
Stochastic Transcription with Alterable Synthesis Rates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
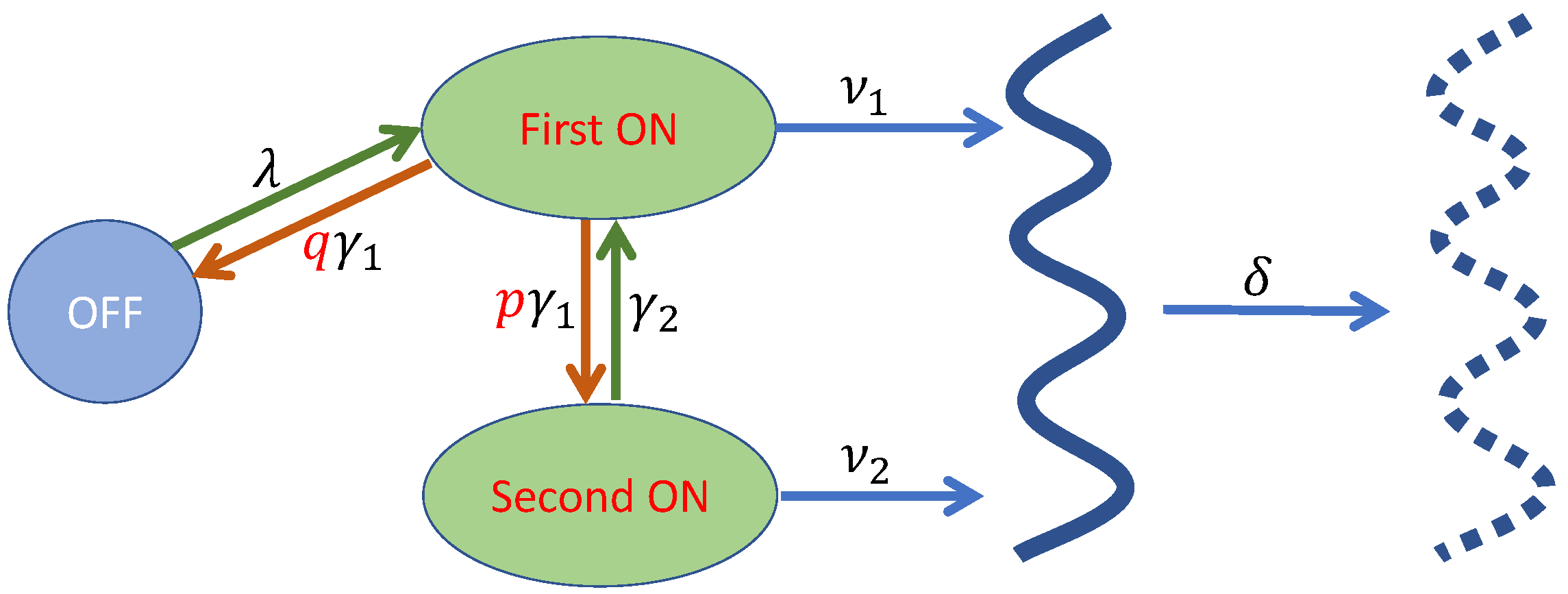
2. Model Specification
2.1. The Characterization of the Gene Transcription Mechanism
2.2. The Master Equations
- 1.
- , with no production or elimination of transcripts and transfer of the system states taking place during the time interval . This event has a probability .
- 2.
- , with one transcript being eliminated during . This event has a probability .
- 3.
- , with one transcript being produced during . This event has a probability .
- 4.
- , the system being transferred from the second ON state to the first one during . This event has a probability .
- 5.
- , the system being transferred from the OFF state to the first ON state during . This event has a probability .
