What Room for Two-Dimensional Gel-Based Proteomics in a Shotgun Proteomics World?



Abstract
1. Introduction
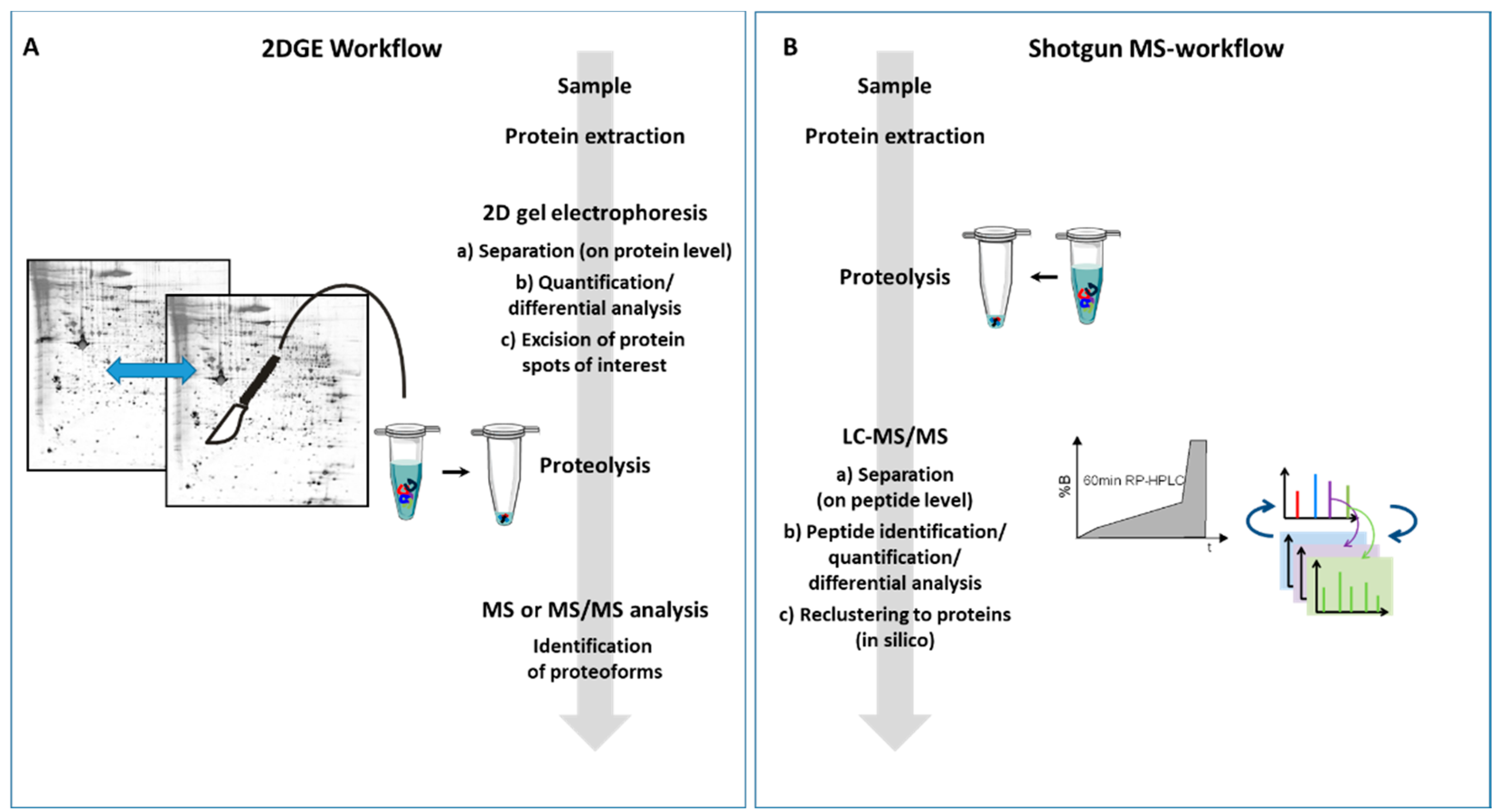
2. Peptides or Proteins—That Is the Question
3. When an Unpredictable Subset of Proteins Is to Be Analyzed: The Example of Immunome/Allergome Studies
4. Going to the Essence of Proteomics: Proteoforms and Post-Translational Modifications
4.1. Guidelines for Use
4.2. Hypothesis Validation: Getting the Most from the 2DGE Data
4.3. The Quest for PTMs: Unsupervised PTM Analysis as a Strength of 2DGE Proteomics
4.4. The Case of Protein Truncation
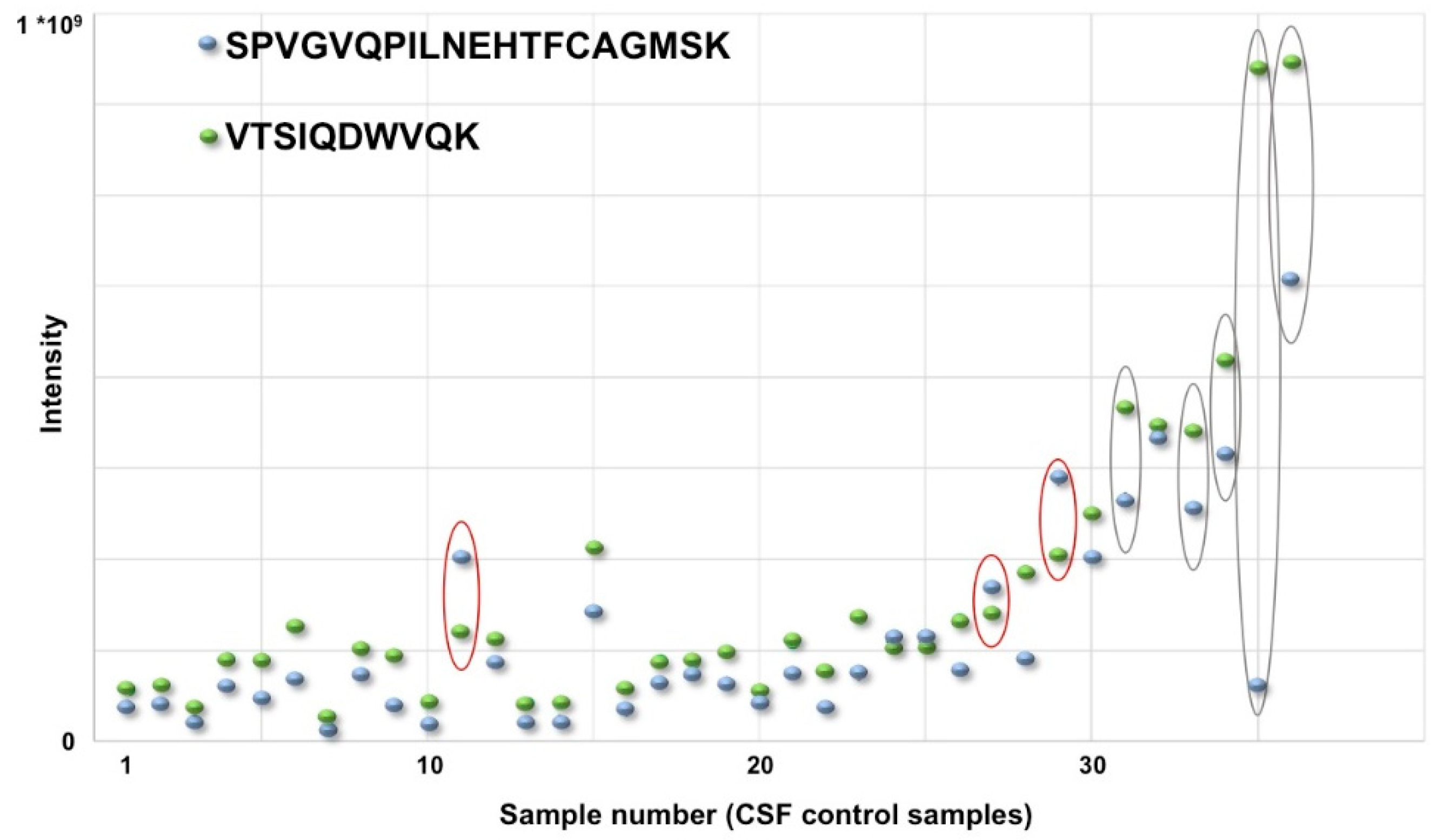
5. 2DGE Proteomics in the Most Difficult Field: Clinical Proteomics
6. As a Conclusion: May Look Slow and Cumbersome, but Still Valuable If Not Irreplaceable
Funding
Conflicts of Interest
Abbreviations
| 2D | two dimensional |
| 2DGE | two-dimensional gel electrophoresis |
| BPH | benign prostate hyperplasia |
| CSF | cerebrospinal fluid |
| DIGE | difference in-gel electrophoresis |
| emPAI | exponentially modified protein abundance index |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| Hp | haptoglobin |
| IEF | isoelectric focusing |
| IgE | immunoglobulin E |
| LC-MS/MS | liquid chromatography tandem mass spectrometry |
| LFQ | label-free quantification |
| MFAP-4 | microfibril–associated protein 4 |
| MS | mass spectrometry |
| MS1 | peptide mass spectrum |
| MS2 or MS/MS | tandem mass spectrum |
| MW | molecular weight |
| PAGE | polyacrylamide gel electrophoresis |
| PCa | prostate cancer |
| pI | isoelectric point |
| PD | Parkinson’s disease |
| PSM | peptide spectrum match |
| PTM | post-translational modification |
| RT-qPCR | real-time quantitative polymerase chain reaction |
| SDS | Sodium-dodecyl-sulfate |
References
- MacGillivray, A.J.; Rickwood, D. The heterogeneity of mouse-chromatin nonhistone proteins as evidenced by two-dimensional polyacrylamide-gel electrophoresis and ion-exchange chromatography. Eur. J. Biochem. 1974, 41, 181–190. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- Rabilloud, T. Paleoproteomics explained to youngsters: How did the wedding of two-dimensional electrophoresis and protein sequencing spark proteomics on: Let there be light. J. Proteom. 2014, 107, 5–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yates, J.R.; McCormack, A.L.; Schieltz, D.; Carmack, E.; Link, A. Direct analysis of protein mixtures by tandem mass spectrometry. J. Protein Chem. 1997, 16, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Rogowska-Wrzesinska, A.; Le Bihan, M.C.; Thaysen-Andersen, M.; Roepstorff, P. 2D gels still have a niche in proteomics. J. Proteom. 2013, 88, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de-Souza, D. 2DE: The phoenix of proteomics. J. Proteom. 2014, 104, 140–150. [Google Scholar] [CrossRef]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the Concept and Practice of Two-Dimensional Gel Electrophoresis in the Analysis of Proteomes at the Proteoform Level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef]
- Hoogland, C.; Mostaguir, K.; Sanchez, J.C.; Hochstrasser, D.F.; Appel, R.D. SWISS-2DPAGE, ten years later. Proteomics 2004, 4, 2352–2356. [Google Scholar] [CrossRef]
- Beck, M.; Schmidt, A.; Malmstroem, J.; Claassen, M.; Ori, A.; Szymborska, A.; Herzog, F.; Rinner, O.; Ellenberg, J.; Aebersold, R. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011, 7, 549. [Google Scholar] [CrossRef]
- Campostrini, N.; Areces, L.B.; Rappsilber, J.; Pietrogrande, M.C.; Dondi, F.; Pastorino, F.; Ponzoni, M.; Righetti, P.G. Spot overlapping in two-dimensional maps: A serious problem ignored for much too long. Proteomics 2005, 5, 2385–2395. [Google Scholar] [CrossRef]
- Hunsucker, S.W.; Duncan, M.W. Is protein overlap in two-dimensional gels a serious practical problem? Proteomics 2006, 6, 1374–1375. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Yang, H.; Peng, F.; Li, J.; Mu, Y.; Long, Y.; Cheng, T.; Huang, Y.; Li, Z.; Lu, M.; et al. How many proteins can be identified in a 2DE gel spot within an analysis of a complex human cancer tissue proteome? Electrophoresis 2018, 39, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High resolution quantitative proteomics of HeLa cells protein species using stable isotope labeling with amino acids in cell culture (SILAC), two-dimensional gel electrophoresis (2DE) and nano-liquid chromatograpohy coupled to an LTQ-OrbitrapMass spectrometer. Mol. Cell. Proteom. MCP 2013, 12, 529–538. [Google Scholar] [CrossRef]
- Li, J.J.; Bickel, P.J.; Biggin, M.D. System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2014, 2, e270. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Lawless, C.; Holman, S.W.; Brownridge, P.; Lanthaler, K.; Harman, V.M.; Watkins, R.; Hammond, D.E.; Miller, R.L.; Sims, P.F.G.; Grant, C.M.; et al. Direct and Absolute Quantification of over 1800 Yeast Proteins via Selected Reaction Monitoring. Mol. Cell. Proteom. MCP 2016, 15, 1309–1322. [Google Scholar] [CrossRef]
- Thiede, B.; Höhenwarter, W.; Krah, A.; Mattow, J.; Schmid, M.; Schmidt, F.; Jungblut, P.R. Peptide mass fingerprinting. Methods San Diego Calif. 2005, 35, 237–247. [Google Scholar] [CrossRef]
- Butt, R.H.; Coorssen, J.R. Postfractionation for enhanced proteomic analyses: Routine electrophoretic methods increase the resolution of standard 2D-PAGE. J. Proteome Res. 2005, 4, 982–991. [Google Scholar] [CrossRef]
- Colignon, B.; Raes, M.; Dieu, M.; Delaive, E.; Mauro, S. Evaluation of three-dimensional gel electrophoresis to improve quantitative profiling of complex proteomes. Proteomics 2013, 13, 2077–2082. [Google Scholar] [CrossRef]
- Haile, D.J.; Rouault, T.A.; Tang, C.K.; Chin, J.; Harford, J.B.; Klausner, R.D. Reciprocal control of RNA-binding and aconitase activity in the regulation of the iron-responsive element binding protein: Role of the iron-sulfur cluster. Proc. Natl. Acad. Sci. USA 1992, 89, 7536–7540. [Google Scholar] [CrossRef]
- Chait, B.T. CHEMISTRY: Mass Spectrometry: Bottom-Up or Top-Down? Science 2006, 314, 65–66. [Google Scholar] [CrossRef] [PubMed]
- Fornelli, L.; Toby, T.K.; Schachner, L.F.; Doubleday, P.F.; Srzentić, K.; DeHart, C.J.; Kelleher, N.L. Top-down proteomics: Where we are, where we are going? J. Proteom. 2018, 175, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Toby, T.K.; Fornelli, L.; Kelleher, N.L. Progress in Top-Down Proteomics and the Analysis of Proteoforms. Annu. Rev. Anal. Chem. 2016, 9, 499–519. [Google Scholar] [CrossRef] [PubMed]
- The Consortium for Top Down Proteomics; Smith, L.M.; Kelleher, N.L. Proteoform: A single term describing protein complexity. Nat. Methods 2013, 10. [Google Scholar] [CrossRef]
- Pappin, D.J.C.; Hojrup, P.; Bleasby, A.J. Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 1993, 3, 327–332. [Google Scholar] [CrossRef]
