
A Review on Initialization Methods for Nonnegative Matrix Factorization: Towards Omics Data Experiments


Abstract
:1. Introduction
2. How Important Are Initializations for NMF?
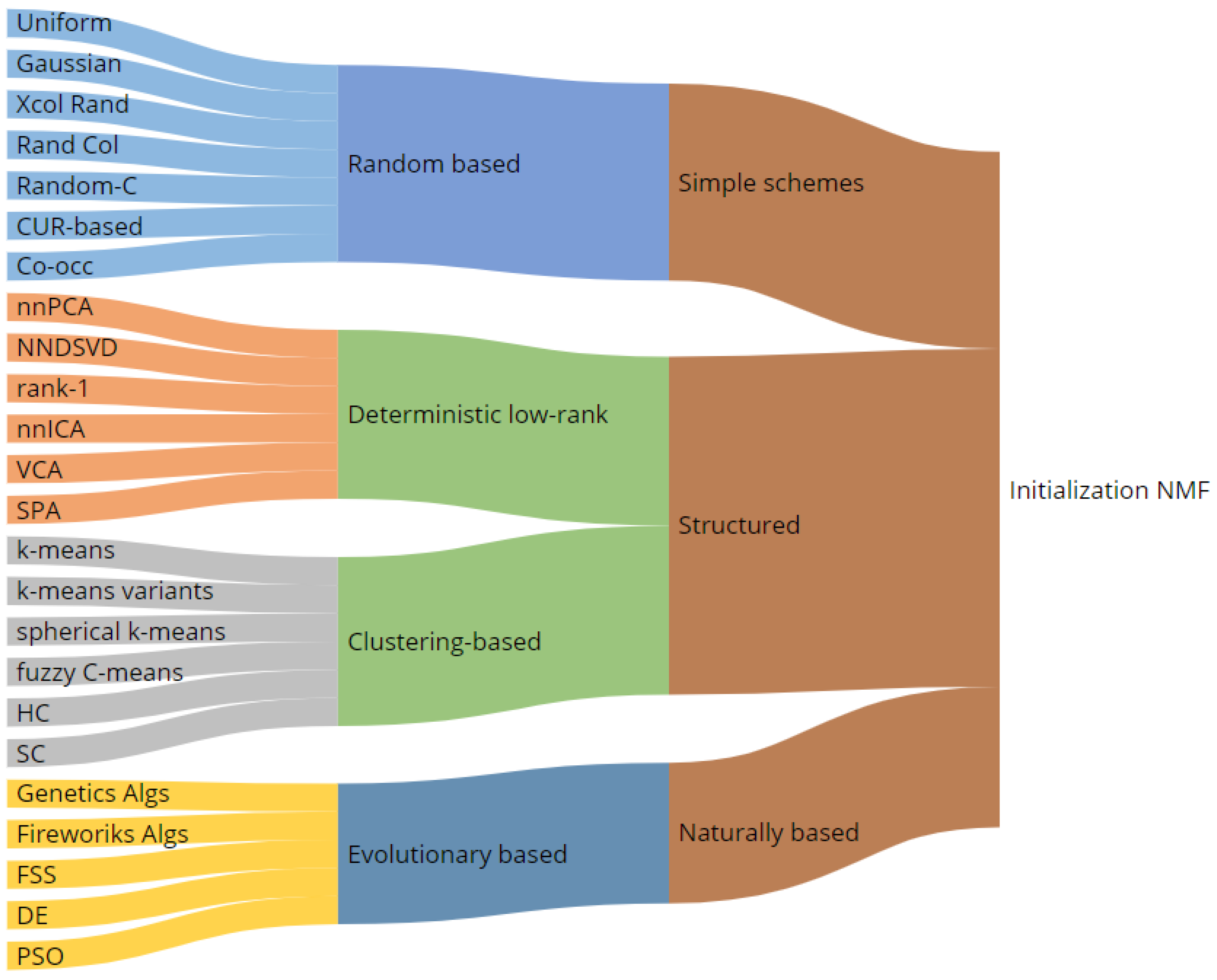
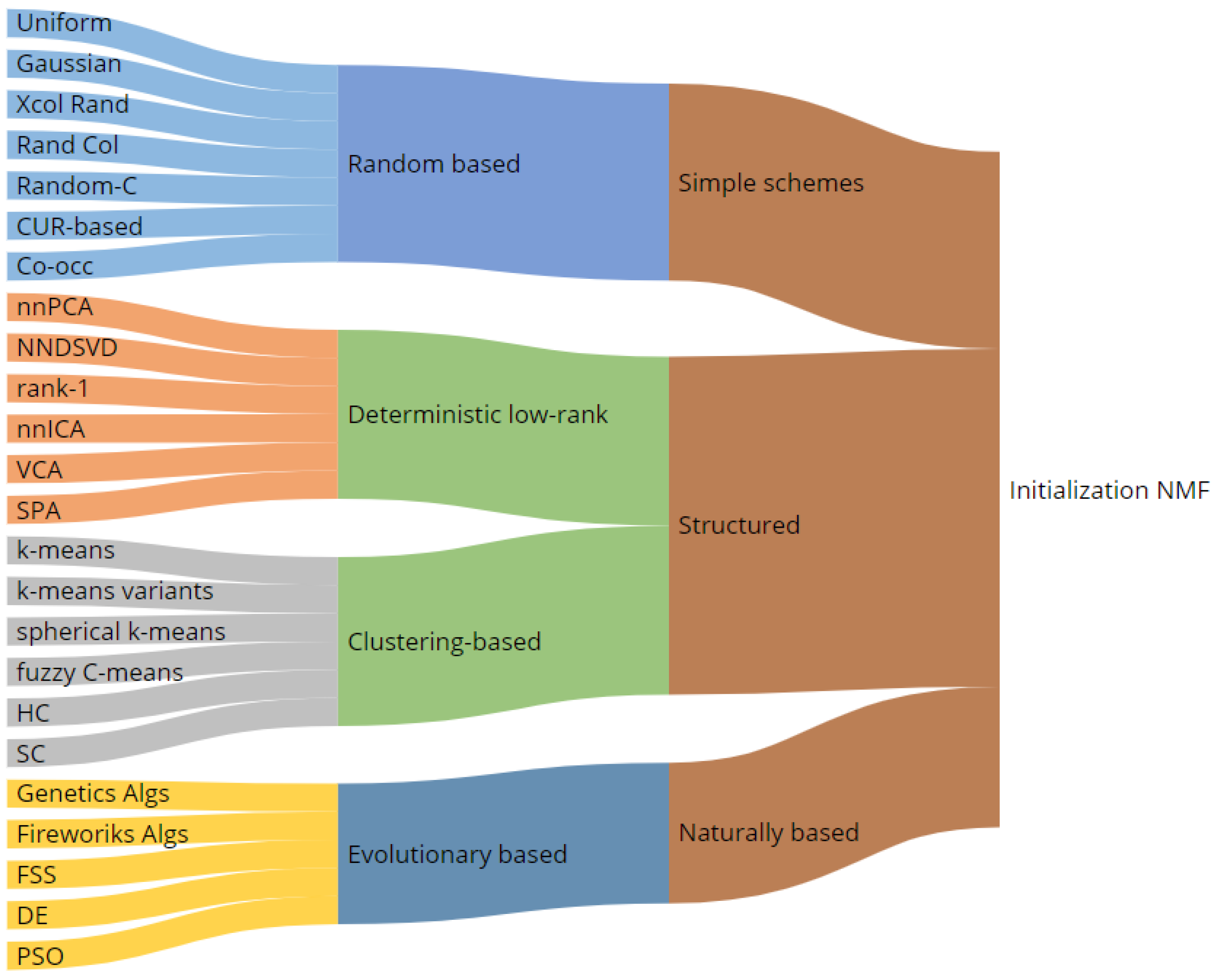
3. NMF Iterative Algorithms and a Complete Taxonomy of Initialization Mechanisms
- -
- Set and W equals to any nonnegative ;
- -
- With fixed , update ;
- -
- With fixed , update ;
- -
- Iterate until a stopping criteria is satisfied.
3.1. Random Based Initializations
- Uniform random: the elements in the matrix W (and in H) are chosen as uniformly distributed numbers in the interval or in the same range as the entries of the target matrix.
- Gaussian random: the elements in the matrix W (and in H) are chosen as , where the number is a Gaussian values.
- Random : the factor matrix W is obtained by averaging over r randomly selected columns of the data matrix X. This scheme is very inexpensive and has the advantage of yielding a sparser factor when the data matrix is already sparse. When dealing with omic data, selected columns of X can be considered as a kind of a priori information that can influence the following update process.
- Random-C initialization: this mechanism is based on a double random selection of columns in the data matrix X as follows:
- -
- Identify p of the longest (in the 2-norm sense) columns of X;
- -
- Randomly choose q columns from the previous p longest;
- -
- Construct each column of W as the average of these q columns.