2.3. The Differential Equations
3. Results
3.1. The Average Transcription Levels
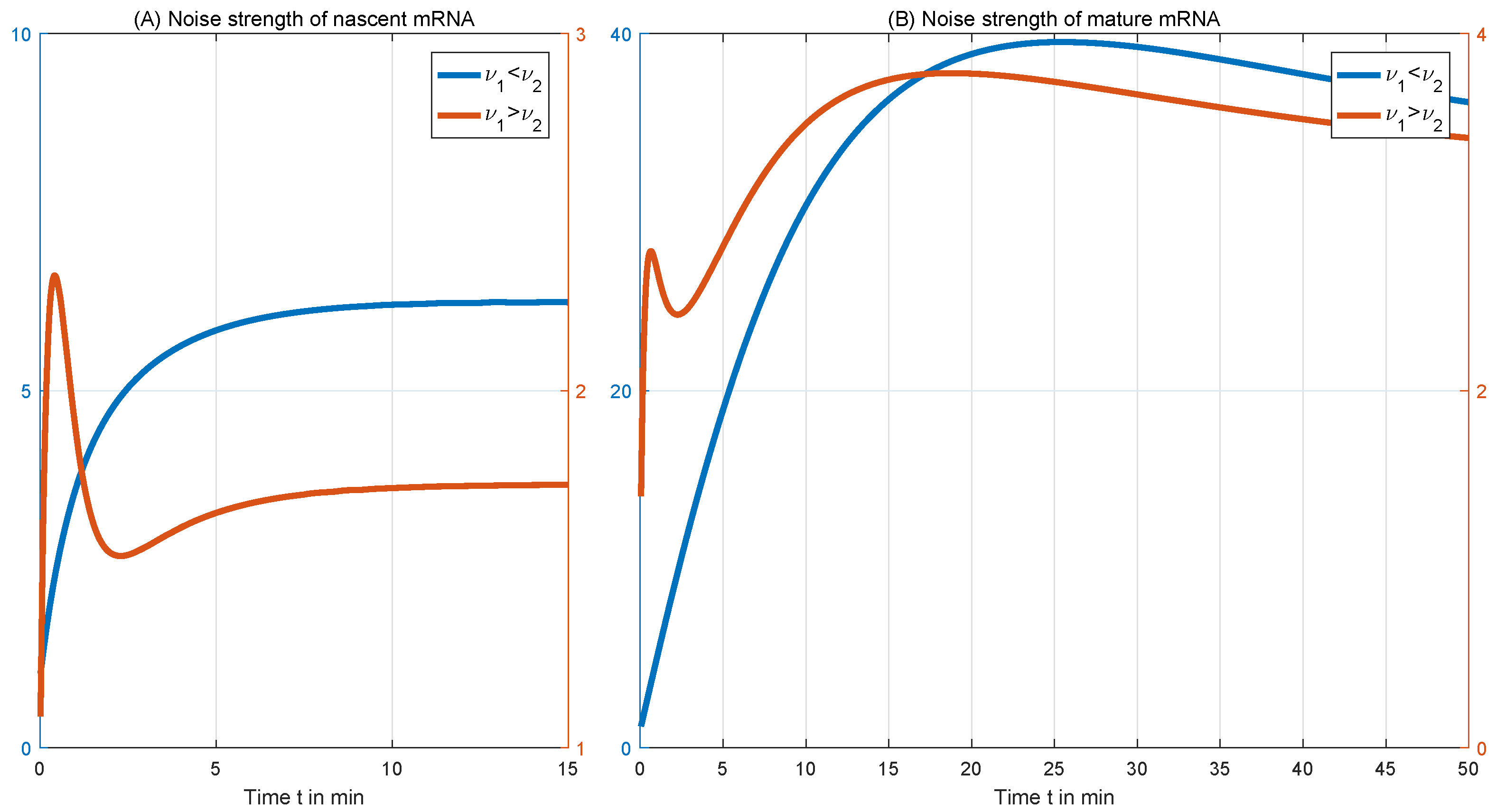
3.2. The Noise and the Skewness of Transcripts
4. Simulation and Discussion
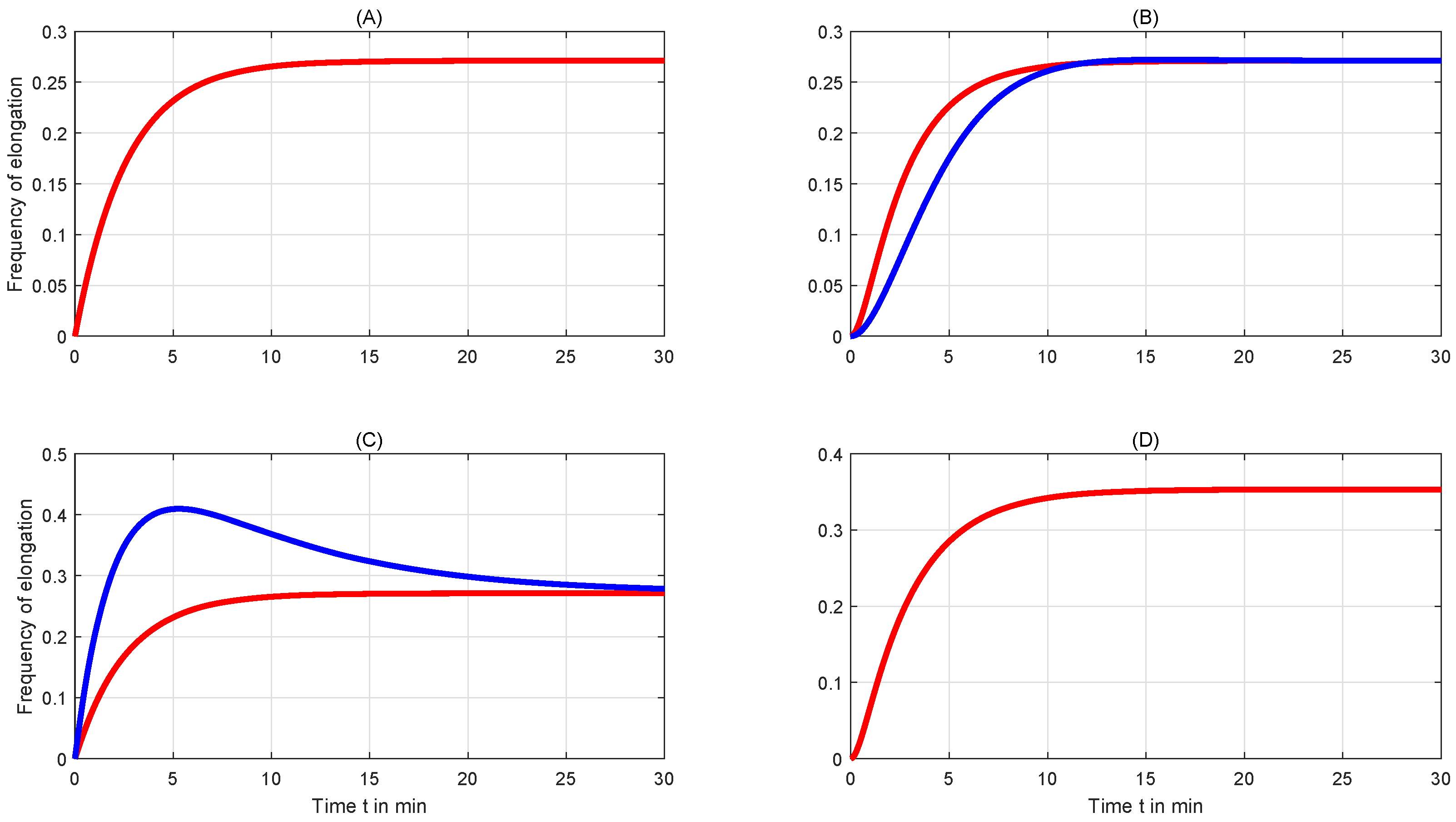
4.1. Comparison of Frequencies in Different Transcription Systems
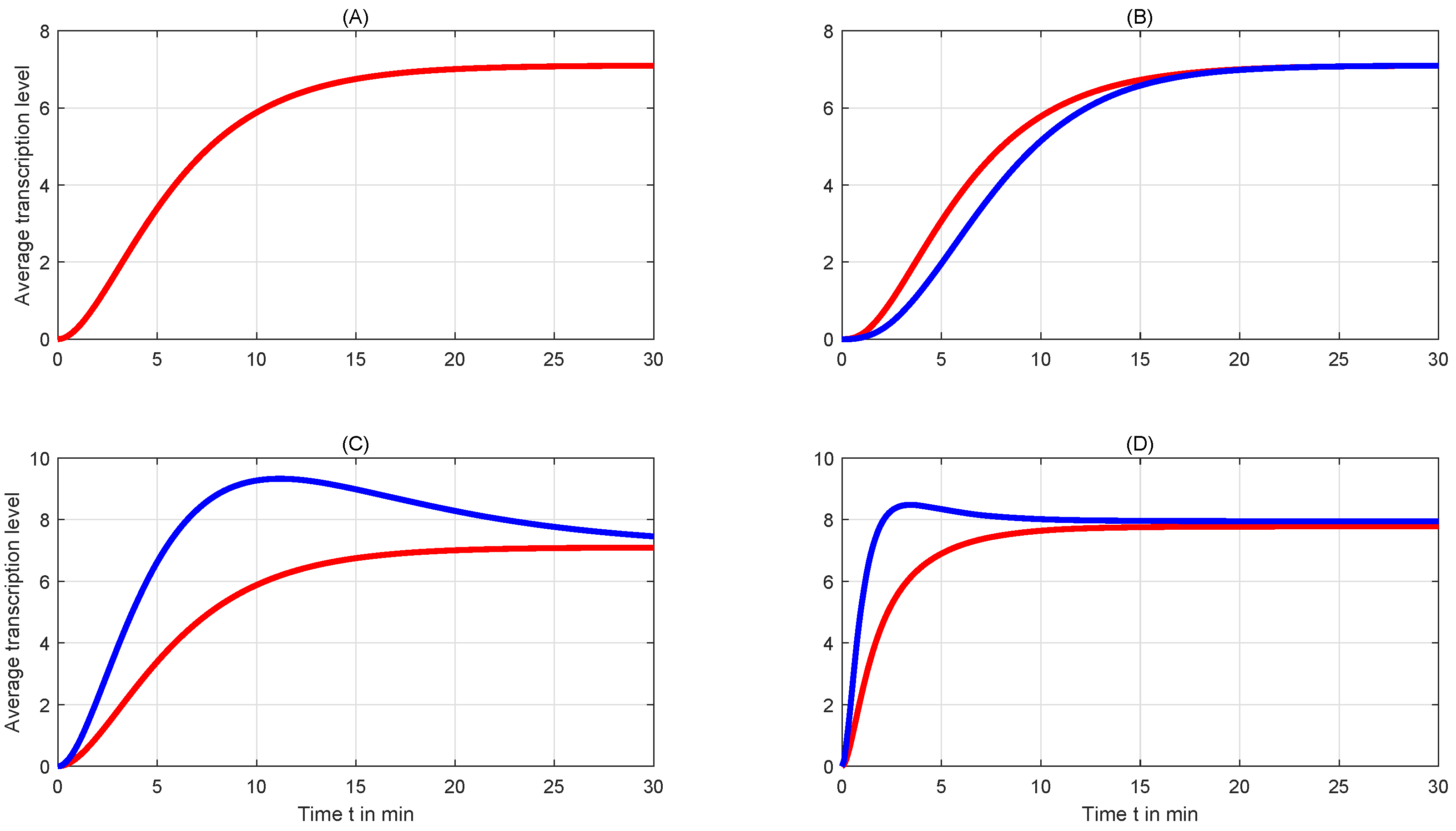
4.2. Comparison of Mean Levels
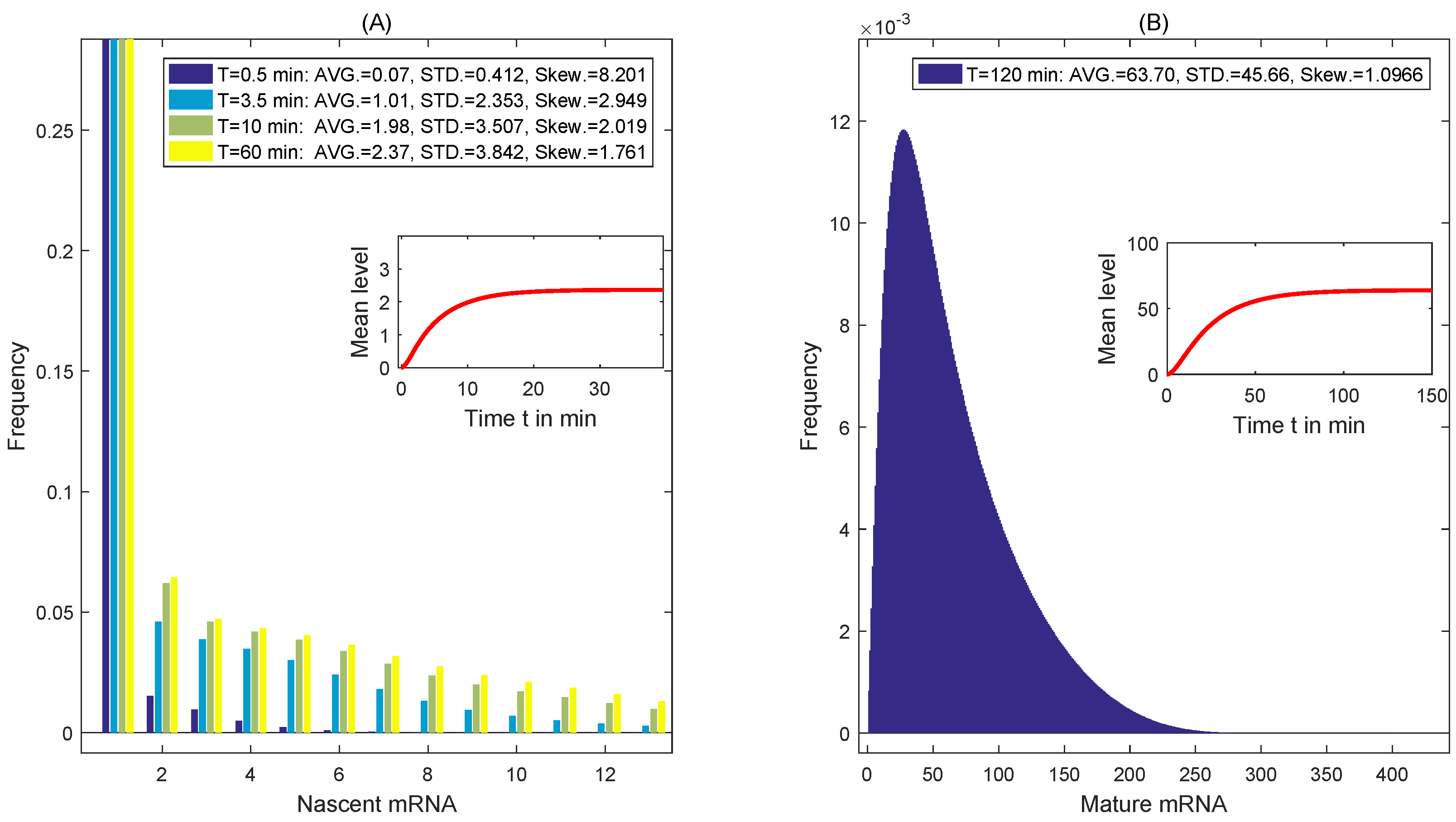
4.3. The Distribution of Transcripts
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic gene expression in a single cell. Science 2002, 297, 1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golding, I.; Paulsson, J.; Zawilski, S.M.; Cox, E.C. Real-time kinetics of gene activity in individual bacteria. Cell 2005, 123, 1025. [Google Scholar] [CrossRef] [Green Version]
- Blake, W.J.; Kaern, M.; Cantor, C.R.; Collins, J.J. Noise in eukaryotic gene expression. Nature 2003, 422, 633. [Google Scholar] [CrossRef] [PubMed]