- Domon, B. Mass Spectrometry and Protein Analysis. Science 2006, 312, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef]
- Perkins, D.N.; Pappin, D.J.C.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of Shotgun Proteomic Data: The Protein Inference Problem. Mol. Cell. Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Sánchez, A.; Ramos, Y.; Schmidt, A.; Müller, M.; Betancourt, L.; González, L.J.; Vera, R.; Padron, G.; Besada, V. In silico analysis of accurate proteomics, complemented by selective isolation of peptides. J. Proteom. 2011, 74, 2071–2082. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Searle, B.C. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. PROTEOMICS 2010, 10, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Yue, Q.X.; Guan, S.H.; Wu, W.Y.; Yang, M.; Jiang, B.H.; Liu, X.; Guo, D.A. Proteomic analysis of possible target-related proteins of cyclophosphamide in mice thymus. Food Chem. Toxicol. 2009, 47, 1841–1847. [Google Scholar] [CrossRef] [PubMed]
- Uszkoreit, J.; Maerkens, A.; Perez-Riverol, Y.; Meyer, H.E.; Marcus, K.; Stephan, C.; Kohlbacher, O.; Eisenacher, M. PIA: An Intuitive Protein Inference Engine with a Web-Based User Interface. J. Proteome Res. 2015, 14, 2988–2997. [Google Scholar] [CrossRef]
- Uszkoreit, J.; Perez-Riverol, Y.; Eggers, B.; Marcus, K.; Eisenacher, M. Protein Inference Using PIA Workflows and PSI Standard File Formats. J. Proteome Res. 2019, 18, 741–747. [Google Scholar] [CrossRef]
- Pfeuffer, J.; Sachsenberg, T.; Dijkstra, T.M.H.; Serang, O.; Reinert, K.; Kohlbacher, O. EPIFANY: A Method for Efficient High-Confidence Protein Inference. J. Proteome Res. 2020, 19, 1060–1072. [Google Scholar] [CrossRef]
- Zhang, B.; Pirmoradian, M.; Zubarev, R.; Käll, L. Covariation of Peptide Abundances Accurately Reflects Protein Concentration Differences. Mol. Cell. Proteom. 2017, 16, 936–948. [Google Scholar] [CrossRef]
- Schilde, L.M.; Kösters, S.; Steinbach, S.; Schork, K.; Eisenacher, M.; Galozzi, S.; Turewicz, M.; Barkovits, K.; Mollenhauer, B.; Marcus, K.; et al. Protein variability in cerebrospinal fluid and its possible implications for neurological protein biomarker research. PLoS ONE 2018, 13, e0206478. [Google Scholar] [CrossRef]
- Abdi, F.; Quinn, J.F.; Jankovic, J.; McIntosh, M.; Leverenz, J.B.; Peskind, E.; Nixon, R.; Nutt, J.; Chung, K.; Zabetian, C.; et al. Detection of biomarkers with a multiplex quantitative proteomic platform in cerebrospinal fluid of patients with neurodegenerative disorders. J. Alzheimers Dis. 2006, 9, 293–348. [Google Scholar] [CrossRef] [PubMed]
- Argüelles, S.; Venero, J.L.; García-Rodriguez, S.; Tomas-Camardiel, M.; Ayala, A.; Cano, J.; Machado, A. Use of haptoglobin and transthyretin as potential biomarkers for the preclinical diagnosis of Parkinson’s disease. Neurochem. Int. 2010, 57, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, S.; Mariosa, D.; Hammar, N.; Andersson, J.; Ingre, C.; Walldius, G.; Fang, F. Peripheral immune biomarkers and neurodegenerative diseases: A prospective cohort study with 20 years of follow-up. Ann. Neurol. 2019, 86, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.N.; Lee, H.W.; Cho, J.-Y.; Suk, K. Neuronal pentraxin receptor in cerebrospinal fluid as a potential biomarker for neurodegenerative diseases. Brain Res. 2009, 1265, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Fernández, M.; Martin, R.; Rivera-Vallvé, S.; Borrás, E.; Chiva, C.; Julià, E.; Rovira, A.; Cantó, E.; Alvarez-Cermeño, J.C.; et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain 2010, 133, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Orenes-Piñero, E.; Hernández-Romero, D.; De Torre, C.; Vilchez, J.A.; Martínez, M.; Romero-Aniorte, A.I.; Climent, V.; García-Honrubia, A.; Valdés, M.; Marín, F. Identification and confirmation of haptoglobin as a potential serum biomarker in hypertrophic cardiomyopathy using proteomic approaches. Ann. Med. 2013, 45, 341–347. [Google Scholar] [CrossRef]
- Garibay-Cerdenares, O.; Hernández-Ramírez, V.; Osorio-Trujillo, J.; Hernández-Ortíz, M.; Gallardo-Rincón, D.; De León, D.C.; Encarnación-Guevara, S.; Villegas-Pineda, J.; Talamás-Rohana, P. Proteomic identification of fucosylated haptoglobin alpha isoforms in ascitic fluids and its localization in ovarian carcinoma tissues from Mexican patients. J. Ovarian Res. 2014, 7, 27. [Google Scholar] [CrossRef]
- Villegas-Pineda, J.C.; Garibay-Cerdenares, O.L.; Hernández-Ramírez, V.I.; Gallardo-Rincón, D.; De León, D.C.; Pérez-Montiel-Gómez, M.D.; Talamás-Rohana, P. Integrins and haptoglobin: Molecules overexpressed in ovarian cancer. Pathol. Res. Pract. 2015, 211, 973–981. [Google Scholar] [CrossRef]
- Halbgebauer, S.; Öckl, P.; Wirth, K.; Steinacker, P.; Otto, M. Protein biomarkers in Parkinson’s disease: Focus on cerebrospinal fluid markers and synaptic proteins: Protein Biomarkers in Parkinson’s Disease. Mov. Disord. 2016, 31, 848–860. [Google Scholar] [CrossRef]
- Fish, R.G.; Gill, T.S.; Adams, M.; Kerby, I. Serum haptoglobin and α1-acid glycoprotein as indicators of the effectiveness of cis-diamminedichloroplatinum (CDDP) in ovarian cancer patients—A preliminary report. Eur. J. Cancer Clin. Oncol. 1984, 20, 625–630. [Google Scholar] [CrossRef]
- Ahmed, N.; Barker, G.; Oliva, K.T.; Hoffmann, P.; Riley, C.; Reeve, S.; Smith, A.I.; Kemp, B.E.; Quinn, M.A.; Rice, G.E. Proteomic-based identification of haptoglobin-1 precursor as a novel circulating biomarker of ovarian cancer. Br. J. Cancer 2004, 91, 129–140. [Google Scholar] [CrossRef] [PubMed]
- May, C.; Brosseron, F.; Chartowski, P.; Meyer, H.E.; Marcus, K. Differential proteome analysis using 2D-DIGE. Methods Mol. Biol. Clifton N.J. 2012, 893, 75–82. [Google Scholar] [CrossRef]
- Riederer, B.M. Non-covalent and covalent protein labeling in two-dimensional gel electrophoresis. J. Proteom. 2008, 71, 231–244. [Google Scholar] [CrossRef]
- Butt, R.H.; Coorssen, J.R. Coomassie blue as a near-infrared fluorescent stain: A systematic comparison with Sypro Ruby for in-gel protein detection. Mol. Cell. Proteom. MCP 2013, 12, 3834–3850. [Google Scholar] [CrossRef] [PubMed]
- Dyballa, N.; Metzger, S. Fast and sensitive coomassie staining in quantitative proteomics. Methods Mol. Biol. Clifton N. J. 2012, 893, 47–59. [Google Scholar] [CrossRef]
- Rabilloud, T. Silver staining of 2D electrophoresis gels. Methods Mol. Biol. Clifton N. J. 2012, 893, 61–73. [Google Scholar] [CrossRef]
- Herrmann, A.G.; Searcy, J.L.; Le Bihan, T.; McCulloch, J.; Deighton, R.F. Total variance should drive data handling strategies in third generation proteomic studies. Proteomics 2013, 13, 3251–3255. [Google Scholar] [CrossRef]
- Sandomenico, A.; Severino, V.; Chambery, A.; Focà, A.; Focà, G.; Farina, C.; Ruvo, M. A comparative structural and bioanalytical study of IVIG clinical lots. Mol. Biotechnol. 2013, 54, 983–995. [Google Scholar] [CrossRef]