This scheme is inspired by the C-matrix of the CUR decomposition and produces the densest W, in which column vectors are closed with the centroids of the data matrix (see spherical-k means initialization) [49]. - CUR-based initialization: the factor matrix W is constructed as a submatrix (with a small number of actual columns) of the data matrix X. In this way, values are obtained that are more interpretable from a biological point of view (and usually to the same extent as the original data) [50,51]. CUR-based mechanisms differ in the “statistical” way of selecting columns (or rows, if we refer to the initialization of the factor H) from the matrix X. This selection is based on computing an “importance score” for each column (row) of X and randomly selecting r of these columns (rows) using this score as the probability distribution for importance sampling. The scoring values of a column in X can be calculated as: normalized statistical leverage score [50], spectral angular distance, or symmetrized KL-divergence [52].
- Co-occurrence initialization: firstly the data co-occurrence matrix (i.e., ) is formed and then r of its columns are selected by the algorithm proposed in [53] to form the factor W. The data co-occurrence matrix contains information about hidden relationships between columns and rows in the data matrix X, but the computational costs required for this make this initialization mechanism expensive and often impractical for use in the context of biomedical data analysis where large datasets are considered.
3.2. Structured Initialization
3.2.1. Non-Negative Double Singular Value Decomposition
- -
- Compute the largest r singular triplets of X (first SVD process);
- -
- Initialize the first column and row vectors in W and H as the nonnegative dominant singular vectors of X weighted by ;
- -
- Compute the positive section of each ;
- -
- Compute the largest r singular triplets of (second SVD process);
- -
- Initialize the j-th columns and rows in W and H, for as the singular dominant vectors of each , weighted by the and normalized.
3.2.2. Nonnegative ICA Initialization
- -
- Compute the ICA on the observed data matrix X;
- -
- Initialized the factor W as the absolute of the independent components source matrix obtained from ICA.
3.2.3. k-Means Initialization
- -
- Initialize k centroids randomly choosing some columns in X and set (this is the initial iteration) ;
- -
- Compute , and ;
- -
- Define the new partition of clusters ;
- -
- Recompute each centroid as: ;
- -
- Initialize the columns of the factor W as the final cluster centroid vectors.
3.3. Evolutionary and Natural Based Initialization
4. How Many Initializations Influence NMF Results for Omic Data Analysis?
5. Conclusive Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NMF | Nonnegative matrix factorizations |
| SVD | Singular Value Decomposition |
| PCA | Principal Component Analysis |
| KL | Kullback-Leiber |
| MU | Mutilplicative Update |
| MM | Majorise-Minimize |
| nnPCA | Nonnegative PCA |
| ICA | Indipendent Component Analysis |
| nnICA | nonnegative ICA |
| NNDSVD | Nonnegative Double SVD |
| VCA | Vertex Complex Analysis |
| SPA | Successive Projection Algorithm |
| HC | Hierarchical Clustering |
| SC | Subtracting Clustering |
| FSS | Fisch School Search |
| DE | Differential Evolution |
| Fireworks Algs | Fireworks Algorithms |
| PSO | Particle Swarm Optimization |
References
- Yamada, R.; Okada, D.; Wang, J.; Basak, T.; Koyama, S. Interpretation of omics data analyses. J. Hum. Genet. 2021, 66, 93–102. [Google Scholar] [CrossRef]
- Nicora, G.; Vitali, F.; Dagliati, A.; Geifman, N.; Bellazzi, R. Integrated Multi-Omics Analyses in Oncology: A Review of Machine Learning Methods and Tools. Front. Oncol. 2020, 10, 1030. [Google Scholar] [CrossRef] [PubMed]
- Stein-O’Brien, G.L.; Arora, R.; Culhane, A.C.; Favorov, A.V.; Garmire, L.X.; Greene, C.S.; Goff, L.A.; Li, Y.; Ngom, A.; Ochs, M.F.; et al. Enter the Matrix: Factorization Uncovers Knowledge from Omics. Trends Genet. 2018, 34, 790–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kossenkov, A.V.; Ochs, M.F. Matrix factorisation methods applied in microarray data analysis. Int. J. Data Min. Bioinform. 2010, 4, 72–90. [Google Scholar] [CrossRef]
