1. Introduction
Sequence alignment (SA) is one of the popular approaches in bioinformatics that is used to arrange the primary sequences of DNA/RNA to identify regions of similarity that may have evolutionary or structural relationships among the sequences [
1]. It helps to locate portions with a common evolutionary history by arranging multiple sequences such that a maximum number of similar or identical residues are aligned or matched in a column [
2]. This can be achieved by aligning the unknown sequences with known sequences from a database [
3,
4]. SA can broadly be divided into Multiple Sequence Alignment (MSA), where multiple sequences are aligned simultaneously, and pairwise sequence alignment (PSA), where only two sequences are involved in the alignment process. Generally, MSA is the most commonly used tool that is capable of precisely identifying a sequence’s functional and structural information, as it can deal with several sequences of a family at a time [
5,
6].
MSA can be achieved globally [
7], where the similarity over the entire sequence length is generally considered, or locally [
8], in which the local best-scoring parts of similar characters are considered. An alignment generally considers scoring functions to measure the alignment quality [
9,
10]. However, in MSA, it is a challenging task to identify an optimum scoring function since the statistically optimized functions are not biologically optimal [
11]. Moreover, in MSA, computational complexity requires many resources [
12]. Recently, to improve the MSA optimization process, a dynamic programming approach was applied [
13]. However, the dynamic programming-based approaches in MSA generally experience high dimensionality due to the increasing number of sequences, which results in exponential growth of the time requirement [
14]. Essentially, the MSA process is NP-complete [
15], and thus, all real-world MSA techniques consider heuristic methods that are approximate in real-world situations.
Two different heuristic methods are popularly used for MSA solutions, namely, the iterative and progressive approaches. In the progressive alignment [
16], the PSA method addresses an MSA process in which all of the possible sequence pairs are first aligned, and a guide tree based on the pairwise distance values is then developed. Then, eventually, the MSA is generated stepwise via the gradual arrangement of all of the sequences based on the guide tree, in which, mainly, the best alignment pair is considered [
17]. A major shortcoming of the progressive approach is that the initial sequence pair alignment usually affects the resultant alignments. Thus, changing the position of the gap in the later stages is practically impossible [
18]. To mitigate these issues, among others, iterative approaches are used in the literature [
19,
20,
21,
22,
23] for the MSA problem. Iterative methods are used to iteratively change the building of the guide tree by adjusting the alignment pairs. One of the popular iterative approaches is the Genetic Algorithm (GA) [
24], which is inspired by natural genetics [
25,
26,
27,
28].
Several approaches have been introduced to utilize GA to solve MSA problems [
27]. For example, SAGA, introduced in [
25], uses 22 various GA operators for the MSA. Naznin et al. [
26] used a GA-based approach to improve an MSA solution by vertically demarcating the sequences into several sub-sequences. In reference [
27], the GA was also applied to identify the best guide tree by iteratively altering the guide trees. In similar methods, GA was integrated with other optimization methods like ant colony optimization (ACO) [
29] and the rubber band technique (RBT) to optimize sequence alignment. Currently, the MSA problem is considered a multi-objective process, where each condition can represent a distinct objective function. However, in multi-objective functions, there is a tendency for the accuracy of one objective function to be affected by the optimization of one or more objective functions. Thus, in a real-world situation, a set of non-dominated solutions is generally considered, known as Pareto optimal solutions [
30]. Apart from the non-dominated set, no other means is feasible to improve any one of the objective functions without affecting the others [
31].
For better MSA optimization, recently, some multi-objective GA-based methods were proposed [
12]. One method was introduced in [
32], which considered three objective functions: Totally Conserved Columns (TCCs), STRIKE score, and non-gaps percentage. One shortcoming of this method is the inadequate availability of structures. In a similar method [
33], three objectives were introduced to derive the non-dominated Pareto alignment solutions: similarity maximization, affine gap penalty minimization, and support maximization. In references [
34,
35], a shuffled frog-leaping optimization method [
36] and an artificial bee colony method [
37] were applied, respectively. Both approaches applied two commonly used fitness functions, the sum of pairs (SOP) and TCC, to obtain a Pareto optimal set. These methods utilized another effective Kalign [
38] as a local search strategy to improve the solutions’ quality. However, in the multi-objective Pareto optimal method, one needs to specify the dominant and non-dominant solutions to obtain the set of non-dominated solutions. This is hard to conduct in real-world situations [
39].
This paper proposes an Enhanced Genetic Method for Optimizing Multiple Sequence Alignment (EGMSA) to address the issues as mentioned earlier. The suggested approach essentially uses a multi-objective optimization technique to achieve a better-quality solution. To mitigate the computational complexity, we ignore any non-dominated set of solutions. In this case, the two popularly applied objective functions, SOP and TCC, are used for the optimization process. More specifically, the entire population of solutions is divided into two different portions, and the population section is achieved based on the two objective functions.
The major contribution of the proposed work is that, different from the existing approaches, an integer-based codification strategy is used, in which chromosomes are coded according to their locations within the corresponding sequence, and gaps are coded according to the final chromosomes’ positions within the corresponding sequence, albeit with a negative value. Moreover, modified crossover and mutation methods are suggested. Lastly, using the sum of pairs and total columns as the fitness functions for population selection is suggested. To demonstrate the efficacy of the suggested approach, a series of experiments was conducted using real-world datasets. The experimental outcomes indicate the power of the suggested approach compared to the baseline methods in terms of both the SOP and TCC.