- Nebija, D.; Noe, C.; Urban, E.; Lachmann, B. Quality Control and Stability Studies with the Monoclonal Antibody, Trastuzumab: Application of 1D- vs. 2D-Gel Electrophoresis. Int. J. Mol. Sci. 2014, 15, 6399–6411. [Google Scholar] [CrossRef]
- Jawa, V.; Joubert, M.K.; Zhang, Q.; Deshpande, M.; Hapuarachchi, S.; Hall, M.P.; Flynn, G.C. Evaluating Immunogenicity Risk Due to Host Cell Protein Impurities in Antibody-Based Biotherapeutics. AAPS J. 2016, 18, 1439–1452. [Google Scholar] [CrossRef]
- Al Shweiki, M.R.; Mönchgesang, S.; Majovsky, P.; Thieme, D.; Trutschel, D.; Hoehenwarter, W. Assessment of Label-Free Quantification in Discovery Proteomics and Impact of Technological Factors and Natural Variability of Protein Abundance. J. Proteome Res. 2017, 16, 1410–1424. [Google Scholar] [CrossRef]
- Trinkle-Mulcahy, L.; Boulon, S.; Lam, Y.W.; Urcia, R.; Boisvert, F.M.; Vandermoere, F.; Morrice, N.A.; Swift, S.; Rothbauer, U.; Leonhardt, H.; et al. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 2008, 183, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef]
- Chevallet, M.; Procaccio, V.; Rabilloud, T. A nonradioactive double detection method for the assignment of spots in two-dimensional blots. Anal. Biochem. 1997, 251, 69–72. [Google Scholar] [CrossRef]
- Kusch, K.; Uecker, M.; Liepold, T.; Möbius, W.; Hoffmann, C.; Neumann, H.; Werner, H.B.; Jahn, O. Partial Immunoblotting of 2D-Gels: A Novel Method to Identify Post-Translationally Modified Proteins Exemplified for the Myelin Acetylome. Proteomes 2017, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Barrera, C.; Millon, L.; Rognon, B.; Quadroni, M.; Roussel, S.; Dalphin, J.C.; Court-Fortune, I.; Caillaud, D.; Jouneau, S.; Fellrath, J.M.; et al. Immunoreactive proteins of Saccharopolyspora rectivirgula for farmer’s lung serodiagnosis. Proteom. Clin. Appl. 2014, 8, 971–981. [Google Scholar] [CrossRef]
- Connolly, J.P.; Comerci, D.; Alefantis, T.G.; Walz, A.; Quan, M.; Chafin, R.; Grewal, P.; Mujer, C.V.; Ugalde, R.A.; DelVecchio, V.G. Proteomic analysis of Brucella abortus cell envelope and identification of immunogenic candidate proteins for vaccine development. Proteomics 2006, 6, 3767–3780. [Google Scholar] [CrossRef]
- Delvecchio, V.G.; Connolly, J.P.; Alefantis, T.G.; Walz, A.; Quan, M.A.; Patra, G.; Ashton, J.M.; Whittington, J.T.; Chafin, R.D.; Liang, X.; et al. Proteomic profiling and identification of immunodominant spore antigens of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis. Appl. Environ. Microbiol. 2006, 72, 6355–6363. [Google Scholar] [CrossRef] [PubMed]
- Fajardo Bonin, R.; Chapeaurouge, A.; Perales, J.; Da Silva, J.G.; Do Nascimento, H.J.; D’Alincourt Carvalho Assef, A.P.; Moreno Senna, J.P. Identification of immunogenic proteins of the bacterium Acinetobacter baumannii using a proteomic approach. Proteom. Clin. Appl. 2014, 8, 916–923. [Google Scholar] [CrossRef]
- Gaur, R.; Alam, S.I.; Kamboj, D.V. Immunoproteomic Analysis of Antibody Response of Rabbit Host Against Heat-Killed Francisella tularensis Live Vaccine Strain. Curr. Microbiol. 2017, 74, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Pitarch, A.; Jimenez, A.; Nombela, C.; Gil, C. Decoding serological response to Candida cell wall immunome into novel diagnostic, prognostic, and therapeutic candidates for systemic candidiasis by proteomic and bioinformatic analyses. Mol. Cell. Proteom. 2006, 5, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Pellon, A.; Ramirez-Garcia, A.; Buldain, I.; Antoran, A.; Rementeria, A.; Hernando, F.L. Immunoproteomics-Based Analysis of the Immunocompetent Serological Response to Lomentospora prolificans. J. Proteome Res. 2016, 15, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Buldain, I.; Ramirez-Garcia, A.; Pellon, A.; Antoran, A.; Sevilla, M.J.; Rementeria, A.; Hernando, F.L. Cyclophilin and enolase are the most prevalent conidial antigens of Lomentospora prolificans recognized by healthy human salivary IgA and cross-react with Aspergillus fumigatus. Proteom. Clin. Appl. 2016, 10, 1058–1067. [Google Scholar] [CrossRef]
- Garcia-Lunar, P.; Regidor-Cerrillo, J.; Gutierrez-Exposito, D.; Ortega-Mora, L.; Alvarez-Garcia, G. First 2-DE approach towards characterising the proteome and immunome of Besnoitia besnoiti in the tachyzoite stage. Vet. Parasitol. 2013, 195, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Reamtong, O.; Rujimongkon, K.; Sookrung, N.; Saeung, A.; Thiangtrongjit, T.; Sakolvaree, Y.; Thammapalo, S.; Loymek, S.; Chaicumpa, W. Immunome and immune complex-forming components of Brugia malayi identified by microfilaremic human sera. Exp. Parasitol. 2019, 200, 92–98. [Google Scholar] [CrossRef]
- Pitarch, A.; Nombela, C.; Gil, C. Prediction of the clinical outcome in invasive candidiasis patients based on molecular fingerprints of five anti-Candida antibodies in serum. Mol. Cell. Proteom. 2011, 10, M110.004010. [Google Scholar] [CrossRef]
- Pitarch, A.; Nombela, C.; Gil, C. Seroprofiling at the Candida albicans protein species level unveils an accurate molecular discriminator for candidemia. J. Proteom. 2016, 134, 144–162. [Google Scholar] [CrossRef]
- Singh, B.; Sharma, G.L.; Oellerich, M.; Kumar, R.; Singh, S.; Bhadoria, D.P.; Katyal, A.; Reichard, U.; Asif, A.R. Novel cytosolic allergens of Aspergillus fumigatus identified from germinating conidia. J. Proteome Res. 2010, 9, 5530–5541. [Google Scholar] [CrossRef]
- Ghosh, N.; Sircar, G.; Saha, B.; Pandey, N.; Bhattacharya, S.G. Search for Allergens from the Pollen Proteome of Sunflower (Helianthus annuus L.): A Major Sensitizer for Respiratory Allergy Patients. PLoS ONE 2015, 10, e0138992. [Google Scholar] [CrossRef]
- Saha, B.; Bhattacharya, S.G. Charting novel allergens from date palm pollen (Phoenix sylvestris) using homology driven proteomics. J. Proteom. 2017, 165, 1–10. [Google Scholar] [CrossRef]
- Park, K.H.; Lee, J.; Lee, J.Y.; Lee, S.C.; Sim, D.W.; Shin, J.U.; Park, C.O.; Lee, J.H.; Lee, K.H.; Jeong, K.Y.; et al. Sensitization to various minor house dust mite allergens is greater in patients with atopic dermatitis than in those with respiratory allergic disease. Clin. Exp. Allergy 2018, 48, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, M.; Di Cagno, R.; Minervini, F.; Rizzello, C.G.; Gobbetti, M. Two-dimensional electrophoresis and IgE-mediated food allergy. Electrophoresis 2010, 31, 2126–2136. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Xiang, Y.; Nakamura, H.; Masuko, K.; Yudoh, K.; Noyori, K.; Nishioka, K.; Saito, T.; Kato, T. Identification of novel citrullinated autoantigens of synovium in rheumatoid arthritis using a proteomic approach. Arthritis Res. Ther. 2006, 8, R175. [Google Scholar] [CrossRef]
