
Mathematical Modelling of Leptin-Induced Effects on Electrophysiological Properties of Rat Cardiomyocytes and Cardiac Arrhythmias

Abstract
1. Introduction
2. Materials and Methods
2.1. Updated Gattoni 2016 Ionic Models of Rat Ventricular Cardiomyocytes
2.2. Leptin-Related Remodeling in the Cellular Model
2.3. Simulation Protocol for Single Cell Models
2.4. Myocardial Tissue Models
3. Results
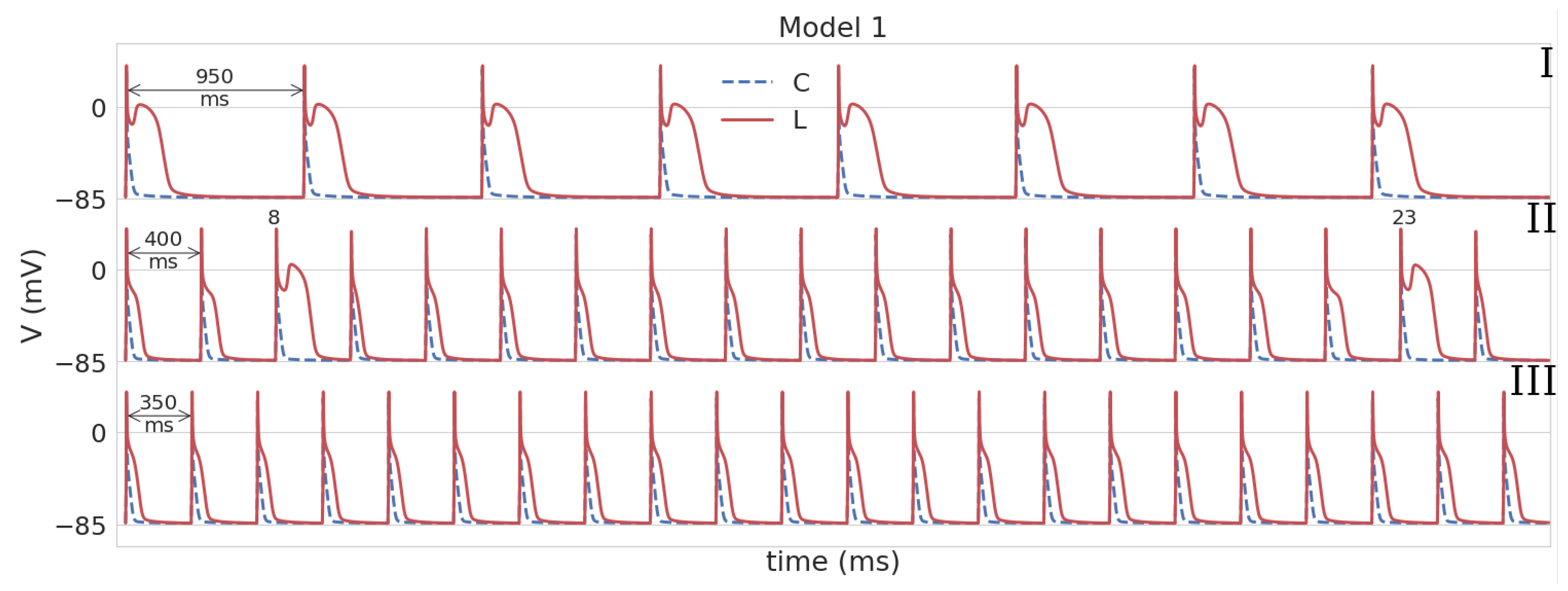
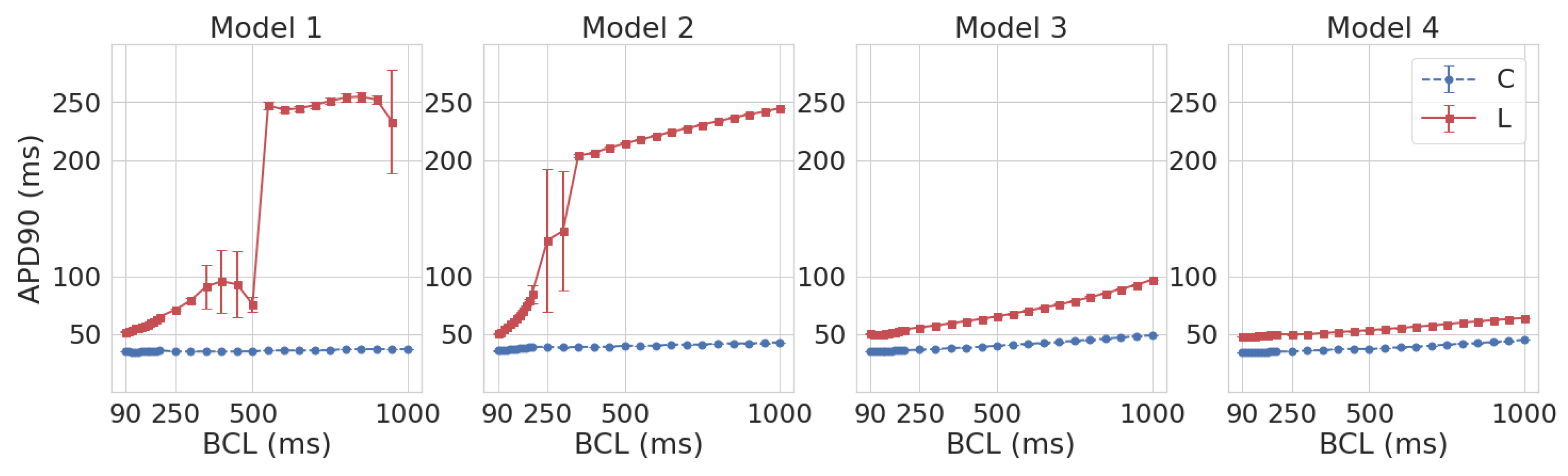
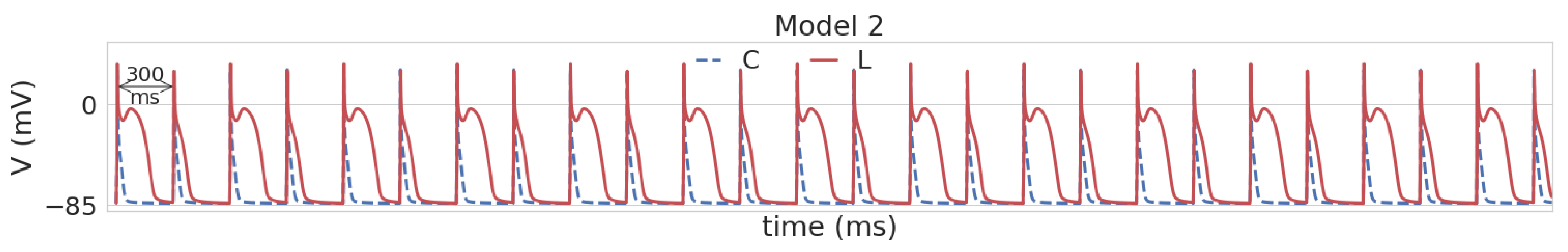
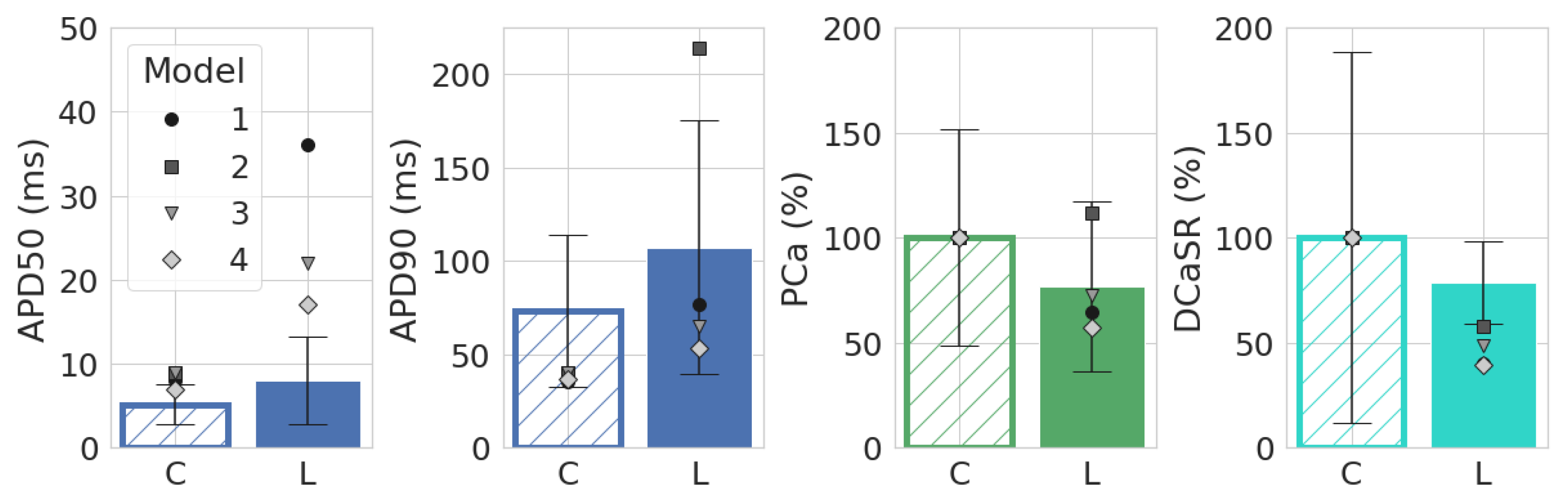
3.1. Leptin Effects in Single Cell Models
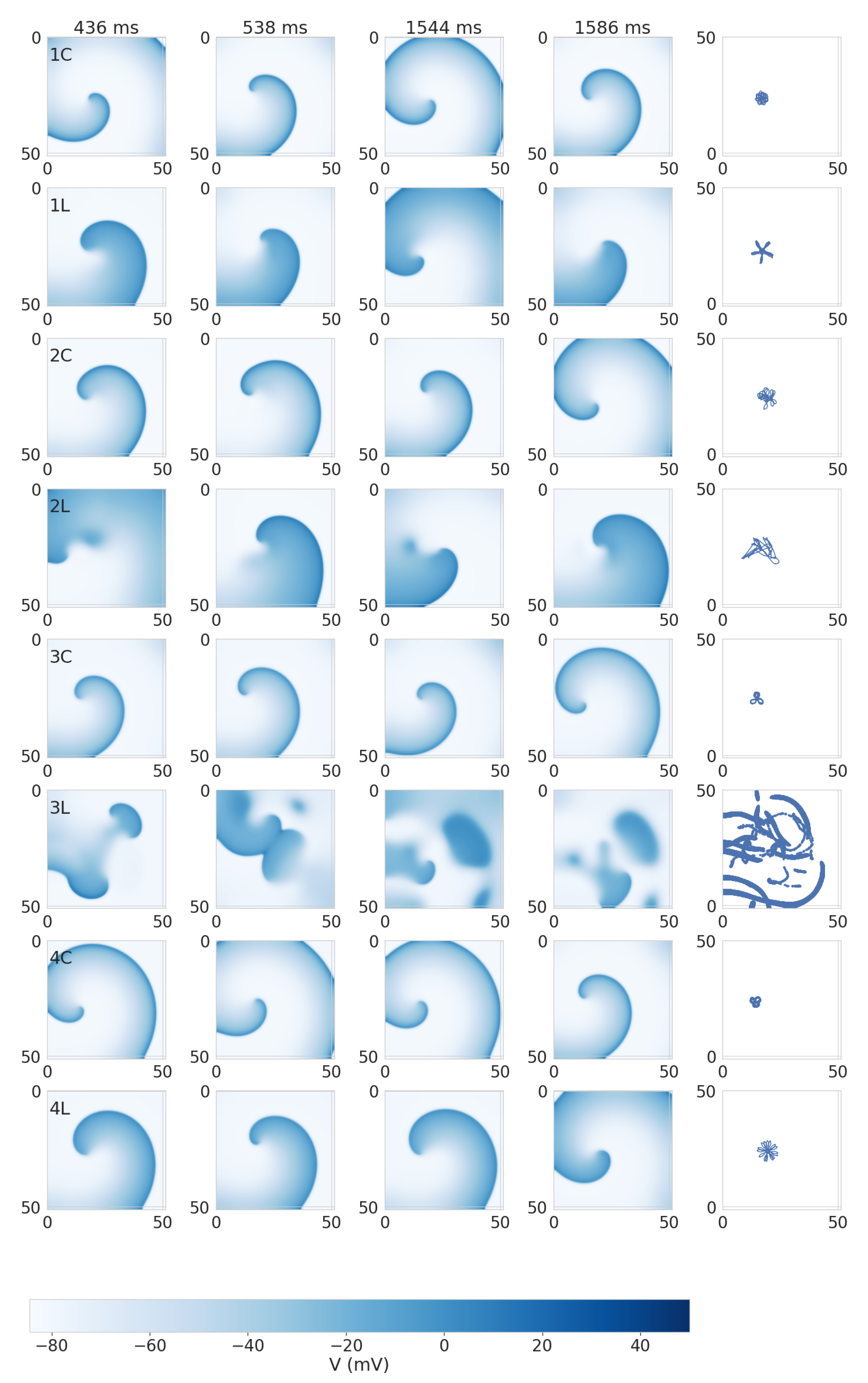
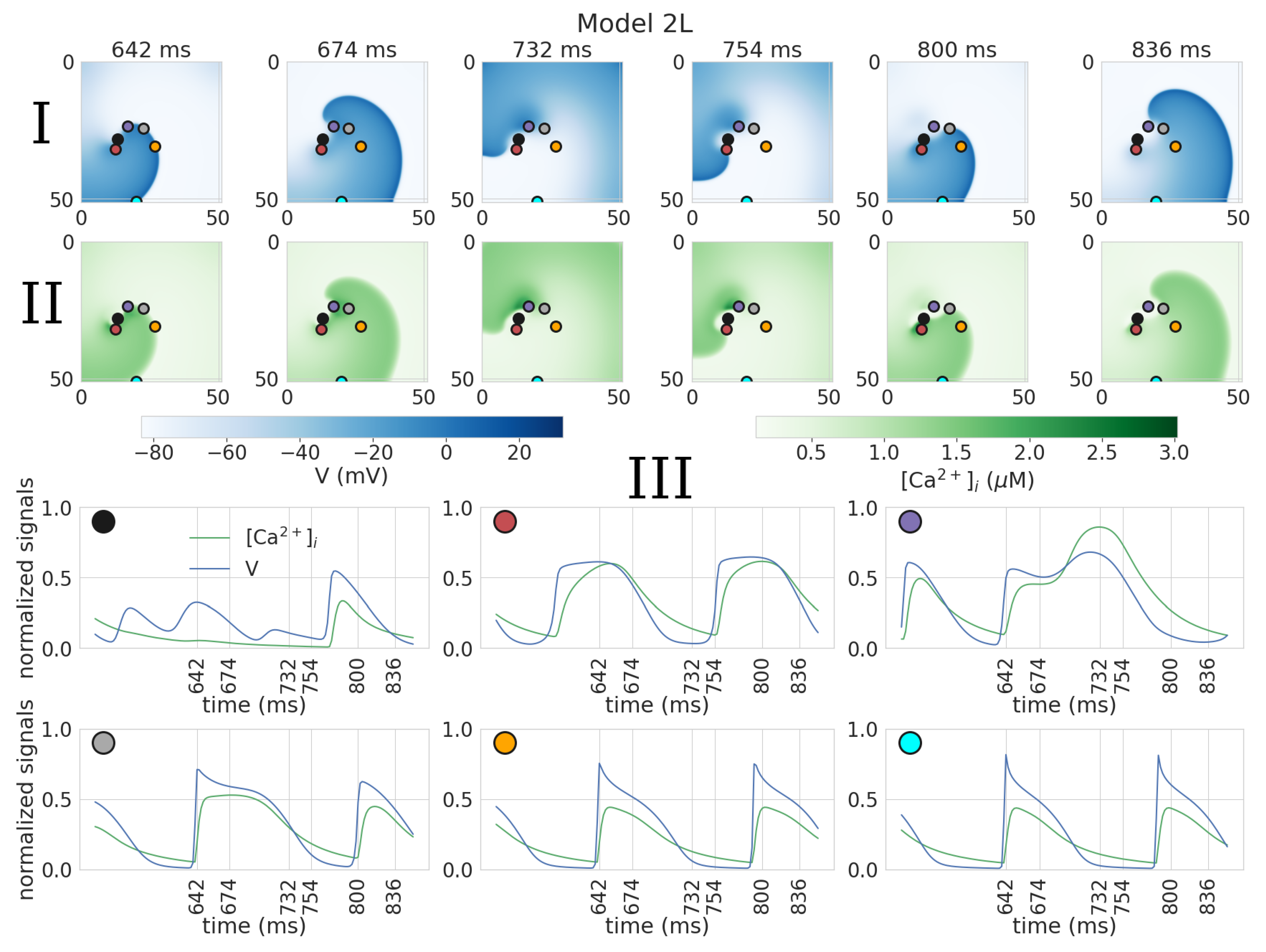
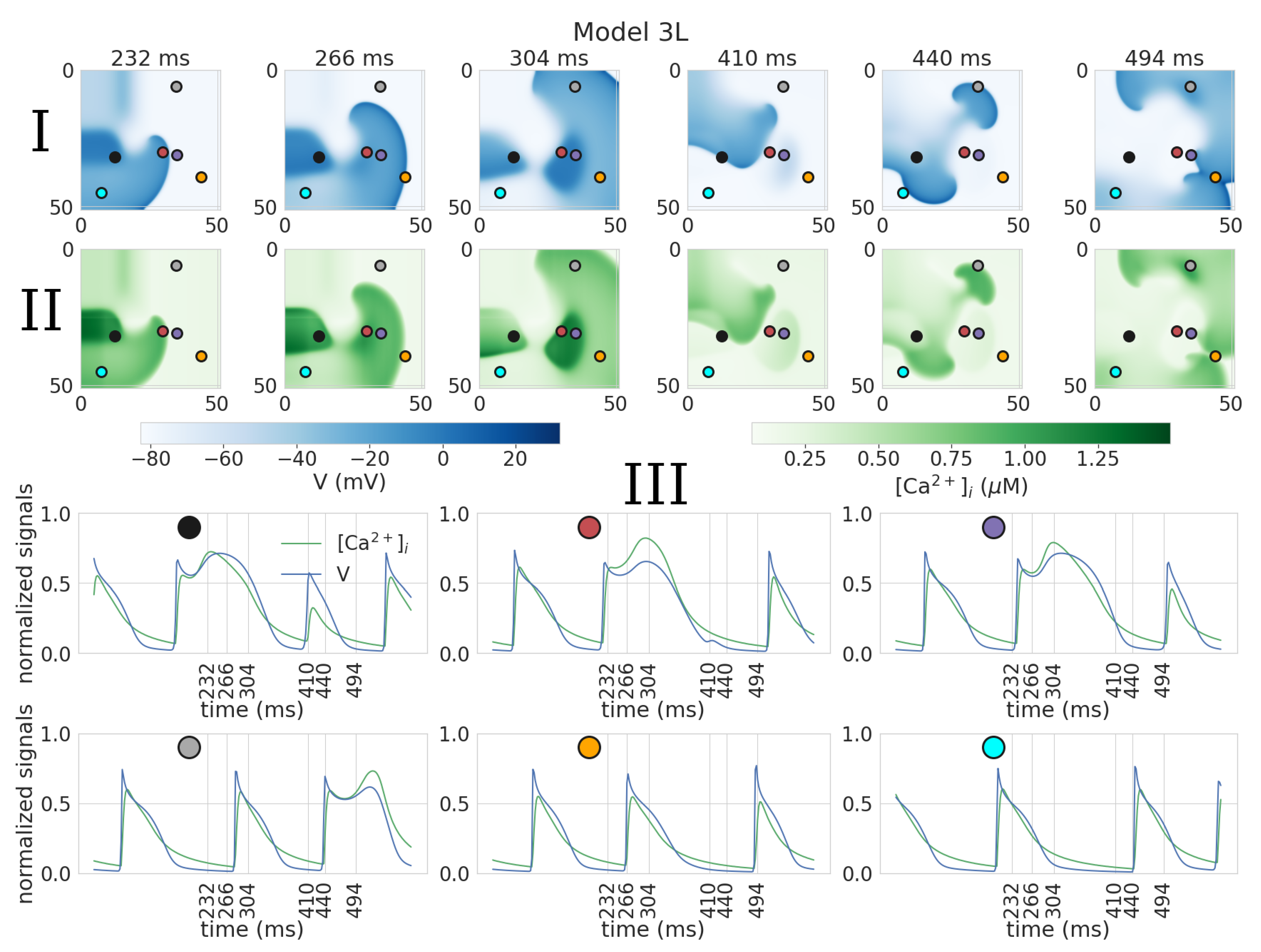
3.2. Dynamics of Spiral Waves in 2D Tissue Models
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AP | action potential |
| APD | action potential duration |
| APD50 | APD at 50% repolarization |
| APD90 | APD at 90% repolarization |
| BCL | basic cycle length |
| DCaSR | diastolic Ca level in the sarcoplasmic reticulum |
| EAD | early after-depolarization |
| SR | sarcoplasmic reticulum |
| PCa | Ca transient peak |
| TdP | Torsade de Pointes |
| I | Na-Ca exchange current |
| I | transient outward potassium current |
| g | pump rate of I |
| g | the maximal velocity of the sarcoplasmic reticulum Ca-ATPase pump |
| g | maximum conductance parameter of I |
References
- Timmis, A.; Vardas, P.; Townsend, N.; Torbica, A.; Katus, H.; De Smedt, D.; Gale, C.P.; Maggioni, A.P.; Petersen, S.E.; Huculeci, R.; et al. European Society of Cardiology: Cardiovascular disease statistics 2021. Eur. Heart J. 2022, 43, 716–799. [Google Scholar] [CrossRef] [PubMed]
- Anumonwo, J.M.; Pandit, S.V. Ionic mechanisms of arrhythmogenesis. Trends Cardiovasc. Med. 2015, 25, 487–496. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Obesity and Overweight Fact-Sheets. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 4 November 2022).