- Devarajan, K. Nonnegative matrix factorization: An analytical and interpretive tool in computational biology. PLoS Comput. Biol. 2008, 4, e1000029. [Google Scholar] [CrossRef] [PubMed]
- Moloshok, T.; Klevecz, R.; Grant, J.; Manion, F.; Speier, W.; Ochs, M. Application of Bayesian Decomposition for analysing microarray data. Bioinformatics 2002, 18, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Saidi, S.A.; Holland, C.M.; Kreil, D.P.; MacKay, D.J.; Charnock-Jones, D.S.; Print, C.G.; Smith, S.K. Independent component analysis of microarray data in the study of endometrial cancer. Oncogene 2004, 23, 6677. [Google Scholar] [CrossRef] [Green Version]
- Alter, O.; Brown, P.O.; Botstein, D. Singular value decomposition for genome-wide expression data processing and modeling. Proc. Natl. Acad. Sci. USA 2000, 97, 10101–10106. [Google Scholar] [CrossRef] [Green Version]
- Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. USA 2004, 101, 4164–4169. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.J.; Lieu, L.; Rocke, D. Dimension reduction for classification with gene expression microarray data. Stat. Appl. Genet. Mol. Biol. 2006, 5, 6. [Google Scholar] [CrossRef]
- Devarajan, K.; Ebrahimi, N. Class Discovery via Nonnegative Matrix Factorization. Am. J. Math. Manag. Sci. 2008, 28, 457–467. [Google Scholar] [CrossRef]
- Kong, W.; Mou, X.; Hu, X. Exploring Matrix Factorization Techniques for Significant Genes Identification of Alzheimer’s Disease Microarray Gene Expression Data; BMC bioinformatics; BioMed Central: London, UK, 2011; Volume 12, p. S7. [Google Scholar]
- Ochs, M.F.; Fertig, E.J. Matrix Factorization for Transcriptional Regulatory Network Inference. In Proceedings of the IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology, San Diego, CA, USA, 9–12 May 2012; pp. 387–396. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Zeleznik, O.A.; Thallinger, G.G.; Kuster, B.; Gholami, A.M.; Culhane, A.C. Dimension reduction techniques for the integrative analysis of multi-omics data. Briefings Bioinform. 2016, 17, 628–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Wang, D.; Gao, Y.L.; Zheng, C.H.; Xu, Y.; Yu, J. Regularized non-negative matrix factorization for identifying differential genes and clustering samples: A survey. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 15, 974–987. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, F.-X.; Ngom, A. A review on machine learning principles for multi-view biological data integration. Brief Bioinform 2018, 19, 325–340. [Google Scholar] [CrossRef]
- Wall, M.E.; Rechtsteiner, A.; Rocha, L.M. Singular Value Decomposition and Principal Component Analysis. In A Practical Approach to Microarray Data Analysis; Berrar, D.P., Dubitzky, W., Granzow, M., Eds.; Springer: Boston, MA, USA, 2003; pp. 91–109. [Google Scholar] [CrossRef] [Green Version]
- Sompairac, N.; Nazarov, P.V.; Czerwinska, U.; Cantini, L.; Biton, A.; Molkenov, A.; Zhumadilov, Z.; Barillot, E.; Radvanyi, F.; Gorban, A.; et al. Independent Component Analysis for Unraveling the Complexity of Cancer Omics. Int. J. Mol. Sci. 2019, 20, 4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Michailidis, G. A Non-negative Matrix Factorization Method for Detecting Modules in Heterogeneous Omics Multi-modal Data. Bioinformatics 2015, 32. [Google Scholar] [CrossRef] [Green Version]
- Boccarelli, A.; Esposito, F.; Coluccia, M.; Frassanito, M.A.; Vacca, A.; Del Buono, N. Improving knowledge on the activation of bone marrow fibroblasts in MGUS and MM disease through the automatic extraction of genes via a Nonnegative Matrix Factorization approach on gene expression profiles. J. Transl. Med. 2018, 16, 217. [Google Scholar] [CrossRef]
- Rappoport, N.; Shamir, R. Multi-omic and multi-view clustering algorithms: Review and cancer benchmark. Nucleic Acids Res. 2018, 46, 10546–10562. [Google Scholar] [CrossRef]