- Kinloch, A.; Lundberg, K.; Wait, R.; Wegner, N.; Lim, N.H.; Zendman, A.J.W.; Saxne, T.; Malmström, V.; Venables, P.J. Synovial fluid is a site of citrullination of autoantigens in inflammatory arthritis. Arthritis Rheum. 2008, 58, 2287–2295. [Google Scholar] [CrossRef]
- Goëb, V.; Thomas-L’Otellier, M.; Daveau, R.; Charlionet, R.; Fardellone, P.; Le Loët, X.; Tron, F.; Gilbert, D.; Vittecoq, O. Candidate autoantigens identified by mass spectrometry in early rheumatoid arthritis are chaperones and citrullinated glycolytic enzymes. Arthritis Res. Ther. 2009, 11, R38. [Google Scholar] [CrossRef] [PubMed]
- Bussone, G.; Dib, H.; Tamby, M.C.; Broussard, C.; Federici, C.; Woimant, G.; Camoin, L.; Guillevin, L.; Mouthon, L. Identification of new autoantibody specificities directed at proteins involved in the transforming growth factor β pathway in patients with systemic sclerosis. Arthritis Res. Ther. 2011, 13, R74. [Google Scholar] [CrossRef]
- Biswas, S.; Sharma, S.; Saroha, A.; Bhakuni, D.S.; Malhotra, R.; Zahur, M.; Oellerich, M.; Das, H.R.; Asif, A.R. Identification of novel autoantigen in the synovial fluid of rheumatoid arthritis patients using an immunoproteomics approach. PLoS ONE 2013, 8, e56246. [Google Scholar] [CrossRef] [PubMed]
- Mathey, E.K.; Derfuss, T.; Storch, M.K.; Williams, K.R.; Hales, K.; Woolley, D.R.; Al-Hayani, A.; Davies, S.N.; Rasband, M.N.; Olsson, T.; et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J. Exp. Med. 2007, 204, 2363–2372. [Google Scholar] [CrossRef]
- Derfuss, T.; Parikh, K.; Velhin, S.; Braun, M.; Mathey, E.; Krumbholz, M.; Kümpfel, T.; Moldenhauer, A.; Rader, C.; Sonderegger, P.; et al. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc. Natl. Acad. Sci. USA 2009, 106, 8302–8307. [Google Scholar] [CrossRef]
- Privitera, D.; Corti, V.; Alessio, M.; Volontè, M.A.; Volontè, A.; Lampasona, V.; Comi, G.; Martino, G.; Franciotta, D.; Furlan, R.; et al. Proteomic identification of aldolase A as an autoantibody target in patients with atypical movement disorders. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2013, 34, 313–320. [Google Scholar] [CrossRef]
- Kuwabara, Y.; Katayama, A.; Kurihara, S.; Orimo, H.; Takeshita, T. Immunoproteomic identification of anti-C9 autoimmune antibody in patients with seronegative obstetric antiphospholipid syndrome. PLoS ONE 2018, 13, e0198472. [Google Scholar] [CrossRef] [PubMed]
- Beadle, G.W.; Tatum, E.L. Genetic Control of Biochemical Reactions in Neurospora. Proc. Natl. Acad. Sci. USA 1941, 27, 499–506. [Google Scholar] [CrossRef] [PubMed]
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef]
- Bischoff, R.; Schlüter, H. Amino acids: Chemistry, functionality and selected non-enzymatic post-translational modifications. J. Proteom. 2012, 75, 2275–2296. [Google Scholar] [CrossRef]
- Wagner, G.R.; Hirschey, M.D. Nonenzymatic protein acylation as a carbon stress regulated by sirtuin deacylases. Mol. Cell 2014, 54, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Trub, A.G.; Hirschey, M.D. Reactive Acyl-CoA Species Modify Proteins and Induce Carbon Stress. Trends Biochem. Sci. 2018, 43, 369–379. [Google Scholar] [CrossRef]
- Thaysen-Andersen, M.; Packer, N.H. Advances in LC-MS/MS-based glycoproteomics: Getting closer to system-wide site-specific mapping of the N- and O-glycoproteome. Biochim. Biophys. Acta 2014, 1844, 1437–1452. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, Y.; Shi, X. Emerging roles of lysine methylation on non-histone proteins. Cell. Mol. Life Sci. CMLS 2015, 72, 4257–4272. [Google Scholar] [CrossRef]
- Wesche, J.; Kühn, S.; Kessler, B.M.; Salton, M.; Wolf, A. Protein arginine methylation: A prominent modification and its demethylation. Cell. Mol. Life Sci. CMLS 2017, 74, 3305–3315. [Google Scholar] [CrossRef]
- Xiong, Y.; Guan, K.-L. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 2012, 198, 155–164. [Google Scholar] [CrossRef]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [PubMed]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Zhao, Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol. Cell. Proteom. MCP 2015, 14, 2308–2315. [Google Scholar] [CrossRef]
- Hentschel, A.; Zahedi, R.P.; Ahrends, R. Protein lipid modifications—More than just a greasy ballast. Proteomics 2016, 16, 759–782. [Google Scholar] [CrossRef]
- Carrico, C.; Meyer, J.G.; He, W.; Gibson, B.W.; Verdin, E. The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab. 2018, 27, 497–512. [Google Scholar] [CrossRef]
- Seo, J.; Jeong, J.; Kim, Y.M.; Hwang, N.; Paek, E.; Lee, K.-J. Strategy for Comprehensive Identification of Post-translational Modifications in Cellular Proteins, Including Low Abundant Modifications: Application to Glyceraldehyde-3-phosphate Dehydrogenase. J. Proteome Res. 2008, 7, 587–602. [Google Scholar] [CrossRef]
- Niimori-Kita, K.; Tamamaki, N.; Koizumi, D.; Niimori, D. Matrin-3 is essential for fibroblast growth factor 2-dependent maintenance of neural stem cells. Sci. Rep. 2018, 8, 13412. [Google Scholar] [CrossRef]
- Dalzon, B.; Torres, A.; Diemer, H.; Ravanel, S.; Collin-Faure, V.; Pernet-Gallay, K.; Jouneau, P.-H.; Bourguignon, J.; Cianfrani, S.; Carrire, M.; et al. How reversible are the effects of silver nanoparticles on macrophages? A proteomic-instructed view. Environ. Sci. Nano 2019, 6, 3133–3157. [Google Scholar] [CrossRef]
- Sun, H.H.; Fukao, Y.; Ishida, S.; Yamamoto, H.; Maekawa, S.; Fujiwara, M.; Sato, T.; Yamaguchi, J. Proteomics Analysis Reveals a Highly Heterogeneous Proteasome Composition and the Post-translational Regulation of Peptidase Activity under Pathogen Signaling in Plants. J. Proteome Res. 2013, 12, 5084–5095. [Google Scholar] [CrossRef]
- Martins-de-Souza, D.; Maccarrone, G.; Wobrock, T.; Zerr, I.; Gormanns, P.; Reckow, S.; Falkai, P.; Schmitt, A.; Turck, C.W. Proteome analysis of the thalamus and cerebrospinal fluid reveals glycolysis dysfunction and potential biomarkers candidates for schizophrenia. J. Psychiatr. Res. 2010, 44, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Chevallet, M.; Luche, S.; Diemer, H.; Strub, J.M.; Van Dorsselaer, A.; Rabilloud, T. Sweet silver: A formaldehyde-free silver staining using aldoses as developing agents, with enhanced compatibility with mass spectrometry. Proteomics 2008, 8, 4853–4861. [Google Scholar] [CrossRef] [PubMed]
- Ryšlavá, H.; Doubnerová, V.; Kavan, D.; Vaněk, O. Effect of posttranslational modifications on enzyme function and assembly. J. Proteom. 2013, 92, 80–109. [Google Scholar] [CrossRef] [PubMed]
- Petrak, J.; Ivanek, R.; Toman, O.; Cmejla, R.; Cmejlova, J.; Vyoral, D.; Zivny, J.; Vulpe, C.D. Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics 2008, 8, 1744–1749. [Google Scholar] [CrossRef]
- Wang, P.; Bouwman, F.G.; Mariman, E.C. Generally detected proteins in comparative proteomics—A matter of cellular stress response? Proteomics 2009, 9, 2955–2966. [Google Scholar] [CrossRef]