2. Related Work
This section reviews previous works related to MSA that use multi-objective and metaheuristic methods. Handl et al. introduced one of the earlier approaches to study the multi-objective method in MSA [
40]. Since then, several other approaches have been introduced [
1]. Seluangsawat et al. [
41] introduced an evolutionary method to solve the MSA problem based on outputs obtained from the Clustal X method by utilizing multiple objective functions, which include the gap penalty and the sum of pairs. This method uses three mutation operators and a two-point crossover. Ortuño et al. [
32] proposed a multi-objective optimization-based approach that uses the classical metaheuristic NSGA based on structural evaluations to solve MSA problems. The proposed approach optimizes multiple objective functions: non-gap percentage, structural information, and TCC. This method applies the hyper-volume [
30] as the quality evaluation measure. Kaya et al. [
33] introduced another approach based on the NSGA-II algorithm, which considers three objective functions: similarity, affine gap penalty minimization, and support maximization. This approach uses three mutations and two crossover genetic strategies. Soto and Becera [
42] proposed a multi-objective approach based on the genetic technique to optimize pre-aligned sequences. Their suggested model uses three operators: random insertion, two-point crossover, and shift mutation.
Silva et al. [
43] proposed Parallel Niche Pareto by using two different objective functions, including the number of totally identical columns and the sum of pairs. Six mutation operators and three crossover strategies were used in this method. Abbasi et al. [
44] proposed different local search methods for MSA solutions to minimize the number of indels and maximize the substitution score. The suggested method uses several neighborhood definitions and perturbations. A multi-objective-based approach based on decomposition applied to solve MSA was introduced by Zhu et al. [
45]. This model applies a gap insertion operation to generate the initial population. Several existing evolutionary alignment algorithms were compared with the tool to evaluate the model performances.
For better MSA optimization, recently, some multi-objective GA-based methods were proposed [
12]. One method was introduced in [
32], which considers three objective functions: Totally Conserved Columns (TCCs), the STRIKE score, and non-gaps percentage. One shortcoming of this method is the inadequate availability of structures. In a similar method [
33], three objectives were introduced to derive the non-dominated Pareto alignment solutions: similarity maximization, affine gap penalty minimization, and support maximization. Recently, Rubio-Largo et al. [
35,
46] introduced two different methods to improve the MSA solution: the hybrid multi-objective Memetic Metaheuristic approach [
46] and the hybrid multi-objective artificial bee colony method [
35]. These two approaches use the conserved columns and weighted sum-of-pairs function (WSP) with affine gap penalties integrated with the Kalign method [
47]. Finally, Rani et al. [
48] introduced two approaches, the Bacterial Foraging Optimization method and the Hybrid GA with Artificial Bee Colony Algorithm. The authors notably utilized four objective functions: the maximization of similarity, conserved blocks, non-gap percentage, and minimization of gap penalty. However, in the multi-objective Pareto optimal approach, one needs to specify the dominant and non-dominant solutions to obtain the set of non-dominated solutions. This is hard to conduct in real-world situations.
Despite the good performances of the above GA-based methods, they experience several shortcomings. Firstly, some existing algorithms use only one criterion in their objective function, and improving one objective may deteriorate one or more other objectives. It is impossible to optimize a single objective to achieve all objectives simultaneously. Secondly, the conventional GA generally represents a solution or a chromosome with a binary string. However, the binary coding in MSA increases the chromosome/string length, computational complexity, and memory space.
Table 1 summarizes some popular state-of-the-art metaheuristic methods (closer to our proposed model) for the MSA, and
Table 2 compares our proposed model with existing methods to justify its novelty.
4. The Proposed Model
In this section, details of the suggested EGMSA approach are reported. We propose a genetic algorithm-based multi-objective sequence alignment method. Here, we attempt to improve the SOP and TCC, the two most popular objective functions. The suggested approach lowers the computing complexity compared to the existing multi-objective optimization strategies since it does not consider any non-dominated set of solutions. This creates two halves out of the total population of solutions. Next, a portion of the population is selected using the SOP objective function, while a separate portion is selected using the TCC objection function. An ideal MSA solution for the proposed model can be obtained using this selection procedure in conjunction with the suggested crossover and mutation operations.
In the suggested framework, alignments are depicted as a matrix where two prerequisites are met: (i) chromosomes are coded according to their locations within the corresponding sequence, and (ii) gaps are coded according to the final chromosomes’ positions within the corresponding sequence, albeit with a negative value. Prior to being incorporated into this program, the input alignments are coded. To complete the optimization, coded alignments are used. Individuals are decoded and subsequently returned to the standard alignment representation once the optimization is complete.
In the following sub-section, we present the evolutionary method based on a genetic algorithm. Then, the individual representation of the individuals is presented, followed by a description of the procedure of the model for the MSA solution.