- Esposito, F.; Gillis, N.; Del Buono, N. Orthogonal joint sparse NMF for microarray data analysis. J. Math. Biol. 2019, 79, 223–247. [Google Scholar] [CrossRef]
- Esposito, F.; Del Buono, N.; Selicato, L. Nonnegative Matrix Factorization models for knowledge extraction from biomedical and other real world data. PAMM 2021, 20, e202000032. [Google Scholar] [CrossRef]
- Lee, D.D.; Seung, H.S. Algorithms for Non-negative Matrix Factorization. In Proceedings of the Advances in Neural Information Processing Systems Conference; MIT Press: Cambridge, MA, USA, 2000; Volume 13, pp. 556–562. [Google Scholar]
- Del Buono, N.; Esposito, F.; Fumarola, F.; Boccarelli, A.; Coluccia, M. Breast Cancer’s Microarray Data: Pattern Discovery Using Nonnegative Matrix Factorizations. In International Workshop on Machine Learning, Optimization and Big Data; Springer: Berlin, Germany, 2016; pp. 281–292. [Google Scholar]
- Gillis, N. Nonnegative Matrix Factorization; SIAM: Philadelphia, PA, USA, 2020. [Google Scholar]
- Del Buono, N.; Esposito, F. Investigating initialization techniques for Nonnegative Matrix Factorization: A survey and a case of study of microarray. In Molecular and Mathematical Biology, Chemistry, Medicine and Medical Statistics, Bioinformatics and Numerical Analysi (Series in Applied Sciences); Carletti, M., Spaletta, G., Eds.; Universitas Studiorum: Mantova, Italy, 2019; Volume 2. [Google Scholar] [CrossRef]
- Casalino, G.; Del Buono, N.; Mencar, C. Subtractive clustering for seeding non-negative matrix factorizations. Inf. Sci. 2014, 257, 369–387. [Google Scholar] [CrossRef]
- Vavasis, S. On the Complexity of Nonnegative Matrix Factorization. SIAM J. Optim. 2010, 20, 1364–1377. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef]
- Yang, Z.; Oja, E. Unified Development of Multiplicative Algorithms for Linear and Quadratic Nonnegative Matrix Factorization. IEEE Trans. Neural Netw. 2011, 22, 1878–1891. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Tan, V.Y.F. A Unified Convergence Analysis of the Multiplicative Update Algorithm for Regularized Nonnegative Matrix Factorization. IEEE Trans. Signal Process. 2018, 66, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Park, H. Nonnegative Matrix Factorization Based on Alternating Nonnegativity Constrained Least Squares and Active Set Method. SIAM J. Matrix Anal. Appl. 2008, 30, 713–730. [Google Scholar] [CrossRef]
- Chen, D.; Plemmons, R. Nonnegativity constraints in numerical analysis. In Symposium on the Birth of Numerical Analysis; Bultheel, A., Cools, R., Eds.; World Scientific Press: Singapore, 2009. [Google Scholar]
- Gillis, N.; Glineur, F. A multilevel approach for nonnegative matrix factorization. J. Comput. Appl. Math. 2012, 236, 1708–1723. [Google Scholar] [CrossRef] [Green Version]
- Lin, C. Projected Gradient Methods for Nonnegative Matrix Factorization. Neural Comput. 2007, 19, 2756–2779. [Google Scholar] [CrossRef] [Green Version]
- Donoho, D.; Stodden, V. When Does Non-negative Matrix Factorization Give a Correct Decomposition into Parts? In NIPS’03 Proceedings of the 16th International Conference on Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2003; pp. 1141–1148. [Google Scholar]
- Fogel, P.; Hawkins, D.M.; Beecher, C.; Luta, G.; Young, S.S. A Tale of Two Matrix Factorizations. Am. Stat. 2013, 67, 207–218. [Google Scholar] [CrossRef]
- Zhaoqiang, L.; Tan, V.Y.F. Rank-One NMF-Based Initialization for NMF and Relative Error Bounds Under a Geometric Assumption. IEEE Trans. Signal Process. 2017, 65, 4717–4731. [Google Scholar]
- Rezaei, M.; Boostani, R.; Rezaei, M. An Efficient Initialization Method for Nonnegative Matrix Factorization. J. Appl. Sci. 2011, 11, 354–359. [Google Scholar] [CrossRef]
- Kitamura, D.; Ono, N. Efficient initialization for nonnegative matrix factorization based on nonnegative independent component analysis. In Proceedings of the 2016 IEEE International Workshop on Acoustic Signal Enhancement (IWAENC), Xi’an, China, 13–16 September 2016; pp. 1–5. [Google Scholar] [CrossRef]