- Suter, D.M.; Molina, M.; Gatfield, D.; Schneider, K.; Schibler, U.; Naef, F. Mammalian genes are transcribed with widely different bursting kinetics. Science 2011, 332, 472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartman, C.R.; Hamagami, N.; Keller, C.A.; Giardine, B.; Hardison, R.C.; Blobel, G.A.; Raj, A. Transcriptional burst initiation and polymerase pause release are key control points of transcriptional regulation. Mol. Cell 2019, 79, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senecal, A.; Munsky, B.; Proux, F.; Ly, N.; Braye, F.E.; Zimmer, C.; Mueller, F.; Darzacq, X. Transcription factors modulate c-Fos transcriptional bursts. Cell Rep. 2014, 8, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Zuin, J.; Roth, G.; Zhan, Y.; Cramard, J.; Redolfi, J.; Piskadlo, E.; Mach, P.; Kryzhanovska, M.; Tihanyi, G.; Kohler, H.; et al. Nonlinear control of transcription through enhancer-promoter interactions. Nature 2022, 604, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Peccoud, J.; Ycart, B. Markovian modeling of gene-product synthesis. Theor. Popul. Biol. 1995, 48, 222. [Google Scholar] [CrossRef]
- Tang, M. The mean and noise of stochastic gene transcription. J. Theor. Biol. 2008, 253, 271–280. [Google Scholar] [CrossRef]
- Cao, Z.; Filatova, T.; Oyarzun, D.A.; Grima, R. A stochastic model of gene expression with polymerase recruitment and pause release. Biophys. J. 2020, 119, 1002–1014. [Google Scholar] [CrossRef]
- Sun, Q.; Cai, Z.; Zhu, C. A novel dynamical regulation of mRNA distribution by cross-talking pathways. Mathematics 2022, 10, 1515. [Google Scholar] [CrossRef]
- Zhu, C.; Han, G.; Jiao, F. Dynamical regulation of mRNA distribution by cross-talking signaling pathways. Complexity 2020, 2020, 6402703. [Google Scholar] [CrossRef]
- Chen, J.; Jiao, F. A novel approach for calculating exact forms of mRNA distribution in single-cell measurements. Mathematics 2022, 10, 27. [Google Scholar] [CrossRef]
- Cao, Z.; Grima, R. Analytical distributions for detailed models of stochastic gene expression in eukaryotic cells. Proc. Natl. Acad. Sci. USA 2020, 117, 4682–4692. [Google Scholar] [CrossRef] [Green Version]