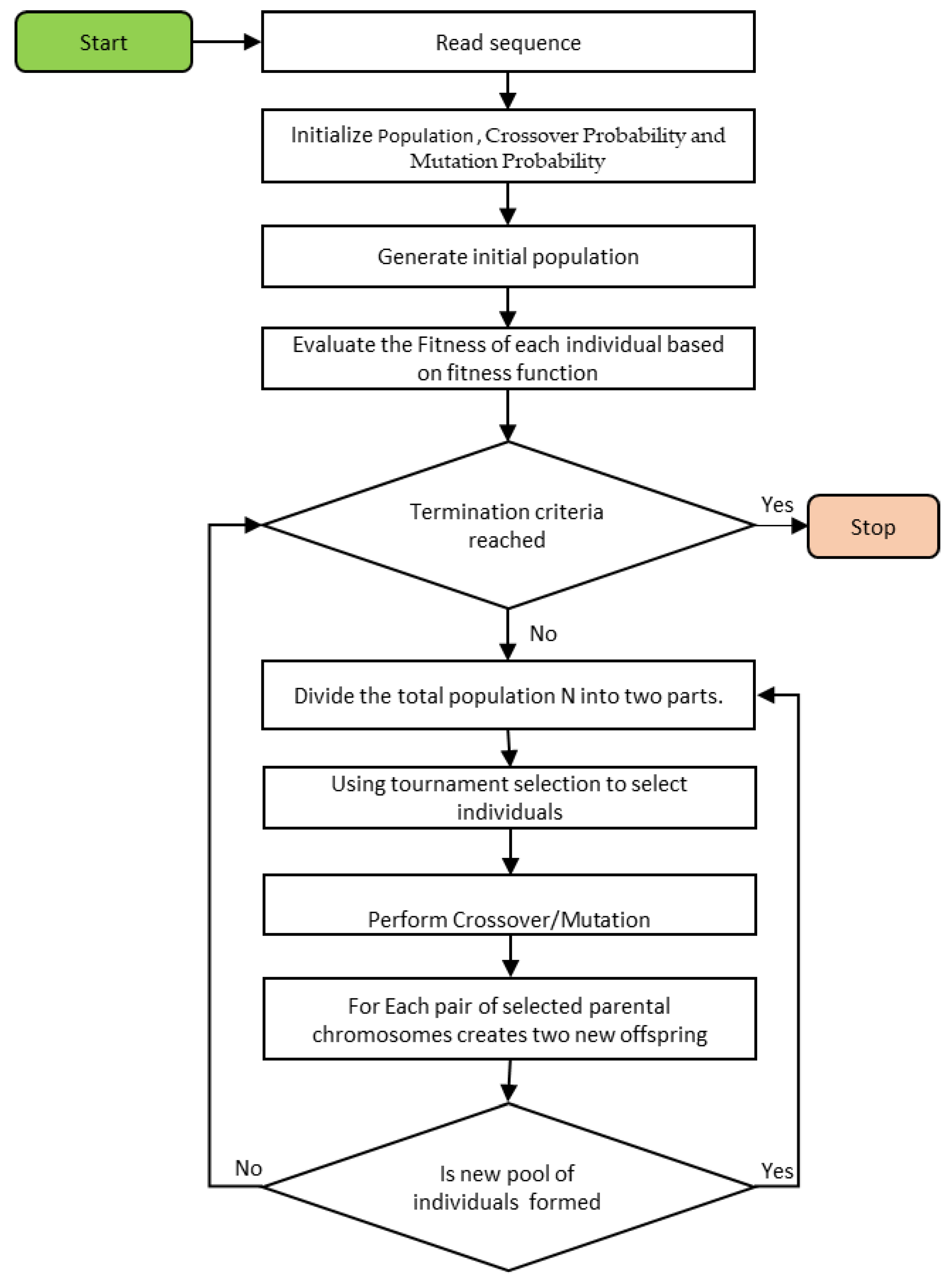
4.1. Genetic Algorithm (GA)
GA is an evolutionary method that imitates the “survival of the fittest” strategy. GA uses a stochastic process that considers a set of probable ways to solve a problem. In this case, each solution represents a chromosome, and the chromosome set is termed a population. To determine the quality of the solution, objective functions or fitness functions are associated with each chromosome. Based on the fitness function, the chromosomes that best fit the generation will be selected using the selection operation. After that, other genetic operations, such as mutation and crossover, are carried out on the mainly chosen chromosomes to change their fitness and produce a new generation. The operators, namely, the selection, crossover, and mutation processes, iteratively proceed until convergence to the best fitness value. All of these steps of the basic genetic algorithm can be found in reference [
21]. The flowchart of the proposed GA-based model is illustrated in
Figure 1.
4.2. Alignment Representations
The population representation process is essential when using a metaheuristic method to solve an MSA problem. The encoding process generally impacts the behaviors of algorithms, and it determines the operation strategies and their efficiency. In this study, a GA [
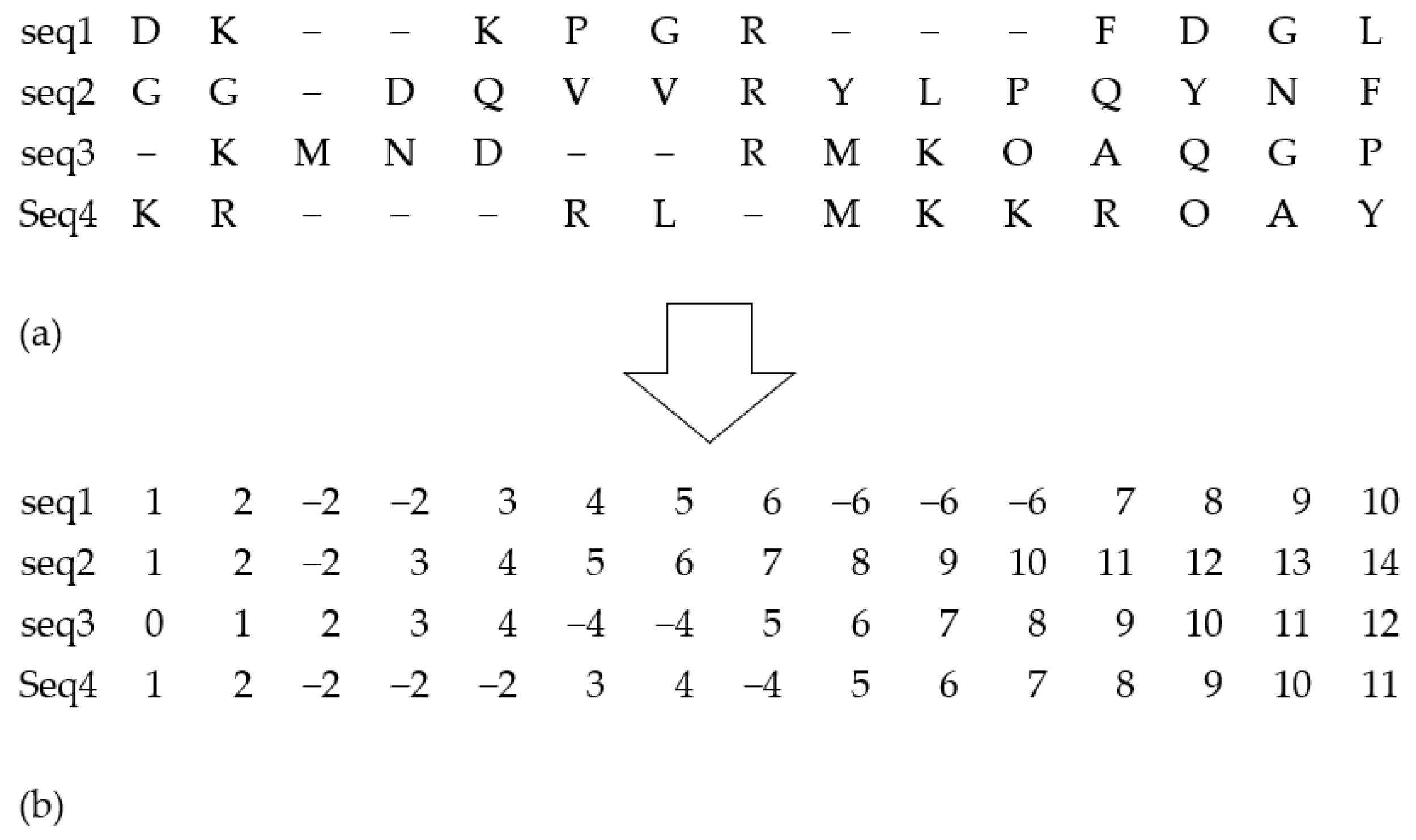
24] was used, which is one of the popular iterative techniques inspired by natural genetics. Most of the existing GA-based approaches for MSA generally focus on the binary string for the chromosomes’ (solution) representation, where the standard alphabet is used for amino acids and the “-” symbol is used for the gaps. However, the existing binary representation in MSA generally increases the length of the string, complexity, and space requirements. To overcome these drawbacks, in this study, an improved integer coding for the string representation is opted to be applied instead of the binary coding process.