- Lenglet, G.; Depauw, S.; Mendy, D.; David-Cordonnier, M.-H. Protein recognition of the S23906-1–DNA adduct by nuclear proteins: Direct involvement of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Biochem. J. 2013, 452, 147–159. [Google Scholar] [CrossRef]
- Angeloni, C.; Turroni, S.; Bianchi, L.; Fabbri, D.; Motori, E.; Malaguti, M.; Leoncini, E.; Maraldi, T.; Bini, L.; Brigidi, P.; et al. Novel targets of sulforaphane in primary cardiomyocytes identified by proteomic analysis. PLoS ONE 2013, 8, e83283. [Google Scholar] [CrossRef]
- Armand, L.; Biola-Clier, M.; Bobyk, L.; Collin-Faure, V.; Diemer, H.; Strub, J.-M.; Cianferani, S.; Van Dorsselaer, A.; Herlin-Boime, N.; Rabilloud, T.; et al. Molecular responses of alveolar epithelial A549 cells to chronic exposure to titanium dioxide nanoparticles: A proteomic view. J. Proteom. 2016, 134, 163–173. [Google Scholar] [CrossRef]
- Luche, S.; Eymard-Vernain, E.; Diemer, H.; Van Dorsselaer, A.; Rabilloud, T.; Lelong, C. Zinc oxide induces the stringent response and major reorientations in the central metabolism of Bacillus subtilis. J. Proteom. 2016, 135, 170–180. [Google Scholar] [CrossRef]
- Chiasserini, D.; Davidescu, M.; Orvietani, P.L.; Susta, F.; Macchioni, L.; Petricciuolo, M.; Castigli, E.; Roberti, R.; Binaglia, L.; Corazzi, L. 3-Bromopyruvate treatment induces alterations of metabolic and stress-related pathways in glioblastoma cells. J. Proteom. 2017, 152, 329–338. [Google Scholar] [CrossRef]
- Dalzon, B.; Aude-Garcia, C.; Collin-Faure, V.; Diemer, H.; Béal, D.; Dussert, F.; Fenel, D.; Schoehn, G.; Cianférani, S.; Carrière, M.; et al. Differential proteomics highlights macrophage-specific responses to amorphous silica nanoparticles. Nanoscale 2017, 9, 9641–9658. [Google Scholar] [CrossRef] [PubMed]
- Heeb, M.J.; Gabriel, O. Enzyme localization in gels. Methods Enzymol. 1984, 104, 416–439. [Google Scholar] [CrossRef]
- Gabriel, O.; Gersten, D.M. Staining for enzymatic activity after gel electrophoresis, I. Anal. Biochem. 1992, 203, 1–21. [Google Scholar] [CrossRef]
- Bischoff, K.M.; Shi, L.; Kennelly, P.J. The detection of enzyme activity following sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Anal. Biochem. 1998, 260, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Manrow, R.E.; Dottin, R.P. Demonstration, by renaturation in O’Farrell gels, of heterogeneity in Dictyostelium uridine diphosphoglucose pyrophosphorylase. Anal. Biochem. 1982, 120, 181–188. [Google Scholar] [CrossRef]
- Durocher, Y.; Chapdelaine, A.; Chevalier, S. Identification of cytosolic protein tyrosine kinases of human prostate by renaturation after SDS/PAGE. Biochem. J. 1992, 284, 653–658. [Google Scholar] [CrossRef]
- Brochu, G.; Shah, G.M.; Poirier, G.G. Purification of poly (ADP-ribose) glycohydrolase and detection of its isoforms by a zymogram following one- or two-dimensional electrophoresis. Anal. Biochem. 1994, 218, 265–272. [Google Scholar] [CrossRef]
- Chen, S.; Meng, F.; Chen, Z.; Tomlinson, B.N.; Wesley, J.M.; Sun, G.Y.; Whaley-Connell, A.T.; Sowers, J.R.; Cui, J.; Gu, Z. Two-Dimensional Zymography Differentiates Gelatinase Isoforms in Stimulated Microglial Cells and in Brain Tissues of Acute Brain Injuries. PLoS ONE 2015, 10, e0123852. [Google Scholar] [CrossRef]
- Stroud, L.J.; Šlapeta, J.; Padula, M.P.; Druery, D.; Tsiotsioras, G.; Coorssen, J.R.; Stack, C.M. Comparative proteomic analysis of two pathogenic Tritrichomonas foetus genotypes: There is more to the proteome than meets the eye. Int. J. Parasitol. 2017, 47, 203–213. [Google Scholar] [CrossRef]
- Triboulet, S.; Aude-Garcia, C.; Armand, L.; Collin-Faure, V.; Chevallet, M.; Diemer, H.; Gerdil, A.; Proamer, F.; Strub, J.M.; Habert, A.; et al. Comparative proteomic analysis of the molecular responses of mouse macrophages to titanium dioxide and copper oxide nanoparticles unravels some toxic mechanisms for copper oxide nanoparticles in macrophages. PLoS ONE 2015, 10, e124496. [Google Scholar] [CrossRef]
- Aude-Garcia, C.; Dalzon, B.; Ravanat, J.L.; Collin-Faure, V.; Diemer, H.; Strub, J.M.; Cianferani, S.; Van Dorsselaer, A.; Carriere, M.; Rabilloud, T. A combined proteomic and targeted analysis unravels new toxic mechanisms for zinc oxide nanoparticles in macrophages. J. Proteom. 2016, 134, 174–185. [Google Scholar] [CrossRef] [PubMed]
- D’Anna, C.; Cigna, D.; Di Sano, C.; Di Vincenzo, S.; Dino, P.; Ferraro, M.; Bini, L.; Bianchi, L.; Di Gaudio, F.; Gjomarkaj, M.; et al. Exposure to cigarette smoke extract and lipopolysaccharide modifies cytoskeleton organization in bronchial epithelial cells. Exp. Lung Res. 2017, 43, 347–358. [Google Scholar] [CrossRef]
- Matsuda, Y.; Ishiwata, T.; Yoshimura, H.; Hagio, M.; Arai, T. Inhibition of nestin suppresses stem cell phenotype of glioblastomas through the alteration of post-translational modification of heat shock protein HSPA8/HSC. Cancer Lett. 2015, 357, 602–611. [Google Scholar] [CrossRef]
- Ostrowski, S.M.; Johnson, K.; Siefert, M.; Shank, S.; Sironi, L.; Wolozin, B.; Landreth, G.E.; Ziady, A.G. Simvastatin inhibits protein isoprenylation in the brain. Neuroscience 2016, 329, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Gamberi, T.; Massai, L.; Magherini, F.; Landini, I.; Fiaschi, T.; Scaletti, F.; Gabbiani, C.; Bianchi, L.; Bini, L.; Nobili, S.; et al. Proteomic analysis of A2780/S ovarian cancer cell response to the cytotoxic organogold (III) compound Aubipy (c). J. Proteom. 2014, 103, 103–120. [Google Scholar] [CrossRef]
- Beltran, L.; Cutillas, P.R. Advances in phosphopeptide enrichment techniques for phosphoproteomics. Amino Acids 2012, 43, 1009–1024. [Google Scholar] [CrossRef]
- Carlson, S.M.; Gozani, O. Emerging technologies to map the protein methylome. J. Mol. Biol. 2014, 426, 3350–3362. [Google Scholar] [CrossRef]
- Ke, M.; Shen, H.; Wang, L.; Luo, S.; Lin, L.; Yang, J.; Tian, R. Identification, Quantification, and Site Localization of Protein Posttranslational Modifications via Mass Spectrometry-Based Proteomics. Adv. Exp. Med. Biol. 2016, 919, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Diallo, I.; Seve, M.; Cunin, V.; Minassian, F.; Poisson, J.-F.; Michelland, S.; Bourgoin-Voillard, S. Current trends in protein acetylation analysis. Expert Rev. Proteomics 2019, 16, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Chen, Y.; Sprung, R.; Tang, Y.; Ball, H.; Sangras, B.; Kim, S.C.; Falck, J.R.; Peng, J.; Gu, W.; Zhao, Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteom. MCP 2007, 6, 812–819. [Google Scholar] [CrossRef]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteom. MCP 2012, 11, 100–107. [Google Scholar] [CrossRef]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef]
