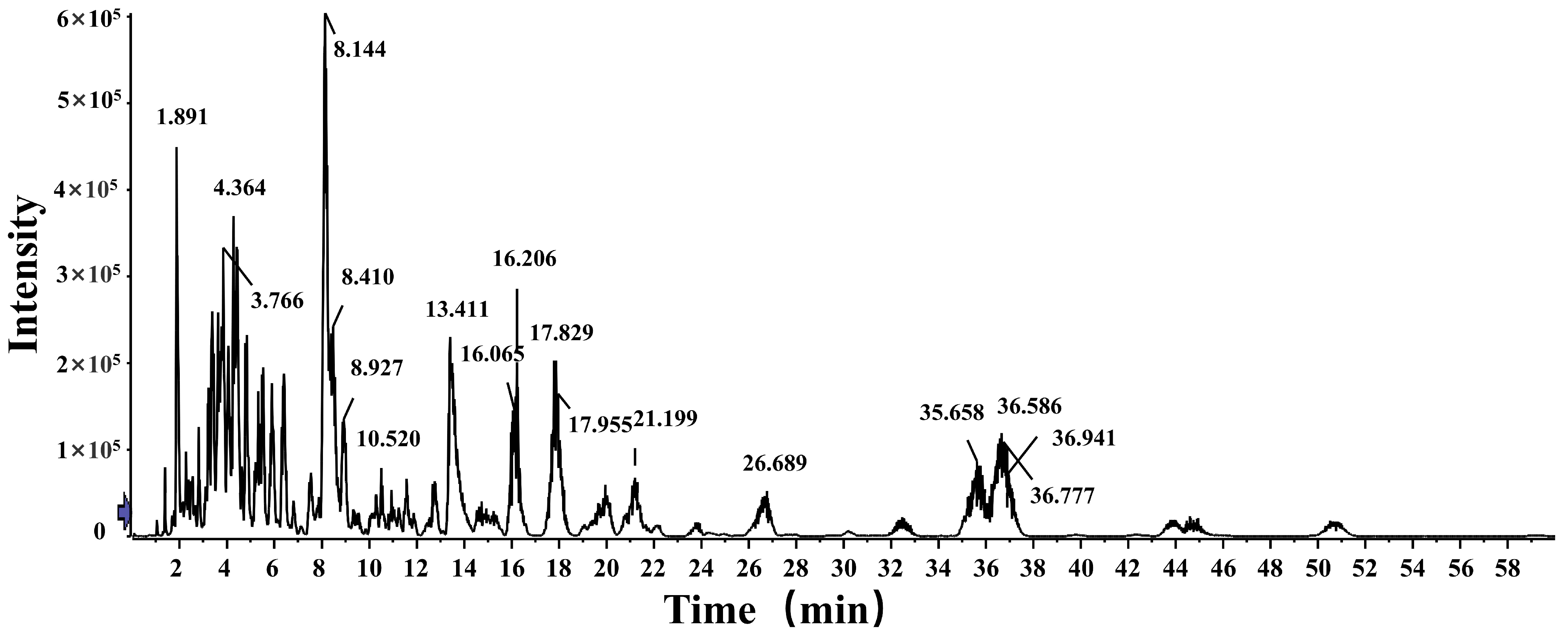
4.6. Fingerprint Analysis and Content Determination Results of Free Fatty Acids in Crude Lanolin
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Basso, J.; Mendes, M.; Cova, T.F.; Sousa, J.J.; Pais, A.A.; Vitorino, C. Analytical Quality by Design (AQbD) as a multiaddressable platform for co-encapsulating drug assays. Anal. Methods 2018, 10, 5659–5671. [Google Scholar]
- Ding, F.; Liu, S.; Wu, G.; Qu, H.; Gong, P.; Xie, Y.; Zhou, P.; Gong, X. A novel process for developing a quantitative chromatographic fingerprint analysis method based on analytical quality by design: A case study of Xiaochaihu capsules. Microchem. J. 2023, 194, 109253. [Google Scholar]
- Tai, Y.; Ren, D.; Zhao, W.; Qu, H.; Xiong, H.; Gong, X. Analytical quality by design oriented development of the UPLC method for analysing multiple pharmaceutical process intermediates: A case study of Compound Danshen Dripping Pills. Microchem. J. 2023, 187, 108438. [Google Scholar]
- Zhang, S.; Wu, L.; Wang, X.; Gong, X.; Qu, H. Development of an HPLC-MS method for the determination of four terpene trilactones in Ginkgo biloba leaf extract via quality by design. Biomed. Chromatogr. BMC 2021, 35, e5170. [Google Scholar]
- Lan, J.; Wu, G.; Wu, L.; Qu, H.; Gong, P.; Xie, Y.; Zhou, P.; Gong, X. Development of a Quantitative Chromatographic Fingerprint Analysis Method for Sugar Components of Xiaochaihu Capsules Based on Quality by Design Concept. Separations 2023, 10, 13. [Google Scholar]
- Zhao, M.; Tai, Y.; Wu, G.; Ding, F.; Qu, H.; Gong, X. Integrated Application of Risk Management Techniques in Developing an Analysis Method for Traditional Chinese Medicine: A Case Study of a Percolation Solution for Xiaochaihu Capsules. Chemosensors 2024, 12, 161. [Google Scholar] [CrossRef]
- Tambare, R.S.; Shahi, S.R.; Gurumukhi, V.C.; Kakade, S.M.; Tapadiya, G.G. Quality by design (QbD) based development and validation of RP-HPLC method for buserelin acetate in polymeric nanoparticles: Release study. Heliyon 2024, 10, e39172. [Google Scholar] [PubMed]
- Mohyeldin, S.M.; Daabees, H.G.; Talaat, W.; Kamal, M.F. Efficient integration of analytical quality-by-design and tri-hued HPLC-DAD approach for synchronized assessment of gemigliptin and rosuvastatin in their fixed-dose pills: Application to stability study. Sustain. Chem. Pharm. 2024, 42, 101777. [Google Scholar]