In the proposed method, alignments are coded as a matrix, where residues or amino acids are coded according to their positions in their respective sequences. At the same time, the gaps are represented by the last residue positions of their respective sequences but with a negative value. Essentially, the input alignments are coded before their processing. The entire optimization process is conducted by applying coded alignments. After finalizing the optimization process, individuals are decoded and consequently revert to their standard representations.
Figure 2 illustrates an example of the suggested representation process. The main benefit of this representation strategy is that it allows for the simple identification of positions for crossover operation. Moreover, it can help to avoid mistakes that are likely to occur in the subsequent crossover operations, thereby ensuring better improvements in alignment management.
4.3. Genetic Operators
The proposed model comprises three different GA operators, which include selection, crossover, and mutation. On the one hand, the selection operator is used to identify the best individuals from the population; conversely, the crossover and mutation operators act on the chosen individuals in the population based on the probabilities, and , accordingly.
4.3.1. Selection Operator
The selection operator is aimed at randomly identifying a pair of best chromosomes as parents (
and
) from the population generation of
to take part in the crossover and mutation processes to generate offspring
and
for the new generation. The chromosomes with the best fitness scores are expected to be chosen for the next generation. In the suggested model, each chromosome is linked with two different functions,
and
, as shown in Equations (1) and (2). However, selecting one chromosome with the best fitness scores for all of the functions is rarely possible. Thus, in the proposed approach, the entire population size is divided into two portions. Essentially,
is used to obtain one portion of the population, while the
is used for the other portions. In this way, the selection operation proceeds along with mutation and crossover until the next generation
is obtained. Several selection methods can be used [
54,
55] to select the best individuals. Mainly, in this work, the tournament selection approach with a tournament size of 2 is employed [
54], which can consider the different selection pressures without any alterations. In this selection process, two randomly selected individuals compete in the tournament. The fittest among the two is chosen as a parent and takes part in the crossover operation.
4.3.2. Crossover Operator
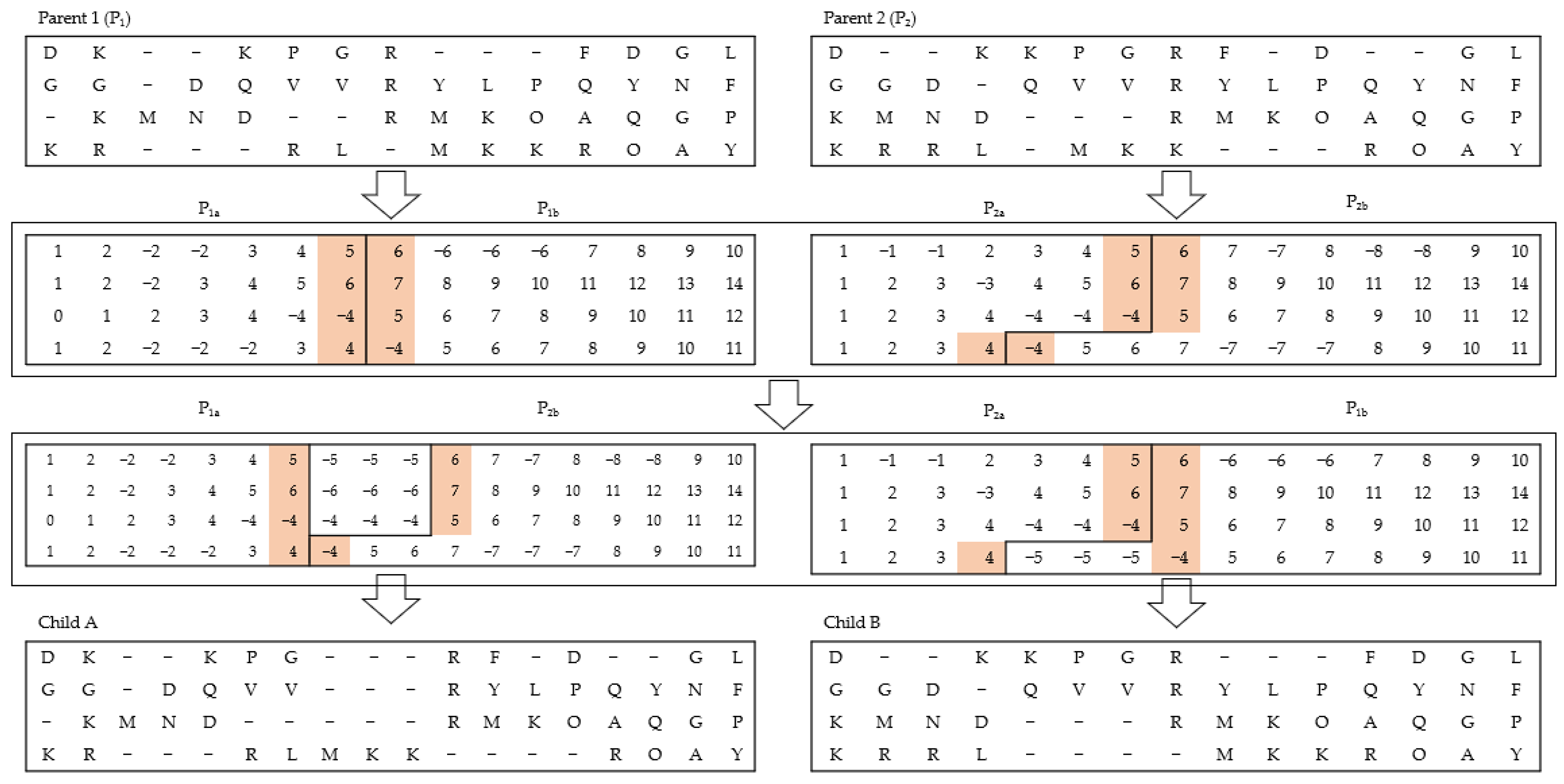
The primary purpose of the crossover strategy is to achieve an interactive exchange of information between a pair of different chromosomes [
10] to generate a new one. During the crossover process, the two parents (say
and
) that are randomly selected exchange their genetic information between themselves to form new populations (
and
for the new generation. In the proposed work, an enhanced one-point crossover is introduced to consider each existing gap. In this approach, two-parent alignments are joined by using a single exchange procedure. More specifically, the first parent
is divided at a randomly selected position. The second parent
is processed so that the right part can be combined with the left part of the first parent and vice versa. Null signs are used to fill any vacant position at the junction point. An illustration of this process is shown in
Figure 3. More specifically, this operator combines the classical properties of a local arrangement mutation and crossover. However, in each case, the occurrence of this operation is specifically guided based on the crossover probability
.
4.3.3. Mutation Operator
The main aim of the mutation operation in the evolutionary process is to help maintain the diversity in the population. Mutation operators randomly transmit the evolutionary information among other individuals and help recover the missing genetic data. Mutation operation also helps in preventing algorithm trapping at a local minimum. In the suggested method, different types of mutation operations are used, which include gap merging, gap insertion, single gap, and block gap mutation. Following the completion of the crossover operation, an offspring continues with the mutation process accordingly. However, each of the operation processes is randomly guided by the mutation probability,