- Sarioglu, H.; Lottspeich, F.; Walk, T.; Jung, G.; Eckerskorn, C. Deamidation as a widespread phenomenon in two-dimensional polyacrylamide gel electrophoresis of human blood plasma proteins. Electrophoresis 2000, 21, 2209–2218. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, G.; Dong, Y.; Zhou, H.H.; Kong, B.; Aleksunes, L.M.; Richardson, J.R.; Li, F.; Guo, G.L. Modulation of farnesoid X receptor results in post-translational modification of poly (ADP-ribose) polymerase 1 in the liver. Toxicol. Appl. Pharmacol. 2013, 266, 260–266. [Google Scholar] [CrossRef]
- Blundon, M.A.; Schlesinger, D.R.; Parthasarathy, A.; Smith, S.L.; Kolev, H.M.; Vinson, D.A.; Kunttas-Tatli, E.; McCartney, B.M.; Minden, J.S. Proteomic analysis reveals APC-dependent post-translational modifications and identifies a novel regulator of β-catenin. Development 2016, 143, 2629–2640. [Google Scholar] [CrossRef]
- Prudent, R.; Demoncheaux, N.; Diemer, H.; Collin-Faure, V.; Kapur, R.; Paublant, F.; Lafanechere, L.; Cianferani, S.; Rabilloud, T. A quantitative proteomic analysis of cofilin phosphorylation in myeloid cells and its modulation using the LIM kinase inhibitor Pyr. PLoS ONE 2018, 13, e0208979. [Google Scholar] [CrossRef]
- Iwai, K.; Shibukawa, Y.; Yamazaki, N.; Wada, Y. Transglutaminase 2-dependent Deamidation of Glyceraldehyde-3-phosphate Dehydrogenase Promotes Trophoblastic Cell Fusion. J. Biol. Chem. 2014, 289, 4989–4999. [Google Scholar] [CrossRef]
- Rabilloud, T.; Heller, M.; Gasnier, F.; Luche, S.; Rey, C.; Aebersold, R.; Benahmed, M.; Louisot, P.; Lunardi, J. Proteomics analysis of cellular response to oxidative stress. Evidence for in vivo overoxidation of peroxiredoxins at their active site. J. Biol. Chem. 2002, 277, 19396–19401. [Google Scholar] [CrossRef]
- Riquier, S.; Breton, J.; Abbas, K.; Cornu, D.; Bouton, C.; Drapier, J.-C. Peroxiredoxin post-translational modifications by redox messengers. Redox Biol. 2014, 2, 777–785. [Google Scholar] [CrossRef]
- Weber, H.; Engelmann, S.; Becher, D.; Hecker, M. Oxidative stress triggers thiol oxidation in the glyceraldehyde-3-phosphate dehydrogenase of Staphylococcus aureus. Mol. Microbiol. 2004, 52, 133–140. [Google Scholar] [CrossRef]
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.-S.; Li, L. Oxidative modifications and aggregation of Cu, Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J. Biol. Chem. 2005, 280, 11648–11655. [Google Scholar] [CrossRef]
- Hwang, N.R.; Yim, S.-H.; Kim, Y.M.; Jeong, J.; Song, E.J.; Lee, Y.; Lee, J.H.; Choi, S.; Lee, K.-J. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 2009, 423, 253–264. [Google Scholar] [CrossRef]
- Poschmann, G.; Seyfarth, K.; Besong Agbo, D.; Klafki, H.W.; Rozman, J.; Wurst, W.; Wiltfang, J.; Meyer, H.E.; Klingenspor, M.; Stuhler, K. High-fat diet induced isoform changes of the Parkinson’s disease protein DJ-1. J. Proteome Res. 2014, 13, 2339–2351. [Google Scholar] [CrossRef]
- Choi, J.E.; Lee, J.J.; Kang, W.; Kim, H.J.; Cho, J.H.; Han, P.L.; Lee, K.J. Proteomic Analysis of Hippocampus in a Mouse Model of Depression Reveals Neuroprotective Function of Ubiquitin C-terminal Hydrolase L1 (UCH-L1) via Stress-induced Cysteine Oxidative Modifications. Mol. Cell Proteom. 2018, 17, 1803–1823. [Google Scholar] [CrossRef]
- John, J.P.P.; Pollak, A.; Lubec, G. Complete sequencing and oxidative modification of manganese superoxide dismutase in medulloblastoma cells. Electrophoresis 2009, 30, 3006–3016. [Google Scholar] [CrossRef]
- Codreanu, S.G.; Liebler, D.C. Novel approaches to identify protein adducts produced by lipid peroxidation. Free Radic. Res. 2015, 49, 881–887. [Google Scholar] [CrossRef][Green Version]
- Toyama, T.; Shinkai, Y.; Yazawa, A.; Kakehashi, H.; Kaji, T.; Kumagai, Y. Glutathione-mediated reversibility of covalent modification of ubiquitin carboxyl-terminal hydrolase L1 by 1,2-naphthoquinone through Cys152, but not Lys. Chem. Biol. Interact. 2014, 214, 41–48. [Google Scholar] [CrossRef]
- Asif, A.R.; Armstrong, V.W.; Voland, A.; Wieland, E.; Oellerich, M.; Shipkova, M. Proteins identified as targets of the acyl glucuronide metabolite of mycophenolic acid in kidney tissue from mycophenolate mofetil treated rats. Biochimie 2007, 89, 393–402. [Google Scholar] [CrossRef]
- Luo, J.; Hill, B.G.; Gu, Y.; Cai, J.; Srivastava, S.; Bhatnagar, A.; Prabhu, S.D. Mechanisms of acrolein-induced myocardial dysfunction: Implications for environmental and endogenous aldehyde exposure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3673–H3684. [Google Scholar] [CrossRef]
- Isbell, M.A.; Morin, D.; Boland, B.; Buckpitt, A.; Salemi, M.; Presley, J. Identification of proteins adducted by reactive naphthalene metabolites in vitro. Proteomics 2005, 5, 4197–4204. [Google Scholar] [CrossRef]
- Koen, Y.M.; Gogichaeva, N.V.; Alterman, M.A.; Hanzlik, R.P. A proteomic analysis of bromobenzene reactive metabolite targets in rat liver cytosol in vivo. Chem. Res. Toxicol. 2007, 20, 511–519. [Google Scholar] [CrossRef]
- Koen, Y.M.; Hajovsky, H.; Liu, K.; Williams, T.D.; Galeva, N.A.; Staudinger, J.L.; Hanzlik, R.P. Liver protein targets of hepatotoxic 4-bromophenol metabolites. Chem. Res. Toxicol. 2012, 25, 1777–1786. [Google Scholar] [CrossRef]
- Koen, Y.M.; Sarma, D.; Hajovsky, H.; Galeva, N.A.; Williams, T.D.; Staudinger, J.L.; Hanzlik, R.P. Protein targets of thioacetamide metabolites in rat hepatocytes. Chem. Res. Toxicol. 2013, 26, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Ikehata, K.; Duzhak, T.G.; Galeva, N.A.; Ji, T.; Koen, Y.M.; Hanzlik, R.P. Protein targets of reactive metabolites of thiobenzamide in rat liver in vivo. Chem. Res. Toxicol. 2008, 21, 1432–1442. [Google Scholar] [CrossRef]
- Moro, S.; Chipman, J.K.; Antczak, P.; Turan, N.; Dekant, W.; Falciani, F.; Mally, A. Identification and Pathway Mapping of Furan Target Proteins Reveal Mitochondrial Energy Production and Redox Regulation as Critical Targets of Furan Toxicity. Toxicol. Sci. 2012, 126, 336–352. [Google Scholar] [CrossRef]
- Lame, M.W.; Jones, A.D.; Wilson, D.W.; Segall, H.J. Monocrotaline pyrrole targets proteins with and without cysteine residues in the cytosol and membranes of human pulmonary artery endothelial cells. Proteomics 2005, 5, 4398–4413. [Google Scholar] [CrossRef]
- Lame, M.W.; Jones, A.D.; Wilson, D.W.; Segall, H.J. Protein targets of 1,4-benzoquinone and 1,4-naphthoquinone in human bronchial epithelial cells. Proteomics 2003, 3, 479–495. [Google Scholar] [CrossRef]
- Van Laar, V.S.; Mishizen, A.J.; Cascio, M.; Hastings, T.G. Proteomic identification of dopamine-conjugated proteins from isolated rat brain mitochondria and SH-SY5Y cells. Neurobiol. Dis. 2009, 34, 487–500. [Google Scholar] [CrossRef]
- Gaviard, C.; Cosette, P.; Jouenne, T.; Hardouin, J. LasB and CbpD Virulence Factors of Pseudomonas aeruginosa Carry Multiple Post-Translational Modifications on Their Lysine Residues. J. Proteome Res. 2019, 18, 923–933. [Google Scholar] [CrossRef]