- Babaker, M.A.; Algohary, A.M.; Ibrahim, A.M. Robust analytical method for the enantiomeric separation of lurasidone hydrochloride: Integrating analytical quality by design and green chemistry principles. Sustain. Chem. Pharm. 2024, 42, 101788. [Google Scholar]
- Basso, J.; Ramos, M.L.; Pais, A.; Vitorino, R.; Fortuna, A.; Vitorino, C. Expediting Disulfiram Assays through a Systematic Analytical Quality by Design Approach. Chemosensors 2021, 9, 172. [Google Scholar] [CrossRef]
- Nompari, L.; Orlandini, S.; Pasquini, B.; Fontana, L.; Rovini, M.; Masi, F.; Gotti, R.; Furlanetto, S. Optimization of hydrolysis conditions of amino acid analysis for UHPLC-UV antigens content determination: Bexsero vaccine a case study. J. Pharm. Biomed. 2024, 241, 115997. [Google Scholar]
- Kochling, J.; Wu, W.; Hua, Y.; Guan, Q.; Castaneda-Merced, J. A platform analytical quality by design (AQbD) approach for multiple UHPLC-UV and UHPLC–MS methods development for protein analysis. J. Pharm. Biomed. Anal. 2016, 125, 130–139. [Google Scholar] [PubMed]
- Simeoni, P.; Deissler, M.; Bienert, R.; Gritsch, M.; Nerkamp, J.; Kirsch, S.; Roesli, C.; Pohl, T.; Anderka, O.; Gellermann, G. Using enhanced development tools offered by analytical Quality by Design to support switching of a quality control method. Biotechnol. Bioeng. 2023, 120, 3299–3310. [Google Scholar]
- Parr, M.K.; Schmidt, A.H. Life cycle management of analytical methods. J. Pharm. Biomed. Anal. 2018, 147, 506–517. [Google Scholar]
- International Conference on Harmonization (ICH). Continuous Manufacturing of Drug Substances and Drug Products Q13. Available online: https://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0 (accessed on 23 March 2025).
- International Conference on Harmonization (ICH). Analytical Procedure Development Q14. Available online: https://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0 (accessed on 23 March 2025).
- International Conference on Harmonization (ICH). Quality Risk Management Q9. Available online: https://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0 (accessed on 23 March 2025).