. Each defined mutation operator is applied one at a time to see which one produces a higher score. The operator that yields the best results for that specific sequence is considered, while the others are rejected. Each defined mutation operator is chosen randomly to solve a specific set of problems with a specific probability. Here, in the suggested method, a different mutation operator from the stated set is chosen and applied to solve the provided problem if one of the randomly chosen mutation operators cannot provide an optimal outcome. The following describes every suggested mutation operator for the experimental analysis. A demonstrative example of the mutation procedure is presented in
Figure 3.
Merging Mutation
This operation process merges multiple spaces of a sequence. This can be achieved by randomly selecting multiple consecutive gaps of a sequence, which may or may not be adjacent, and then merging these gaps. Consequently, the gaps are moved to a randomly selected position in the same sequence. An illustration of this mutation operation is depicted in
Figure 4a.
Insertion Mutation
This variety of mutation operation is used to produce the mutation if the fitness of the mutated alignments is more than the fitness of the original ones. A random position in the alignment sequences (column and line indexes) is randomly selected to achieve this. Then, a chosen number of gaps are added to that position, and all of the other lines are filled with gaps until they all have the same size.
Figure 4b depicts an illustration of the insertion mutation.
Block-Gap Mutation
In this operation, a random row is chosen among others, and a block of gap positions that contain continuous gap values is selected. Finally, the whole block of gap values is shifted to a particular random position. The chosen block of gaps must have multiple continuous gap positions.
Figure 4c illustrates a typical example of this operation.
4.4. Termination Criteria
Termination criteria are essential for stopping the algorithm when it has achieved a satisfactory solution or when a further optimization is not yielding substantial improvements. We established a termination criterion to reach the optimal score of the leading or most outstanding chromosome. In our suggested approach, which involves considering multiple objective criteria during selection, we end up with two elite chromosomes, each excelling in one of the two objective functions. Suppose the fitness scores of these top-performing solutions persist without change for 100 consecutive generations. In that case, we permit the process to conclude to save computational time and reduce memory usage.
6. Results and Discussion
This section presents a comparison of the results of the proposed EGMSA model against the existing approaches. In the first place, the comparison results of our model based on BALiBASE 2.0 and BALiBASE 3.0 are reported. Then, later, the results obtained based on the SABmark datasets are presented.
To evaluate our model with the BAlibase 2.0 datasets, only the results for the sum-of-pairs (SOP) score are reported, as all of the compared approaches considered only the SOP scores of the datasets. The bolded values indicate the highest scores in each instance. A blank entry signifies the absence of scores.
Table 4 displays the results of the sequence alignments obtained using our proposed method and the compared evolutionary-based approaches published in the literature, which include GAPAM, MSA-GA [
19], MO-SAStrE, VDGA, RBT-GA, SAGA, and HMOABC [
46]. These approaches were evaluated by using a subset of the BALiBASE2.0 datasets. Essentially, for the VDGA method, there are three various settings of the model, which include VDGA D-2, VDGA D-3, and VDGA D-4; additionally, the MSA-GA variant with a pre-aligned strategy (MSA-GA w/p) is also used in the comparison.