- Fonfara, S.; Kitz, S.; Hetzel, U.; Kipar, A. Myocardial leptin transcription in feline hypertrophic cardiomyopathy. Res. Vet. Sci. 2017, 112, 105–108. [Google Scholar] [CrossRef]
- Zeidan, A.; Purdham, D.M.; Rajapurohitam, V.; Javadov, S.; Chakrabarti, S.; Karmazyn, M. Leptin induces vascular smooth muscle cell hypertrophy through angiotensin II-and endothelin-1-dependent mechanisms and mediates stretch-induced hypertrophy. J. Pharmacol. Exp. Ther. 2005, 315, 1075–1084. [Google Scholar] [PubMed]
- Matsui, H.; Motooka, M.; Koike, H.; Inoue, M.; Iwasaki, T.; Suzuki, T.; Kurabayashi, M.; Yokoyama, T. Ischemia/reperfusion in rat heart induces leptin and leptin receptor gene expression. Life Sci. 2007, 80, 672–680. [Google Scholar] [PubMed]
- Bravo, P.E.; Morse, S.; Borne, D.M.; Aguilar, E.A.; Reisin, E. Leptin and hypertension in obesity. Vasc. Health Risk Manag. 2006, 2, 163. [Google Scholar]
- Leifheit-Nestler, M.; Wagner, N.M.; Gogiraju, R.; Didié, M.; Konstantinides, S.; Hasenfuss, G.; Schäfer, K. Importance of leptin signaling and signal transducer and activator of transcription-3 activation in mediating the cardiac hypertrophy associated with obesity. J. Transl. Med. 2013, 11, 170. [Google Scholar] [CrossRef]
- Polyakova, E.A.; Mikhaylov, E.N.; Galagudza, M.M.; Shlyakhto, E.V. Hyperleptinemia results in systemic inflammation and the exacerbation of ischemia-reperfusion myocardial injury. Heliyon 2021, 7, e08491. [Google Scholar]
- Gómez-Hurtado, N.; Fernández-Velasco, M.; Fernández-Alfonso, M.S.; Boscá, L.; Delgado, C. Prolonged leptin treatment increases transient outward K+ current via upregulation of Kv4. 2 and Kv4. 3 channel subunits in adult rat ventricular myocytes. Pflügers Arch.-Eur. J. Physiol. 2014, 466, 903–914. [Google Scholar] [CrossRef]