- Hassan, M.H.; Abou-Elkhair, R.A.; Hassan, A.E.; Mousa, H.S. Analytical quality by design-compliant development of quantitative 1H- and 19F-nuclear magnetic resonance methods for the analytical evaluation of the anti-cancer drug, sorafenib. Microchem. J. 2024, 208, 112544. [Google Scholar]
- Xuan, D.T.; Nguyen, H.M.T.; Hoang, V.D. Recent applications of analytical quality-by-design methodology for chromatographic analysis: A review. Chemom. Intell. Lab. Syst. 2024, 254, 105243. [Google Scholar]
- Rüttler, F.; Hammerschick, T.; Schlag, S.; Vetter, W. Isolation of lanosterol and dihydrolanosterol from the unsaponifiable matter of lanolin by urea complexation and countercurrent chromatography in heart-cut recycling mode. J. Chromatogr. B 2022, 1210, 123470. [Google Scholar]
- Moldovan, Z.; Jover, E.; Bayona, J.M. Gas chromatographic and mass spectrometric methods for the characterisation of long-chain fatty acids: Application to wool wax extracts. Anal. Chim. Acta 2002, 465, 359–378. [Google Scholar]
- Wang, B.; Chen, J.; Yv, Y.; Hu, Z.; Bi, Y. Physicochemical indexes and main components of lanolin. J. China Oils Fats 2022, 47, 22–27. [Google Scholar]
- Wu, J. Study on the refining method of lanolin. Chem. Manag. 2018, 9, 64–65. [Google Scholar]
- Danielle, D.; McCabe, M.; Bourdillon, K. The impact of refinement on the sensory properties of lanolin. J. Sens. Stud. 2023, 38, e12822. [Google Scholar] [CrossRef]
- Kunyk, O.; Leal Filho, W.; Pasichnyi, V. Study of the equivalence of substituting lanolin with a plant-based alternative in cosmetic products. Sustain. Chem. Pharm. 2024, 39, 101555. [Google Scholar] [CrossRef]
- Bourdillon, K.; McCausland, T.; McCabe, M. Multi-residue analysis of certain lanolin nipple care products for trace contaminants. BMC Chem. 2023, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Infante, V.H.P.; Lohan, S.B.; Schanzer, S.; Campos, P.M.B.G.M.; Lademann, J.; Meinke, M.C. Eco-friendly sunscreen formulation based on starches and PEG-75 lanolin increases the antioxidant capacity and the light scattering activity in the visible light. J. Photochem. Photobiol. B 2021, 222, 112264. [Google Scholar] [CrossRef]
- National Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; China Medical Science Press: Beijing, China, 2020. [Google Scholar]
- Cuadros-Rodríguez, L.; Ruiz-Samblás, C.; Valverde-Som, L.; Pérez-Castaño, E.; González-Casado, A. Chromatographic fingerprinting: An innovative approach for food ‘identitation’ and food authentication—A tutorial. Anal. Chim. Acta 2016, 909, 9–23. [Google Scholar] [CrossRef]
- Wolfender, J.L.; Marti, G.; Thomas, A.; Bertrand, S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J. Chromatogr. A 2015, 1382, 136–164. [Google Scholar]
- Liang, Y.-Z.; Xie, P.; Chan, K. Quality control of herbal medicines. J. Chromatogr. B 2004, 812, 53–70. [Google Scholar]
- Shao, J.Y.; Guo, J.L.; Guo, S.X.; Shu, Z.H.; Qu, H.B.; Gong, X.C. Quantitative chromatographic fingerprint analysis of Sanye Tangzhiqing Decoction based on quality by design concept. China J. Chin. Mater. Med. 2019, 44, 4844–4851. [Google Scholar]
- Chen, Z.; Li, Q.; Sun, J.; Cao, F.; Li, C. A New Calculation Method for Peak Width and Resolution of Trailing Peaks in Chromatograms and Its Application. Chin. J. Pharm. 2023, 54, 1374–1381. [Google Scholar]
- Gong, X.; Li, Y.; Chen, H.; Qu, H. Design space development for the extraction process of Danhong injection using a Monte Carlo simulation method. PLoS ONE 2015, 10, e0128236. [Google Scholar]