- Forthun, R.B.; Aasebo, E.; Rasinger, J.D.; Bedringaas, S.L.; Berven, F.; Selheim, F.; Bruserud, O.; Gjertsen, B.T. Phosphoprotein DIGE profiles reflect blast differentiation, cytogenetic risk stratification, FLT3/NPM1 mutations and therapy response in acute myeloid leukaemia. J. Proteom. 2018, 173, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Drougat, L.; Olivier-Van Stichelen, S.; Mortuaire, M.; Foulquier, F.; Lacoste, A.-S.; Michalski, J.-C.; Lefebvre, T.; Vercoutter-Edouart, A.-S. Characterization of O-GlcNAc cycling and proteomic identification of differentially O-GlcNAcylated proteins during G1/S transition. Biochim. Biophys. Acta BBA Gen. Subj. 2012, 1820, 1839–1848. [Google Scholar] [CrossRef]
- Jiang, Z.; Cui, Y.; Wang, L.; Zhao, Y.; Yan, S.; Chang, X. Investigating citrullinated proteins in tumour cell lines. World J. Surg. Oncol. 2013, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, A.; Masutomi, H.; Handa, S.; Nakamura, M.; Nakaya, S.; Uchida, Y.; Saito, Y.; Murayama, S.; Jang, B.; Jeon, Y.-C.; et al. Mass spectrometric identification of citrullination sites and immunohistochemical detection of citrullinated glial fibrillary acidic protein in Alzheimer’s disease brains. J. Neurosci. Res. 2015, 93, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Amici, A.; Levine, R.L.; Tsai, L.; Stadtman, E.R. Conversion of amino acid residues in proteins and amino acid homopolymers to carbonyl derivatives by metal-catalyzed oxidation reactions. J. Biol. Chem. 1989, 264, 3341–3346. [Google Scholar]
- Hatasa, Y.; Chikazawa, M.; Furuhashi, M.; Nakashima, F.; Shibata, T.; Kondo, T.; Akagawa, M.; Hamagami, H.; Tanaka, H.; Tachibana, H.; et al. Oxidative Deamination of Serum Albumins by (-)-Epigallocatechin-3-O-Gallate: A Potential Mechanism for the Formation of Innate Antigens by Antioxidants. PLoS ONE 2016, 11, e153002. [Google Scholar] [CrossRef]
- Refsgaard, H.H.; Tsai, L.; Stadtman, E.R. Modifications of proteins by polyunsaturated fatty acid peroxidation products. Proc. Natl. Acad. Sci. USA 2000, 97, 611–616. [Google Scholar] [CrossRef]
- Hoff, H.F.; O’Neil, J. Structural and functional changes in LDL after modification with both 4-hydroxynonenal and malondialdehyde. J. Lipid Res. 1993, 34, 1209–1217. [Google Scholar]
- De Waal, E.M.; Liang, H.; Pierce, A.; Hamilton, R.T.; Buffenstein, R.; Chaudhuri, A.R. Elevated protein carbonylation and oxidative stress do not affect protein structure and function in the long-living naked-mole rat: A proteomic approach. Biochem. Biophys. Res. Commun. 2013, 434, 815–819. [Google Scholar] [CrossRef]
- Hu, W.; Culloty, S.; Darmody, G.; Lynch, S.; Davenport, J.; Ramirez-Garcia, S.; Dawson, K.A.; Lynch, I.; Blasco, J.; Sheehan, D. Toxicity of copper oxide nanoparticles in the blue mussel, Mytilus edulis: A redox proteomic investigation. Chemosphere 2014, 108, 289–299. [Google Scholar] [CrossRef]
- Perutka, Z.; Šebela, M. Pseudotrypsin: A Little-Known Trypsin Proteoform. Molecules 2018, 23, 2637. [Google Scholar] [CrossRef]
- Keil-Dlouhá, V.; Zylber, N.; Imhoff, J.-M.; Tong, N.-T.; Keil, B. Proteolytic activity of pseudotrypsin. FEBS Lett. 1971, 16, 291–295. [Google Scholar] [CrossRef]
- Fortelny, N.; Pavlidis, P.; Overall, C.M. The path of no return-Truncated protein N-termini and current ignorance of their genesis. PROTEOMICS 2015, 15, 2547–2552. [Google Scholar] [CrossRef]
- Staes, A.; Impens, F.; Van Damme, P.; Ruttens, B.; Goethals, M.; Demol, H.; Timmerman, E.; Vandekerckhove, J.; Gevaert, K. Selecting protein N-terminal peptides by combined fractional diagonal chromatography. Nat. Protoc. 2011, 6, 1130–1141. [Google Scholar] [CrossRef] [PubMed]
- Bertaccini, D.; Vaca, S.; Carapito, C.; Arsène-Ploetze, F.; Van Dorsselaer, A.; Schaeffer-Reiss, C. An Improved Stable Isotope N-Terminal Labeling Approach with Light/Heavy TMPP To Automate Proteogenomics Data Validation: dN-TOP. J. Proteome Res. 2013, 12, 3063–3070. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Eckhard, U.; Overall, C.M. Protein Termini and Their Modifications Revealed by Positional Proteomics. ACS Chem. Biol. 2015, 10, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Schilling, O.; Barré, O.; Huesgen, P.F.; Overall, C.M. Proteome-wide analysis of protein carboxy termini: C terminomics. Nat. Methods 2010, 7, 508–511. [Google Scholar] [CrossRef]
- Lee, A.Y.; Park, B.C.; Jang, M.; Cho, S.; Lee, D.H.; Lee, S.C.; Myung, P.K.; Park, S.G. Identification of caspase-3 degradome by two-dimensional gel electrophoresis and matrix-assisted laser desorption/ionization-time of flight analysis. Proteomics 2004, 4, 3429–3436. [Google Scholar] [CrossRef]
- Kim, C.; Yun, N.; Lee, Y.M.; Jeong, J.Y.; Baek, J.Y.; Song, H.Y.; Ju, C.; Youdim, M.B.H.; Jin, B.K.; Kim, W.-K.; et al. Gel-based protease proteomics for identifying the novel calpain substrates in dopaminergic neuronal cell. J. Biol. Chem. 2013, 288, 36717–36732. [Google Scholar] [CrossRef]
- Kim, C.; Oh, Y.J. A Novel 2-DE-Based Proteomic Analysis to Identify Multiple Substrates for Specific Protease in Neuronal Cells. Methods Mol. Biol. Clifton N.J. 2017, 1598, 229–245. [Google Scholar] [CrossRef]
- Le Naour, F.; Hohenkirk, L.; Grolleau, A.; Misek, D.E.; Lescure, P.; Geiger, J.D.; Hanash, S.; Beretta, L. Profiling Changes in Gene Expression during Differentiation and Maturation of Monocyte-derived Dendritic Cells Using Both Oligonucleotide Microarrays and Proteomics. J. Biol. Chem. 2001, 276, 17920–17931. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, A.U.; Cullen, S.P.; Martin, S.J. Chapter Seventeen Two-Dimensional Gel-Based Analysis of the Demolition Phase of Apoptosis. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 442, pp. 343–354. ISBN 978-0-12-374312-1. [Google Scholar]
- Marino, R.; Albenzio, M.; Della Malva, A.; Santillo, A.; Loizzo, P.; Sevi, A. Proteolytic pattern of myofibrillar protein and meat tenderness as affected by breed and aging time. Meat Sci. 2013, 95, 281–287. [Google Scholar] [CrossRef] [PubMed]
- López-Pedrouso, M.; Pérez-Santaescolástica, C.; Franco, D.; Fulladosa, E.; Carballo, J.; Zapata, C.; Lorenzo, J.M. Comparative proteomic profiling of myofibrillar proteins in dry-cured ham with different proteolysis indices and adhesiveness. Food Chem. 2018, 244, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Terova, G.; Addis, M.F.; Preziosa, E.; Pisanu, S.; Pagnozzi, D.; Biosa, G.; Gornati, R.; Bernardini, G.; Roggio, T.; Saroglia, M. Effects of postmortem storage temperature on sea bass (Dicentrarchus labrax) muscle protein degradation: Analysis by 2-D DIGE and MS. Proteomics 2011, 11, 2901–2910. [Google Scholar] [CrossRef] [PubMed]