- Lin, Y.C.; Huang, J.; Hileman, S.; Martin, K.H.; Hull, R.; Davis, M.; Yu, H.G. Leptin decreases heart rate associated with increased ventricular repolarization via its receptor. Am. J. Physiol.-Heart Circ. Physiol. 2015, 309, H1731–H1739. [Google Scholar] [CrossRef]
- Gómez-Hurtado, N.; Domínguez-Rodríguez, A.; Mateo, P.; Fernández-Velasco, M.; Val-Blasco, A.; Aizpún, R.; Sabourin, J.; Gómez, A.M.; Benitah, J.P.; Delgado, C. Beneficial effects of leptin treatment in a setting of cardiac dysfunction induced by transverse aortic constriction in mouse. J. Physiol. 2017, 595, 4227–4243. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Chen, Y.C.; Huang, J.H.; Lin, Y.J.; Huang, S.S.; Chen, S.A.; Chen, Y.J. Leptin modulates electrophysiological characteristics and isoproterenol-induced arrhythmogenesis in atrial myocytes. J. Biomed. Sci. 2013, 20, 94. [Google Scholar] [CrossRef] [PubMed]
- Ricci, E.; Smallwood, S.; Chouabe, C.; Mertani, H.C.; Raccurt, M.; Morel, G.; Bonvallet, R. Electrophysiological characterization of left ventricular myocytes from obese Sprague-Dawley rat. Obesity 2006, 14, 778–786. [Google Scholar] [CrossRef]
- Qu, Z.; Weiss, J.N.; Garfinkel, A. Cardiac electrical restitution properties and stability of reentrant spiral waves: A simulation study. Am. J. Physiol.-Heart Circ. Physiol. 1999, 276, H269–H283. [Google Scholar]
- Ten Tusscher, K.H.; Panfilov, A.V. Alternans and spiral breakup in a human ventricular tissue model. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H1088–H1100. [Google Scholar]
- Pravdin, S.F.; Dierckx, H.; Katsnelson, L.B.; Solovyova, O.; Markhasin, V.S.; Panfilov, A.V. Electrical wave propagation in an anisotropic model of the left ventricle based on analytical description of cardiac architecture. PLoS ONE 2014, 9, e93617. [Google Scholar] [CrossRef]
- Dusturia, N.; Choi, S.W.; Song, K.S.; Lim, K.M. Effect of myocardial heterogeneity on ventricular electro-mechanical responses: A computational study. Biomed. Eng. Online 2019, 18, 1–18. [Google Scholar] [CrossRef]