- Jia, C.; Grima, R. Frequency domain analysis of fluctuations of mRNA and protein copy numbers within a cell lineage: Theory and experimental validation. Phys. Rev. X 2021, 11, 021032. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, T. Markovian approaches to modeling intracellular reaction processes with molecular memory. Proc. Natl. Acad. Sci. USA 2019, 116, 23542–23550. [Google Scholar] [CrossRef] [Green Version]
- Zopf, C.J.; Quinn, K.; Zeidman, J.; Maheshri, N. Cell-cycle dependence of transcription dominates noise in gene expression. PLoS Comput. Biol. 2013, 9, e1003161. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Jiao, F.; Lin, G.; Yu, J.; Tang, M. The nonlinear dynamics and fluctuations of mRNA levels in cell cycle coupled transcription. PLoS Comput. Biol. 2019, 15, e100701. [Google Scholar] [CrossRef]
- Jiao, F.; Tang, M. Quantification of transcription noise’s impact on cell fate commitment with digital resolutions. Bioinformatics 2022, 38, 3062–3069. [Google Scholar] [CrossRef]
- Sun, Q.; Jiao, F.; Yu, J. The dynamics of gene transcription with a periodic synthesis rate. Nonlinear Dynamcis 2021, 104, 4477–4492. [Google Scholar] [CrossRef]
- Larson, D.R. What do expression dynamics tell us about the mechanism of transcription? Curr. Opin. Genet. Dev 2011, 21, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Yuan, Z.; Liu, P.; Zhou, T. Effects of promoter leakage on dynamics of gene expression. BMC Syst. Biol. 2015, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.; Grima, R. Plasticity of the truth table of low-leakage genetic logic gates. Phys. Rev. E 2018, 98, 062410. [Google Scholar] [CrossRef] [Green Version]
- Weisstein, E.W. Laplace Transform. Available online: https://mathworld.wolfram.com/ (accessed on 8 August 2020).
- Raj, A.; van den Bogaard, P.; Rifkin, S.A.; van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq wholetranscriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- MathWorks. Matlab 9.4.0.813654 (R2018a). Available online: https://ww2.mathworks.cn (accessed on 4 March 2020).