- Chalise, P.; Fridley, L. Integrative clustering of multi-level ‘omic data based on non-negative matrix factorization algorithm. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutsidis, C.; Gallopoulos, E. SVD based initialization: A head start for nonnegative matrix factorization. Pattern Recognit. 2008, 41, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Welch, J.D. Iterative Refinement of Cellular Identity from Single-Cell Data Using Online Learning. In Research in Computational Molecular Biology; Schwartz, R., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 248–250. [Google Scholar]
- Chalise, P.; Ni, Y.; Fridley, B.L. Network-based integrative clustering of multiple types of genomic data using non-negative matrix factorization. Comput. Biol. Med. 2020, 118, 103625. [Google Scholar] [CrossRef]
- Hobolth, A.; Guo, Q.; Kousholt, A.; Jensen, J.L. A Unifying Framework and Comparison of Algorithms for Non-negative Matrix Factorisation. Int. Stat. Rev. 2020, 88, 29–53. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; He, Y.; Park, H. Algorithms for nonnegative matrix and tensor factorizations: A unified view based on block coordinate descent framework. J. Glob. Optimation 2014, 58, 285–319. [Google Scholar] [CrossRef] [Green Version]
- Fevotte, C.; Idier, J. Algorithms for Nonnegative Matrix Factorization with the β-Divergence. Neural Comput. 2011, 23, 2421–2456. [Google Scholar] [CrossRef]
- Langville, A.; Meyer, C.D.; Albright, R. Initializations for the nonnegative matrix factorization. In Proceedings of the Twelfth ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Philadelphia, PA, USA, 20–23 August 2006. [Google Scholar]
- Mahoney, M.; Drineas, P. CUR matrix decompositions for improved data analysi. Proc. Natl. Acad. Sci. USA 2009, 106, 697–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwowar, M.; Kocemba-Pilarczyk, K.; Piwowar, P. Regularization and grouping-omics data by GCA method: A transcriptomic case. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Gengxin, Z.; Xinpeng, D. CUR Based Initialization Strategy for Non-Negative Matrix Factorization in Application to Hyperspectral Unmixing. J. Appl. Math. Phys. 2016, 4, 614. [Google Scholar]
- Sandler, M. On the Use of Linear Programming for Unsupervised Text Classification. In KDD ’05 Proceedings of the Eleventh ACM SIGKDD International Conference on Knowledge Discovery in Data Mining; ACM: New York, NY, USA, 2005; pp. 256–264. [Google Scholar] [CrossRef] [Green Version]
- Ewert, S.; Muller, M. Using score-informed constraints for NMF-based source separation. In Proceedings of the IEEE International Conference on Acoustics, Speech, and Signal Processing (ICASSP), Kyoto, Japan, 25–30 March 2012. [Google Scholar]
- Fritsch, J.; Plumbley, M.D. Score informed audio source separation using constrained nonnegative matrix factorization and score synthesis. In Proceedings of the 2013 IEEE International Conference on Acoustics, Speech and Signal Processing, Vancouver, BC, Canada, 26–31 May 2013; pp. 888–891. [Google Scholar] [CrossRef] [Green Version]
- Rohlfing, C.; Becker, J.M. Extended semantic initialization for NMF-based audio source separation. In Proceedings of the 2015 International Symposium on Intelligent Signal Processing and Communication Systems (ISPACS), Nusa Dua, Bali, Indonesia, 9–12 November 2015; pp. 95–100. [Google Scholar] [CrossRef]
- Zdunek, R. Initialization of Nonnegative Matrix Factorization with Vertices of Convex Polytope. In Artificial Intelligence and Soft Computing; ICAISC 2012; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2012; Volume 7267. [Google Scholar]
- Julian Mathias, B.; Matthias, M.; Christian, R. Complex SVD Initialization for NMF Source Separation on Audio Spectrograms. In Proceedings of the Deutsche Jahrestagung fur Akustik (DAGA), Nuremberg, Germany, 16–19 March 2015. [Google Scholar]
- Atif, S.; Qazi, S.; Gillis, N. Improved SVD-based initialization for nonnegative matrix factorization using low-rank correction. Pattern Recognit. Lett. 2019, 122. [Google Scholar] [CrossRef] [Green Version]