- Konovalov, P.; Mangileva, D.; Dokuchaev, A.; Solovyova, O.; Panfilov, A.V. Rotational activity around an obstacle in 2d cardiac tissue in presence of cellular heterogeneity. Mathematics 2021, 9, 3090. [Google Scholar] [CrossRef]
- Rokeakh, R.; Nesterova, T.; Ushenin, K.; Polyakova, E.; Sonin, D.; Galagudza, M.; De Coster, T.; Panfilov, A.; Solovyova, O. Anatomical model of rat ventricles to study cardiac arrhythmias under infarction injury. Mathematics 2021, 9, 2604. [Google Scholar]
- Antzelevitch, C. Basic mechanisms of reentrant arrhythmias. Curr. Opin. Cardiol. 2001, 16, 1–7. [Google Scholar]
- Gattoni, S.; Røe, Å.T.; Frisk, M.; Louch, W.E.; Niederer, S.A.; Smith, N.P. The calcium–frequency response in the rat ventricular myocyte: An experimental and modelling study. J. Physiol. 2016, 594, 4193–4224. [Google Scholar] [PubMed]
- Pandit, S.V.; Clark, R.B.; Giles, W.R.; Demir, S.S. A mathematical model of action potential heterogeneity in adult rat left ventricular myocytes. Biophys. J. 2001, 81, 3029–3051. [Google Scholar] [CrossRef] [PubMed]
- Lewalle, A.; Niederer, S.A.; Smith, N.P. Species-dependent adaptation of the cardiac Na+/K+ pump kinetics to the intracellular Na+ concentration. J. Physiol. 2014, 592, 5355–5371. [Google Scholar] [CrossRef] [PubMed]
- Hinch, R.; Greenstein, J.; Tanskanen, A.; Xu, L.; Winslow, R. A simplified local control model of calcium-induced calcium release in cardiac ventricular myocytes. Biophys. J. 2004, 87, 3723–3736. [Google Scholar] [CrossRef] [PubMed]
- Handa, B.S.; Roney, C.H.; Houston, C.; Qureshi, N.A.; Li, X.; Pitcher, D.S.; Chowdhury, R.A.; Lim, P.B.; Dupont, E.; Niederer, S.A.; et al. Analytical approaches for myocardial fibrillation signals. Comput. Biol. Med. 2018, 102, 315–326. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.D.; Beckman, R.J.; Conover, W.J. A comparison of three methods for selecting values of input variables in the analysis of output from a computer code. Technometrics 2000, 42, 55–61. [Google Scholar]