The results show that all of the methods that used multi-objective approaches, namely, MO-SAStr, HMOABC, and BSAGA, recorded the best accuracies compared to the other approaches. However, from the results, it can be seen that our suggested approach outperformed all of the compared methods, including the MO-SAStrE, HMOABC, and BSAGA models.
To further confirm the efficacy of the suggested solution, the suggested model was also compared with popular methods that applied the BALiBASE 3.0 datasets. To this end, the results presented in [
46] were used for the comparison. The approaches used for the comparison based on the BALiBASE datasets included Clustal X [
17], DIALIGN-TX [
60], FSA and FSA-max, MAFFT [
62] (LINS-i, EINS-i, and GINS-i), MSAProbs [
63], MUMMALS [
64], MUSCLE [
21], ProbCons [
5], PRANK [
65], HMOABC [
46], ProbAlign [
66], and T-Coffee [
67]. Furthermore, the latest versions of the T-Coffee and BSAGA [
68] models were also included in our comparative study.
Table 5 shows the scores of the compared methods and the suggested model on the BALiBASE datasets. Each case’s highest scores are bolded in both the Q and TC metrics. As can be seen, the proposed model obtained the highest results in most subsets in terms of the Q and TC measures. However, for RV11, which has the lowest identity virtually within the range between 0 and 20%, the proposed model performed much better than the prominent methods, namely, the BSAGA and HMOABC approaches. Essentially, the HMOABC methods used multi-objective methods and generated a particularly complex set of non-dominated solutions. Additionally, this method is based on Kalign, which is a deterministic heuristic method. On the other hand, the suggested approach particularly used the evolutionary method for the optimization, and it produced the two best solutions based on the two different objective functions, instead of generating a non-dominated set of solutions. Overall, from the results, it can be observed that our proposed model shows consistently good results for the other sub-datasets in all cases.
Finally, the suggested EGMSA model results were also compared with the existing approaches that used the SABmark v1.65 datasets. The compared methods and their respective results are taken as presented in [
46].
Table 6 shows the comparison results of the proposed mode model on the SABmark v1.65 datasets. From the results, one can see that the results of our proposed model outperformed all of the existing approaches, including the H4MSA [
46] and BSAGA methods. Essentially, for both of the sub-datasets (superfamily and twilight), the proposed model performed better regarding both the TC and Q measures. Particularly, the second sub-dataset, “twilight”, signifies the worst-case alignment situation as the sequences share below 25% of identity [
57]. In all of the cases, our proposed model outperformed the compared approaches regarding both Q and TC.
6.1. Statistical Test
A significance test was carried out to investigate whether the improvement of our proposed approach was statistically significant. To this end, a pairwise comparison was conducted in which the proposed model was compared with each baseline method from the comparative results, as shown in