- Biggs, M.; Ghodsi, A.; Vavasis, S. Nonnegative Matrix Factorization via Rank-One Downdate. In Proceedings of the 25th International Conference on Machine Learning, Helsinki, Finland, 5–9 July 2008. [Google Scholar] [CrossRef] [Green Version]
- Xuansheng Wang, X.X.; Lu, L. An Effective Initialization for Orthogonal Nonnegative Matrix Factorization. J. Comput. Math. 2012, 30, 34–46. [Google Scholar] [CrossRef]
- Zhao, L.; Zhuang, G.; Xu, X. Facial expression recognition based on PCA and NMF. In Proceedings of the 2008 7th World Congress on Intelligent Control and Automation, Chongqing, China, 25–27 June 2008; pp. 6826–6829. [Google Scholar]
- Xiu-rui, G.; Lu-yan, J.; Kang, S. Non-negative matrix factorization based unmixing for principal component transformed hyperspectral data. Front. Inf. Technol. Electron. Eng. 2016, 17, 403–412. [Google Scholar] [CrossRef]
- Oja, E.; Plumbley, M. Blind Separation of Positive Sources by Globally Convergent Gradient Search. Neural Comput. 2004, 16, 1811–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, J.M.P.; Dias, J.M.B. Vertex component analysis: A fast algorithm to unmix hyperspectral data. IEEE Trans. Geosci. Remote. Sens. 2005, 43, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Zhenwei, S.; Zhenyu, A. Nonnegative matrix factorization for hyperspectral unmixing using prior knowledge of spectral signatures. Opt. Eng. 2012, 51, 1–10. [Google Scholar] [CrossRef]
- Cao, J.; Lilian, Z.; Haiyan, T. An Endmember Initialization Scheme for Nonnegative Matrix Factorization and Its Application in Hyperspectral Unmixing. ISPRS Int. J. Geo-Inf. 2018, 7, 195. [Google Scholar] [CrossRef] [Green Version]
- Sauwen, N.; Acou, M.; Halandur, N.; Bharath, D.M.; Sima, J.V.; Maes, F.; Himmelreich, U.; Achten, E.; Van Huffel, S. The successive projection algorithm as an initialization method for brain tumor segmentation using non-negative matrix factorization. PLoS ONE 2017, 12, e0180268. [Google Scholar] [CrossRef] [PubMed]
- Selicato, L.; Del Buono, N.; Esposito, F. Methods for Hyperparameters Optimization in Learning Approaches: An overview. In Machine Learning, Optimization, and Data Science. LOD 2020. Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2020; Volume 12514. [Google Scholar] [CrossRef]
- Cichocki, A.; Zdunek, R.; Phan, A.H.; Amari, S. Nonnegative Matrix and Tensor Factorizations: Applications to Exploratory Multi-Way Data Analysis and Blind Source Separation; Wiley: New York, NY, USA, 2009. [Google Scholar]
- Gong, L.; Nandi, A.K. An enhanced initialization method for non-negative matrix factorization. In Proceedings of the 2013 IEEE International Workshop on Machine Learning for Signal Processing (MLSP), Southampton, UK, 22–25 September 2013; pp. 1–6. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Sze Tong, C.; Chen, Y.; Chen, W.S. Clustering-based initialization for non-negative matrix factorization. Appl. Math. Comput. 2008, 205, 525–536, Special Issue on Advanced Intelligent Computing Theory and Methodology in Applied Mathematics and Computation. [Google Scholar] [CrossRef]
- Wild, S. Seeding Non-Negative Matrix Factorizations with the Spherical K-Means Clustering. Ph.D. Thesis, University of Colorado, Denver, CO, USA, 2003. [Google Scholar]
- Wild, S.; Curry, J.; Dougherty, A. Improving non-negative matrix factorizations through structured initialization. Pattern Recognit. 2004, 37, 2217–2232. [Google Scholar] [CrossRef]
- Zheng, Z.; Yang, J.; Zhu, Y. Initialization enhancer for non-negative matrix factorization. Eng. Appl. Artif. Intell. 2007, 20, 101–110. [Google Scholar] [CrossRef]
- Kim, Y.D.; Choi, S. A Method of Initialization for Nonnegative Matrix Factorization. In Proceedings of the 2007 IEEE International Conference on Acoustics, Speech and Signal Processing-ICASSP ’07, Honolulu, HI, USA, 15–20 April 2007; Volume 2, pp. 537–540. [Google Scholar]