- Hardy, M.E.; Pervolaraki, E.; Bernus, O.; White, E. Dynamic action potential restitution contributes to mechanical restitution in right ventricular myocytes from pulmonary hypertensive rats. Front. Physiol. 2018, 9, 205. [Google Scholar] [PubMed]
- Annoni, E.M.; Xie, X.; Lee, S.W.; Libbus, I.; KenKnight, B.H.; Osborn, J.W.; Tolkacheva, E.G. Intermittent electrical stimulation of the right cervical vagus nerve in salt-sensitive hypertensive rats: Effects on blood pressure, arrhythmias, and ventricular electrophysiology. Physiol. Rep. 2015, 3, e12476. [Google Scholar] [CrossRef]
- Bébarová, M.; Matejovic, P.; Pásek, M.; Simurdova, M.; Simurda, J. Dual effect of ethanol on inward rectifier potassium current IK1 in rat ventricular myocytes. J. Physiol. Pharmacol. 2014, 65, 497–509. [Google Scholar]
- Hindmarsh, A.C.; Brown, P.N.; Grant, K.E.; Lee, S.L.; Serban, R.; Shumaker, D.E.; Woodward, C.S. SUNDIALS: Suite of nonlinear and differential/algebraic equation solvers. ACM Trans. Math. Softw. TOMS 2005, 31, 363–396. [Google Scholar] [CrossRef]
- Clerx, M.; Collins, P.; de Lange, E.; Volders, P.G. Myokit: A simple interface to cardiac cellular electrophysiology. Prog. Biophys. Mol. Biol. 2016, 120, 100–114. [Google Scholar] [PubMed]
- Lee, S.W.; Li, Q.; Libbus, I.; Xie, X.; KenKnight, B.H.; Garry, M.G.; Tolkacheva, E.G. Chronic cyclic vagus nerve stimulation has beneficial electrophysiological effects on healthy hearts in the absence of autonomic imbalance. Physiol. Rep. 2016, 4, e12786. [Google Scholar] [CrossRef] [PubMed]
- Fenton, F.; Karma, A. Vortex dynamics in three-dimensional continuous myocardium with fiber rotation: Filament instability and fibrillation. Chaos Interdiscip. J. Nonlinear Sci. 1998, 8, 20–47. [Google Scholar]