Table 7. To this end, a non-parametric test was used, considering that the data are abnormally distributed. More specifically, a Wilcoxon signed-rank test was used, which is a non-parametric test that is similar to the
t-test used for the significance study. In the Wilcoxon test, the null hypothesis is used with the assumption that there is no significant difference in two different outcomes for different sources. Thus, the alternative hypothesis is that there is a significant difference between the two outcomes. Hence, the null hypothesis would be rejected to indicate the significant difference between the two examples. In this case, the 5% significance level was used for the statistical test.
As shown in
Table 3, yes and no are assigned (“yes” for
p ≤ 0.05 and “no” for
p > 0.05) for the comparison of any two approaches (as indicated in the sixth column), where “yes” means that our proposed model improvement over the compared one is significant, and “no” means that there is no significant improvement.
6.2. Parameters Sensitivity Analysis
A parameter analysis was also conducted to investigate the influence of the different parameters on the model performances. Thus, an investigation was conducted to measure the impact of the crossover and mutation probabilities , the impact of the population size (N), and the different iteration values.
6.2.1. Impacts of and
and
are two important parameters that largely influence the accuracy of the quality solution of GA-based techniques. Thus, in this subsection, an investigation was conducted to measure the impacts of
and
on the model accuracy to enable us to select the best combination of parameters.
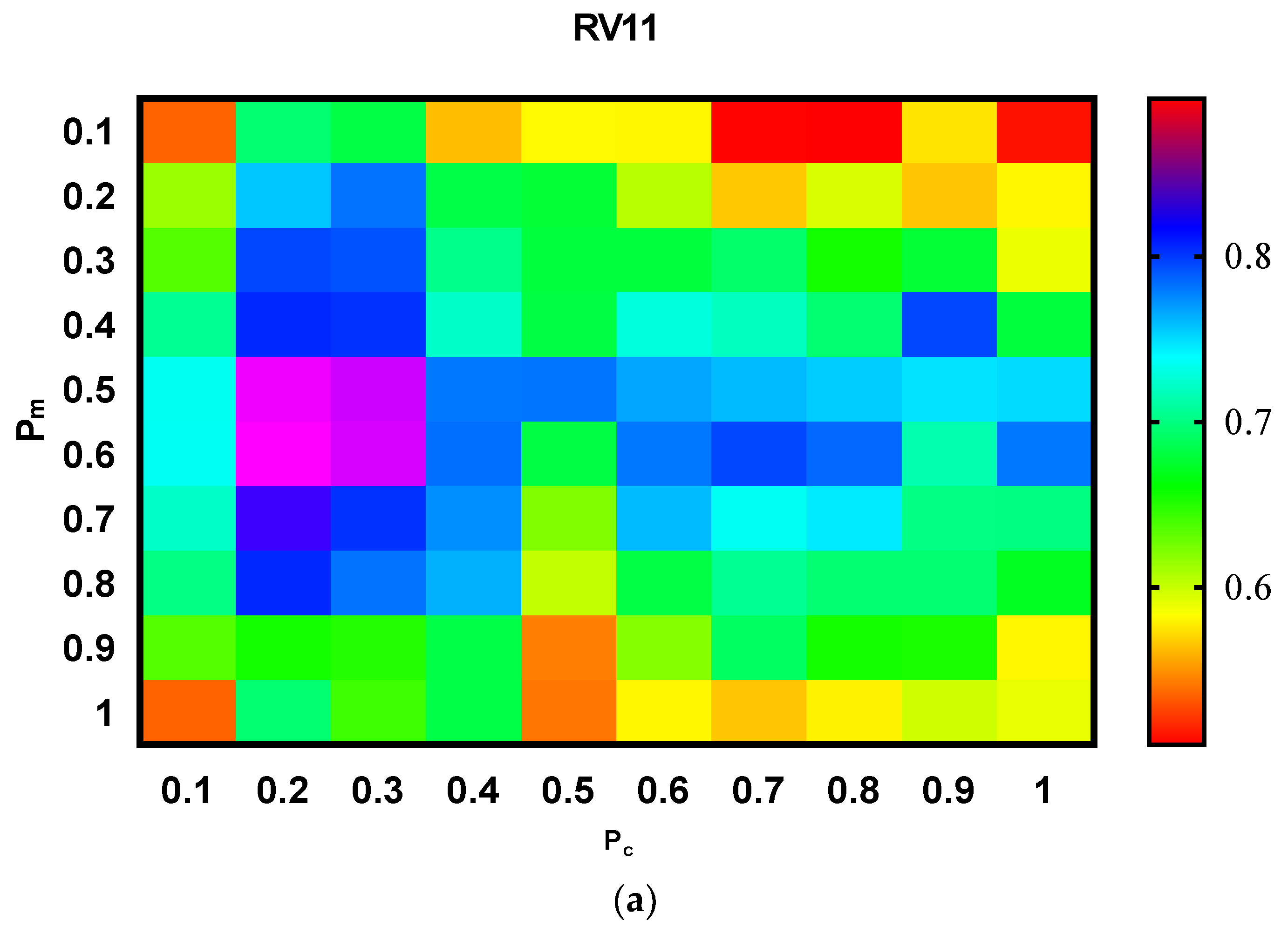
Figure 5 displays the performance of the proposed approach for different values and a combination of
and
respectively. Specifically, adjusted values were used [0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1] for both
and
. From the results, it can be seen that although the
value is not very significant, the proposed model records a stable performance for the value in the range of [0.2–0.5] for both
and
. Thus, to obtain a better setting for the best quality solution of the proposed model, the proposed method selected the range of [0.2–0.5] for both parameters.
6.2.2. Impact of N
To investigate the impact of the population size
, different values of the
N parameter were experimented with, which include [50, 100, 150, 200, and 250] with already chosen (
and
) crossover and mutation probability values. From the results in
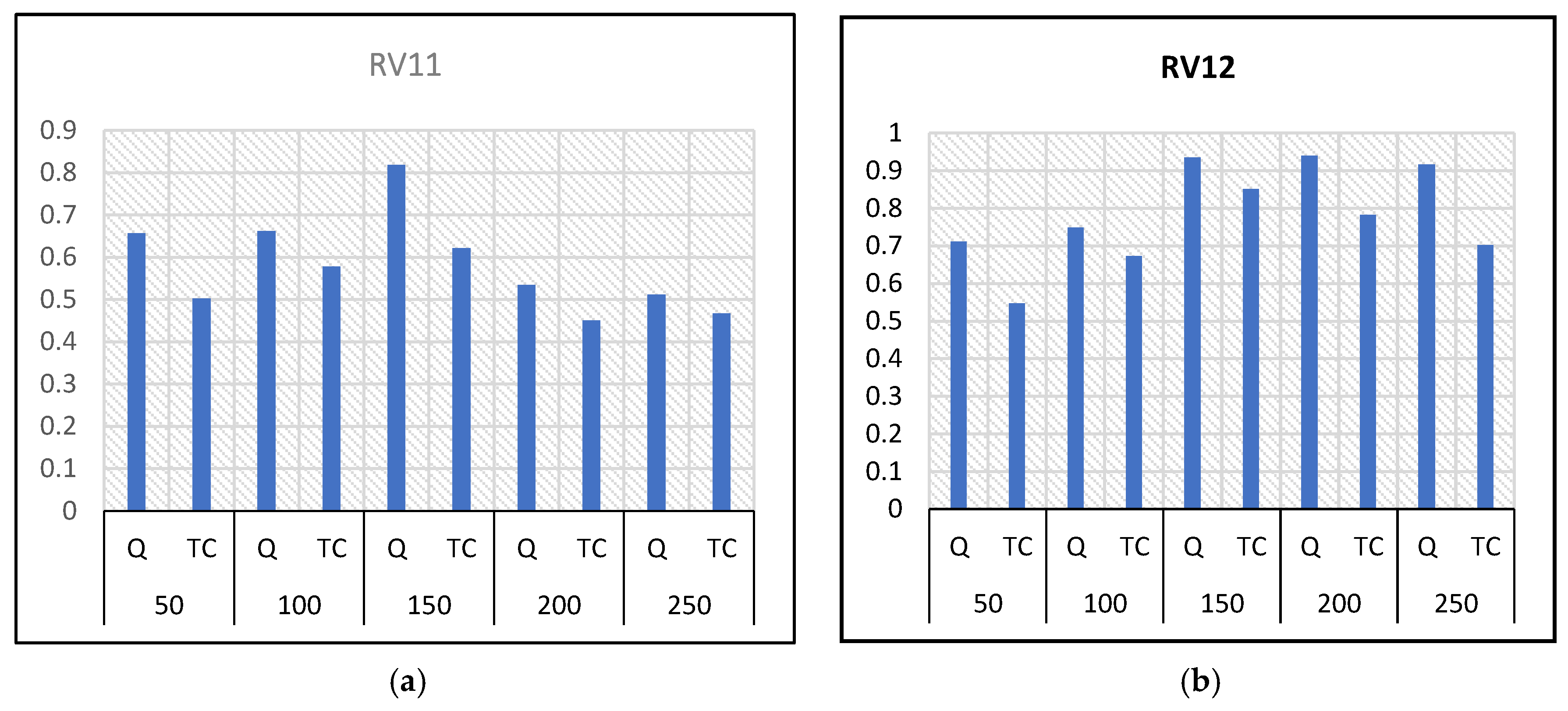
Figure 6a–f, it can be seen that all other values used in the setting do not show a significant improvement between 100 and 150. This means that the model started to improve when the N values reached a value of 150 in most cases. However, increasing the value of N could increase the complexity of the training, including the computation time. The ideal value of the population size for the model configuration was selected as 150.