- Shalliker, R.; Stevenson, P.; Shock, D.; Mnatsakanyan, M.; Dasgupta, P.; Guiochon, G. Application of power functions to chromatographic data for the enhancement of signal to noise ratios and separation resolution. J. Chromatogr. A 2010, 1217, 5693–5699. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor Name | Symbol | Unit | Level | ||
|---|---|---|---|---|---|
| Low | Middle | High | |||
| Column Temperature | X1 | °C | 30 | 35 | 40 |
| Flow Rate | X2 | mL/min | 0.7 | 0.8 | 0.9 |
| Isocratic Time | X3 | min | 25 | 30 | 35 |
| Gradient End Organic Phase Ratio | X4 | % | 78 | 80 | 82 |
| Gradient Time | X5 | min | 30 | 35 | 40 |
| Evaluation Indicators | Low Limit | High Limit |
|---|---|---|
| Y1: Retention time difference between Peak 2 and Peak 1 (min) | 1.100 | — |
| Y2: Retention time difference between Peak 3 and Peak 2 (min) | 1.200 | — |
| Y3: Retention time difference between Peak 5 and Peak 4 (min) | 2.500 | — |
| Y4: Retention time difference between Peak 6 and Peak 5 (min) | 2.500 | — |
| Y5: Retention time of Peak 7 (min) | — | 60.000 |
| Y6: Tailing factor of Peak 1 | 0.9 | 1.1 |
| Y7: Tailing factor of Peak 6 | 0.9 | 1.1 |
| Validation Points | Within the MODR | Outside the MODR | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| Experimental Conditions | X1 (°C) | 35 | 35 | 40 |
| X2 (mL/min) | 0.85 | 0.8 | 0.85 | |
| X3 (min) | 32 | 30 | 26 | |
| X4 (%) | 79 | 81 | 79 | |
| X5 (min) | 30 | 30 | 30 | |
| Experimental Results | Y1 | 1.34 | 1.45 | 1.068 |
| Y2 | 1.355 | 1.475 | 1.252 | |
| Y3 | 2.78 | 2.982 | 2.276 | |
| Y4 | 2.528 | 2.796 | 2.065 | |
| Y5 | 51.965 | 53.005 | 44.978 | |
| Y6 | 1 | 1.04 | 0.9 | |
| Y7 | 1.07 | 0.98 | 1.25 | |
| Batches Number | Palmitic Acid (mg/g Lanolin) | Oleic Acid (mg/g Lanolin) | Stearic Acid (mg/g Lanolin) |
|---|---|---|---|
| S1 | 0.584 | 0.171 | 0.261 |
| S2 | 0.563 | 0.151 | 0.301 |
| S3 | 0.608 | 0.123 | 0.246 |
| S4 | 0.571 | 0.071 | 0.260 |
| S5 | 0.352 | 0.135 | 0.324 |
| S6 | 0.378 | 0.059 | 0.227 |
| S7 | 11.46 | 12.64 | 5.997 |
| S8 | 0.390 | 0.053 | 0.195 |
| S9 | 0.653 | 0.223 | 0.349 |
| S10 | 1.563 | 3.296 | 0.750 |
| S11 | 0.540 | 0.077 | 0.263 |
| S12 | 0.814 | 0.257 | 0.257 |
| S13 | 0.558 | 0.075 | 0.440 |
| S14 | 14.61 | 8.428 | 9.189 |
| S15 | 0.661 | 0.099 | 0.492 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Wu, S.; Zhang, H.; Gong, X. Developing a Quantitative Profiling Method for Detecting Free Fatty Acids in Crude Lanolin Based on Analytical Quality by Design. Chemosensors 2025, 13, 126. https://doi.org/10.3390/chemosensors13040126
Liu S, Wu S, Zhang H, Gong X. Developing a Quantitative Profiling Method for Detecting Free Fatty Acids in Crude Lanolin Based on Analytical Quality by Design. Chemosensors. 2025; 13(4):126. https://doi.org/10.3390/chemosensors13040126
Chicago/Turabian StyleLiu, Sihan, Shaohua Wu, Hao Zhang, and Xingchu Gong. 2025. "Developing a Quantitative Profiling Method for Detecting Free Fatty Acids in Crude Lanolin Based on Analytical Quality by Design" Chemosensors 13, no. 4: 126. https://doi.org/10.3390/chemosensors13040126
APA StyleLiu, S., Wu, S., Zhang, H., & Gong, X. (2025). Developing a Quantitative Profiling Method for Detecting Free Fatty Acids in Crude Lanolin Based on Analytical Quality by Design. Chemosensors, 13(4), 126. https://doi.org/10.3390/chemosensors13040126