- Nickola, M.W.; Wold, L.E.; Colligan, P.B.; Wang, G.J.; Samson, W.K.; Ren, J. Leptin attenuates cardiac contraction in rat ventricular myocytes: Role of NO. Hypertension 2000, 36, 501–505. [Google Scholar]
- Khokhlova, A.; Myachina, T.; Butova, X.; Kochurova, A.; Polyakova, E.; Galagudza, M.; Solovyova, O.; Kopylova, G.; Shchepkin, D. The Acute Effects of Leptin on the Contractility of Isolated Rat Atrial and Ventricular Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 8356. [Google Scholar] [CrossRef]
- Hintz, K.; Aberle, N.; Ren, J. Insulin resistance induces hyperleptinemia, cardiac contractile dysfunction but not cardiac leptin resistance in ventricular myocytes. Int. J. Obes. 2003, 27, 1196–1203. [Google Scholar]
- Wold, L.E.; Relling, D.P.; Duan, J.; Norby, F.L.; Ren, J. Abrogated leptin-induced cardiac contractile response in ventricular myocytes under spontaneous hypertension: Role of Jak/STAT pathway. Hypertension 2002, 39, 69–74. [Google Scholar] [CrossRef]
- Ren, J.; Relling, D.P. Leptin-induced suppression of cardiomyocyte contraction is amplified by ceramide. Peptides 2006, 27, 1415–1419. [Google Scholar] [CrossRef]
- Song, Z.; Ko, C.Y.; Nivala, M.; Weiss, J.N.; Qu, Z. Calcium-voltage coupling in the genesis of early and delayed afterdepolarizations in cardiac myocytes. Biophys. J. 2015, 108, 1908–1921. [Google Scholar] [CrossRef]
- Roden, D.M. A current understanding of drug-induced QT prolongation and its implications for anticancer therapy. Cardiovasc. Res. 2019, 115, 895–903. [Google Scholar]
- Fan, Y.; Huang, S.; Li, S.; Wu, B.; Huang, L.; Zhao, Q.; Zheng, Z.; Xie, X.; Liu, J.; Huang, W.; et al. The adipose-neural axis is critically involved in cardiac arrhythmias. bioRxiv 2022. [Google Scholar] [CrossRef]