6.2.3. Iteration
Different values of successive iteration numbers were used in the experiments to determine the termination criteria of the proposed model after convergence [50, 100, 150, and 200]. The different scores obtained are shown in
Figure 7a–f. From the results in the figures, one can see that the accuracy improved more between 100 and 150 in most of the cases. However, most cases’ model improvements are almost similar for 100 and 150. Thus, 100 was chosen as the ideal value for setting up the proposed model to minimize the computational complexity.
6.2.4. Time Complexity
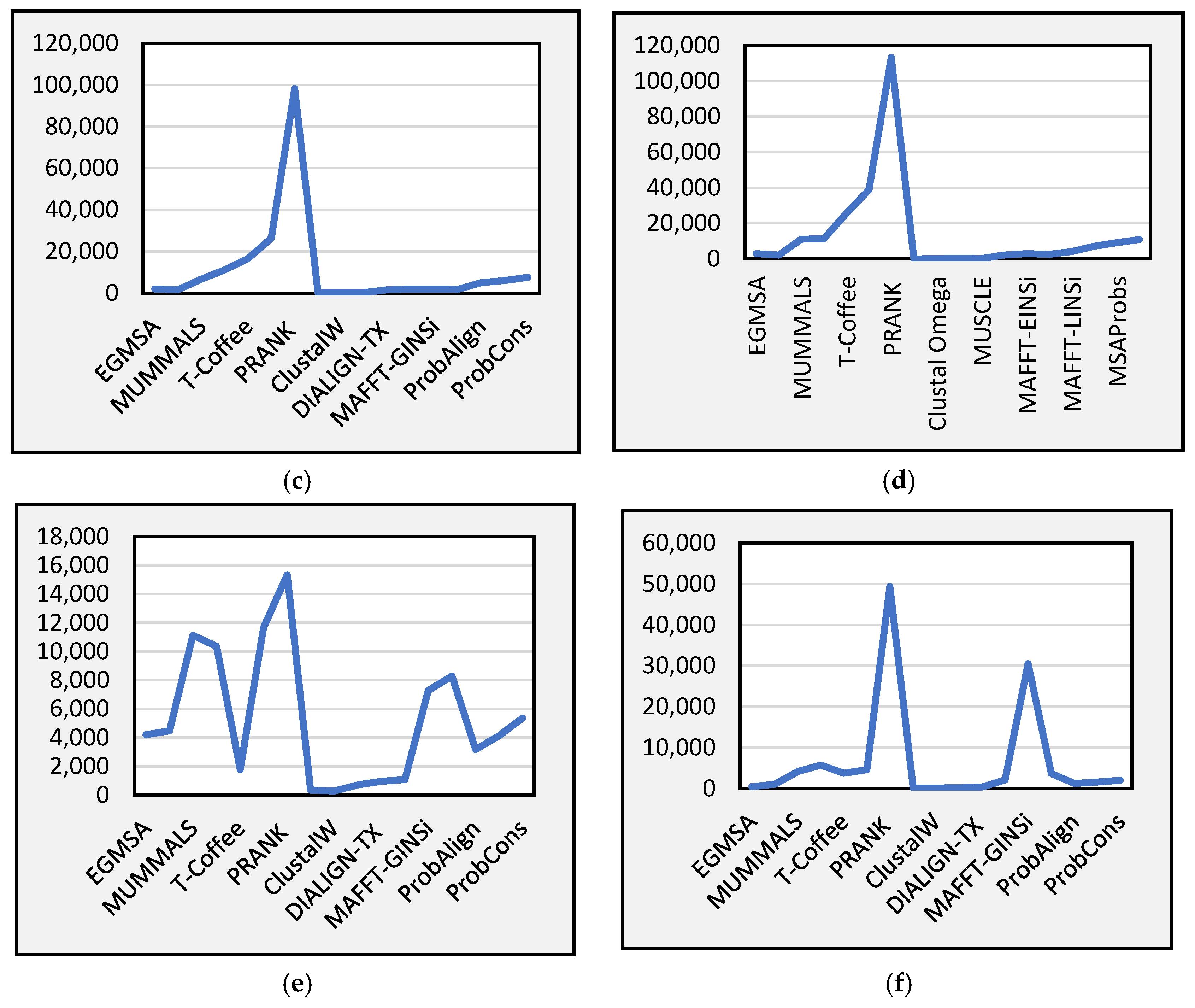
Figure 8 presents the total runtime required by the different comparison methods and our proposed method for the six subsets in BAliBASE v3.0. As can be seen, the runtime required by our proposed model is reasonable. In general, there is a trade-off between the alignment quality performance and computation time across all of the BAliBASE datasets, as can be noted in the existing literature [
46].
Figure 8a–f depict the runtime for the proposed model alongside the baseline methods.
Further, from the results, it can be seen that the runtime of the proposed model is relatively stable in each case. This means that our suggested approach consumes less time compared to other multi-objective approaches. Although the proposed model takes a relatively longer time compared to some baseline approaches from the figures, the runtime of our suggested approach is significantly better than GAPAM, HMOABC, and MOSAStrE algorithms, which are also multi-objective methods used to solve the MSA problem. This indicates that the proposed model is an effective algorithm.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}