- Djaouad, B.; Shahram, H.; Yannick, D.; Moussa, K.; Abdelkader, H. Modified Independent Component Analysis for Initializing Non-negative Matrix Factorization: An approach to Hyperspectral Image Unmixing. In Proceedings of the International Workshop on Electronics, Control, Modelling, Measurement and Signals (ECMS 2013), Toulouse, France, 24–26 June 2013; pp. 1–6. [Google Scholar]
- Alshabrawy, O.S.; Ghoneim, M.E.; Awad, W.A.; Hassanien, A.E. Underdetermined blind source separation based on Fuzzy C-Means and Semi-Nonnegative Matrix Factorization. In Proceedings of the 2012 Federated Conference on Computer Science and Information Systems (FedCSIS), Wroclaw, Poland, 9–12 September 2012; pp. 695–700. [Google Scholar]
- Suleman, A. On ill-conceived initialization in archetypal analysis. Adv. Data Anal. Classif. 2017, 11, 785–808. [Google Scholar] [CrossRef]
- Mejia Roa, E.; Carmona-Saez, P.; Nogales-Cadenas, R.; Vicente, C.; Vazquez, M.; Yang, X.; Garcia, C.; Tirado, F.; Pascual-Montano, A. BioNMF: A web-based tool for nonnegative matrix factorization in biology. Nucleic Acids Res. 2008, 36, W523–W528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef] [Green Version]
- Janecek, A.; Tan, Y. Iterative improvement of the Multiplicative Update NMF algorithm using nature-inspired optimization. In Proceedings of the 2011 Seventh International Conference on Natural Computation, Shanghai, China, 26–28 July 2011; Volume 3, pp. 1668–1672. [Google Scholar] [CrossRef]
- Janecek, A.; Tan, Y. Using Population Based Algorithms for Initializing Nonnegative Matrix Factorization. In Advances in Swarm Intelligence; Tan, Y., Shi, Y., Chai, Y., Wang, G., Eds.; ICSI 2011; Lecture Notes in Computer Science: Berlin/Heidelberg, Germany, 2011; Volume 6729. [Google Scholar]
- Stadlthanner, K.; Lutter, D.; Theis, F.J.; Lang, E.W.; Tome, A.M.; Georgieva, P.; Puntonet, C.G. Sparse Nonnegative Matrix Factorization with Genetic Algorithms for Microarray Analysis. In Proceedings of the 2007 International Joint Conference on Neural Networks, Orlando, FL, USA, 12–17 August 2007; pp. 294–299. [Google Scholar] [CrossRef]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 351–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Main References | Omic Data | |
|---|---|---|---|
| Deterministic low-rank | NNDSVD | [43,57,58] | yes |
| NNDSVD variant | [59] | no | |
| nonnegative PCA | [62,63,71,75] | no | |
| rank-1 | [39] | no | |
| nonnegative ICA | [41,64,77] | yes | |
| Vertex Component Anal. | [65,66,67] | no | |
| Successive Projection Alg. | [68] | no | |
| Clustering-based | k-means | [72] | no |
| k-means variant | [57,71] | yes | |
| spherical k-means | [73,74] | no | |
| fuzzy C-Means | [40,75,78,79] | no | |
| Hierarchical Clustering | [76] | no | |
| Subtracting Clustering | [28] | no |
| Dataset | no. Rows | no. Columns | Type | References |
|---|---|---|---|---|
| (Genes) | (Compounds/Patients) | |||
| MCF7 | 10,331 | 434 | breast cancer | [25] |
| Golub | 5000 | 38 | leukemia | [9,85] |
| Initialization | |||
|---|---|---|---|
| Dataset | Random | nnICA | NDVSD |
| MCF7 | 18 | 15 | 10 |
| 24 | 25 | 10 | |
| 2 | 4 | 0 | |
| Golub | 19 | 30 | 10 |
| 10 | 25 | 10 | |
| 1 | 3 | 0 |
| Percentage of Variance | PC1 | PC2 | PC3 | PC4 |
|---|---|---|---|---|
| MCF7 | ||||
| Golub |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, F. A Review on Initialization Methods for Nonnegative Matrix Factorization: Towards Omics Data Experiments. Mathematics 2021, 9, 1006. https://doi.org/10.3390/math9091006
Esposito F. A Review on Initialization Methods for Nonnegative Matrix Factorization: Towards Omics Data Experiments. Mathematics. 2021; 9(9):1006. https://doi.org/10.3390/math9091006
Chicago/Turabian StyleEsposito, Flavia. 2021. "A Review on Initialization Methods for Nonnegative Matrix Factorization: Towards Omics Data Experiments" Mathematics 9, no. 9: 1006. https://doi.org/10.3390/math9091006
APA StyleEsposito, F. (2021). A Review on Initialization Methods for Nonnegative Matrix Factorization: Towards Omics Data Experiments. Mathematics, 9(9), 1006. https://doi.org/10.3390/math9091006