- Watanabe, Y.; Koide, Y.; Kimura, J. Topics on the Na+/Ca2+ exchanger: Pharmacological characterization of Na+/Ca2+ exchanger inhibitors. J. Pharmacol. Sci. 2006, 102, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Watanabe, Y.; Kita, S.; Blaustein, M.P. Na+/Ca2+ exchange inhibitors: A new class of calcium regulators. Cardiovasc. Haematol. Disord.-Drug Targets Formerly Curr. Drug Targets-Cardiovasc. Hematol. Disord. 2007, 7, 188–198. [Google Scholar]
- Morciano, G.; Rimessi, A.; Patergnani, S.; Vitto, V.A.; Danese, A.; Kahsay, A.; Palumbo, L.; Bonora, M.; Wieckowski, M.R.; Giorgi, C.; et al. Calcium dysregulation in heart diseases: Targeting calcium channels to achieve a correct calcium homeostasis. Pharmacol. Res. 2022, 177, 106119. [Google Scholar]
- Ravens, U. Antiarrhythmic therapy in atrial fibrillation. Pharmacol. Ther. 2010, 128, 129–145. [Google Scholar]
- Quinn, T.A.; Granite, S.; Allessie, M.A.; Antzelevitch, C.; Bollensdorff, C.; Bub, G.; Burton, R.A.B.; Cerbai, E.; Chen, P.S.; Delmar, M.; et al. Minimum Information about a Cardiac Electrophysiology Experiment (MICEE): Standardised reporting for model reproducibility, interoperability, and data sharing. Prog. Biophys. Mol. Biol. 2011, 107, 4–10. [Google Scholar]
- Erdemir, A.; Mulugeta, L.; Ku, J.P.; Drach, A.; Horner, M.; Morrison, T.M.; Peng, G.C.; Vadigepalli, R.; Lytton, W.W.; Myers, J.G. Credible practice of modeling and simulation in healthcare: Ten rules from a multidisciplinary perspective. J. Transl. Med. 2020, 18, 369. [Google Scholar] [CrossRef]
- Joukar, S. A comparative review on heart ion channels, action potentials and electrocardiogram in rodents and human: Extrapolation of experimental insights to clinic. Lab. Anim. Res. 2021, 37, 25. [Google Scholar] [CrossRef]
- Demir, S.S. Computational modeling of cardiac ventricular action potentials in rat and mouse. Jpn. J. Physiol. 2004, 54, 523–530. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Definition | Reference Model | Model 1 | Model 2 | Model 3 | Model 4 |