- Sun, Q.; Tang, M.; Yu, J. Temporal profile of gene transcription noise modulated by cross-talking signal transduction pathways. Bull. Math. Biol. 2012, 74, 375–398. [Google Scholar] [CrossRef]
- Yu, J.; Sun, Q.; Tang, M. The nonlinear dynamics and fluctuations of mRNA levels in cross-talking pathway activated transcription. J. Theor. Biol. 2014, 363, 223–234. [Google Scholar] [CrossRef]
- Jiao, F.; Zhu, C. Regulation of gene activation by competitive cross talking pathways. Biphysical J. 2020, 119, 1204–1214. [Google Scholar] [CrossRef]
- Jiao, F.; Ren, J.; Yu, J. Analytical formula and dynamic profile of mRNA distribution. Discret. Contin. Dyn. Syst. B 2020, 25, 241–257. [Google Scholar] [CrossRef] [Green Version]
- Shyu, A.B.; Greenberg, M.E.; Belasco, J.G. The c-Fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes Dev. 1989, 3, 60–72. [Google Scholar] [CrossRef] [Green Version]
- Munsky, B.; Khammash, M. The finite state projection algorithm for the solution of the chemical master equation. J. Chem. Phys. 2006, 124, 044104. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Stangner, K.; Schmitt, T.; Wintsche, A.; Engeland, K. Timing of transcription during the cell cycle: Protein complexes binding to E2F, E2F/CLE, CDE/CHR, or CHR promoter elements define early and late cell cycle gene expression. Oncotarget 2017, 8, 97736–97748. [Google Scholar] [CrossRef] [Green Version]
- Caveney, P.M.; Norred, S.E.; Chin, C.W.; Boreyko, J.B.; Razooky, B.S.; Retterer, S.T.; Collier, C.P.; Simpson, M.L. Resource sharing controls gene expression bursting. ACS Synth. Biol. 2017, 6, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Jia, C. Simplification of Markov chains with infinite state space and the mathematical theory of random gene expression bursts. Phys. Rev. E 2017, 96, 032402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, C. Kinetic foundation of the zero-inflated negative binomial model for single-cell RNA sequencing data. SIAM J. Appl. Math. 2020, 80, 1336–1355. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.; Zhou, T.; Zhang, J. Exploring dissipative sources of non-Markovian biochemical reaction systems. Phys. Rev. E 2021, 103, 052411. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Kocarev, L. First encounters on Bethe lattices and Cayley trees. Commun. Nonlinear Sci. Numer. Simul. 2021, 95, 105594. [Google Scholar] [CrossRef]
- Gao, L.; Peng, J.; Tang, C. Optimizing the first-passage process on a class of fractal scale-free trees. Fractal Fract. 2021, 5, 184. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, C.; Jiao, F. A generalized moment-based method for estimating parameters of stochastic gene transcription. Math. Biosci. 2022, 345, 108780. [Google Scholar] [CrossRef]
- Cao, Z.; Grima, R. Accuracy of parameter estimation for auto-regulatory transcriptional feedback loops from noisy data. J. R. Soc. Interface 2019, 16, 20180967. [Google Scholar] [CrossRef]
- Larke, M.S.C.; Schwessinger, R.; Nojima, T.; Telenius, J.; Beagrie, R.A.; Downes, D.J.; Oudelaar, A.M.; Truch, J.; Graham, B.; Bender, M.A.; et al. Enhancers predominantly regulate gene expression during differentiation via transcription initiation. Mol. Cell 2021, 81, 983–997.e7. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, C.; Chen, Z.; Sun, Q. Stochastic Transcription with Alterable Synthesis Rates. Mathematics 2022, 10, 2189. https://doi.org/10.3390/math10132189
Zhu C, Chen Z, Sun Q. Stochastic Transcription with Alterable Synthesis Rates. Mathematics. 2022; 10(13):2189. https://doi.org/10.3390/math10132189
Chicago/Turabian StyleZhu, Chunjuan, Zibo Chen, and Qiwen Sun. 2022. "Stochastic Transcription with Alterable Synthesis Rates" Mathematics 10, no. 13: 2189. https://doi.org/10.3390/math10132189
APA StyleZhu, C., Chen, Z., & Sun, Q. (2022). Stochastic Transcription with Alterable Synthesis Rates. Mathematics, 10(13), 2189. https://doi.org/10.3390/math10132189