|---|---|---|---|---|---|---|
| g | Ca flux rate from dyadic space to cytosol | 100% (99 nm/ms) | 40% | 40% | 40% | 40% |
| g (1000 ms) | maximal velocity of SR Ca-ATPase pump | 100% (0.47 M/ms) | 71% | 79% | 116% | 155% |
| g (170 ms) | 145% (0.68 M/ms) | 130% | 143% | 168% | 225% | |
| J | permeability of single L-type Ca channel | 100% (0.7 nm/ms) | 118% | 165% | 100% | 87% |
| g | pump rate of Na-Ca exchange current | 100% (51.5 M/ms) | 69% | 63% | 168% | 154% |
| g | maximum conductance of transient outward K current | 100% (19.6 nS) | 178% | 185% | 196% | 193% |
| g | maximum conductance of steady state outward K current | 100% (12 nS) | 72% | 41% | 26% | 62% |
| i | maximal velocity of Na/K pump | 100% (1.38 nA) | 181% | 140% | 64% | 105% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesterova, T.; Rokeakh, R.; Solovyova, O.; Panfilov, A. Mathematical Modelling of Leptin-Induced Effects on Electrophysiological Properties of Rat Cardiomyocytes and Cardiac Arrhythmias. Mathematics 2023, 11, 874. https://doi.org/10.3390/math11040874
Nesterova T, Rokeakh R, Solovyova O, Panfilov A. Mathematical Modelling of Leptin-Induced Effects on Electrophysiological Properties of Rat Cardiomyocytes and Cardiac Arrhythmias. Mathematics. 2023; 11(4):874. https://doi.org/10.3390/math11040874
Chicago/Turabian StyleNesterova, Tatiana, Roman Rokeakh, Olga Solovyova, and Alexander Panfilov. 2023. "Mathematical Modelling of Leptin-Induced Effects on Electrophysiological Properties of Rat Cardiomyocytes and Cardiac Arrhythmias" Mathematics 11, no. 4: 874. https://doi.org/10.3390/math11040874
APA StyleNesterova, T., Rokeakh, R., Solovyova, O., & Panfilov, A. (2023). Mathematical Modelling of Leptin-Induced Effects on Electrophysiological Properties of Rat Cardiomyocytes and Cardiac Arrhythmias. Mathematics, 11(4), 874. https://doi.org/10.3390/math11040874