- Addis, M.F.; Pisanu, S.; Preziosa, E.; Bernardini, G.; Pagnozzi, D.; Roggio, T.; Uzzau, S.; Saroglia, M.; Terova, G. 2D DIGE/MS to investigate the impact of slaughtering techniques on postmortem integrity of fish filet proteins. J. Proteom. 2012, 75, 3654–3664. [Google Scholar] [CrossRef]
- Ethuin, P.; Marlard, S.; Delosière, M.; Carapito, C.; Delalande, F.; Van Dorsselaer, A.; Dehaut, A.; Lencel, V.; Duflos, G.; Grard, T. Differentiation between fresh and frozen-thawed sea bass (Dicentrarchus labrax) fillets using two-dimensional gel electrophoresis. Food Chem. 2015, 176, 294–301. [Google Scholar] [CrossRef]
- Deng, X.; Lei, Y.; Yu, Y.; Lu, S.; Zhang, J. The Discovery of Proteins Associated with Freshness of Coregonus Peled Muscle During Refrigerated Storage. J. Food Sci. 2019, 84, 1266–1272. [Google Scholar] [CrossRef]
- Pepe, M.S.; Etzioni, R.; Feng, Z.; Potter, J.D.; Thompson, M.L.; Thornquist, M.; Winget, M.; Yasui, Y. Phases of Biomarker Development for Early Detection of Cancer. JNCI J. Natl. Cancer Inst. 2001, 93, 1054–1061. [Google Scholar] [CrossRef]
- Rifai, N.; Gillette, M.A.; Carr, S.A. Protein biomarker discovery and validation: The long and uncertain path to clinical utility. Nat. Biotechnol. 2006, 24, 971–983. [Google Scholar] [CrossRef]
- Mischak, H.; Apweiler, R.; Banks, R.E.; Conaway, M.; Coon, J.; Dominiczak, A.; Ehrich, J.H.H.; Fliser, D.; Girolami, M.; Hermjakob, H.; et al. Clinical proteomics: A need to define the field and to begin to set adequate standards. PROTEOMICS Clin. Appl. 2007, 1, 148–156. [Google Scholar] [CrossRef]
- Hristova, V.A.; Chan, D.W. Cancer biomarker discovery and translation: Proteomics and beyond. Expert Rev. Proteom. 2019, 16, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Charrier, J.P.; Tournel, C.; Michel, S.; Dalbon, P.; Jolivet, M. Two-dimensional electrophoresis of prostate-specific antigen in sera of men with prostate cancer or benign prostate hyperplasia. Electrophoresis 1999, 20, 1075–1081. [Google Scholar] [CrossRef]
- Charrier, J.P.; Tournel, C.; Michel, S.; Comby, S.; Jolivet-Reynaud, C.; Passagot, J.; Dalbon, P.; Chautard, D.; Jolivet, M. Differential diagnosis of prostate cancer and benign prostate hyperplasia using two-dimensional electrophoresis. Electrophoresis 2001, 22, 1861–1866. [Google Scholar] [CrossRef]
- Kondo, T. Cancer biomarker development and two-dimensional difference gel electrophoresis (2D-DIGE). Biochim. Biophys. Acta BBA Proteins Proteom. 2019, 1867, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Nakanishi, Y.; Kato, H.; Saito, S.; Nagino, M.; Hirohashi, S.; Kondo, T. Transglutaminase 3 as a prognostic biomarker in esophageal cancer revealed by proteomics. Int. J. Cancer 2009, 124, 2106–2115. [Google Scholar] [CrossRef]
- Okano, T.; Kondo, T.; Fujii, K.; Nishimura, T.; Takano, T.; Ohe, Y.; Tsuta, K.; Matsuno, Y.; Gemma, A.; Kato, H.; et al. Proteomic Signature Corresponding to the Response to Gefitinib (Iressa, ZD1839), an Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor in Lung Adenocarcinoma. Clin. Cancer Res. 2007, 13, 799–805. [Google Scholar] [CrossRef]
- Yokoo, H.; Kondo, T.; Okano, T.; Nakanishi, K.; Sakamoto, M.; Kosuge, T.; Todo, S.; Hirohashi, S. Protein expression associated with early intrahepatic recurrence of hepatocellular carcinoma after curative surgery. Cancer Sci. 2007, 98, 665–673. [Google Scholar] [CrossRef]
- Orimo, T.; Ojima, H.; Hiraoka, N.; Saito, S.; Kosuge, T.; Kakisaka, T.; Yokoo, H.; Nakanishi, K.; Kamiyama, T.; Todo, S.; et al. Proteomic profiling reveals the prognostic value of adenomatous polyposis coli-end-binding protein 1 in hepatocellular carcinoma. Hepatology 2008, 48, 1851–1863. [Google Scholar] [CrossRef]
- Kimura, K.; Ojima, H.; Kubota, D.; Sakumoto, M.; Nakamura, Y.; Tomonaga, T.; Kosuge, T.; Kondo, T. Proteomic identification of the macrophage-capping protein as a protein contributing to the malignant features of hepatocellular carcinoma. J. Proteom. 2013, 78, 362–373. [Google Scholar] [CrossRef]
- Suehara, Y.; Kondo, T.; Seki, K.; Shibata, T.; Fujii, K.; Gotoh, M.; Hasegawa, T.; Shimada, Y.; Sasako, M.; Shimoda, T.; et al. Pfetin as a prognostic biomarker of gastrointestinal stromal tumors revealed by proteomics. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 1707–1717. [Google Scholar] [CrossRef]
- Kikuta, K.; Gotoh, M.; Kanda, T.; Tochigi, N.; Shimoda, T.; Hasegawa, T.; Katai, H.; Shimada, Y.; Suehara, Y.; Kawai, A.; et al. Pfetin as a prognostic biomarker in gastrointestinal stromal tumor: Novel monoclonal antibody and external validation study in multiple clinical facilities. Jpn. J. Clin. Oncol. 2010, 40, 60–72. [Google Scholar] [CrossRef]
- Kubota, D.; Okubo, T.; Saito, T.; Suehara, Y.; Yoshida, A.; Kikuta, K.; Tsuda, H.; Katai, H.; Shimada, Y.; Kaneko, K.; et al. Validation study on pfetin and ATP-dependent RNA helicase DDX39 as prognostic biomarkers in gastrointestinal stromal tumour. Jpn. J. Clin. Oncol. 2012, 42, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Lescuyer, P.; Allard, L.; Zimmermann-Ivol, C.G.; Burgess, J.A.; Hughes-Frutiger, S.; Burkhard, P.R.; Sanchez, J.-C.; Hochstrasser, D.F. Identification of post-mortem cerebrospinal fluid proteins as potential biomarkers of ischemia and neurodegeneration. Proteomics 2004, 4, 2234–2241. [Google Scholar] [CrossRef]
- Allard, L.; Burkhard, P.R.; Lescuyer, P.; Burgess, J.A.; Walter, N.; Hochstrasser, D.F.; Sanchez, J.-C. PARK7 and nucleoside diphosphate kinase A as plasma markers for the early diagnosis of stroke. Clin. Chem. 2005, 51, 2043–2051. [Google Scholar] [CrossRef] [PubMed]
- Turck, N.; Vutskits, L.; Sanchez-Pena, P.; Robin, X.; Hainard, A.; Gex-Fabry, M.; Fouda, C.; Bassem, H.; Mueller, M.; Lisacek, F.; et al. A multiparameter panel method for outcome prediction following aneurysmal subarachnoid hemorrhage. Intensive Care Med. 2010, 36, 107–115. [Google Scholar] [CrossRef]
- Turck, N.; Robin, X.; Walter, N.; Fouda, C.; Hainard, A.; Sztajzel, R.; Wagner, G.; Hochstrasser, D.F.; Montaner, J.; Burkhard, P.R.; et al. Blood glutathione S-transferase-π as a time indicator of stroke onset. PLoS ONE 2012, 7, e43830. [Google Scholar] [CrossRef]
- Lagerstedt, L.; Egea-Guerrero, J.J.; Bustamante, A.; Rodríguez-Rodríguez, A.; El Rahal, A.; Quintana-Diaz, M.; García-Armengol, R.; Prica, C.M.; Andereggen, E.; Rinaldi, L.; et al. Combining H-FABP and GFAP increases the capacity to differentiate between CT-positive and CT-negative patients with mild traumatic brain injury. PLoS ONE 2018, 13, e0200394. [Google Scholar] [CrossRef]
- Posti, J.P.; Takala, R.S.K.; Lagerstedt, L.; Dickens, A.M.; Hossain, I.; Mohammadian, M.; Ala-Seppälä, H.; Frantzén, J.; van Gils, M.; Hutchinson, P.J.; et al. Correlation of Blood Biomarkers and Biomarker Panels with Traumatic Findings on Computed Tomography after Traumatic Brain Injury. J. Neurotrauma 2019, 36, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Mölleken, C.; Sitek, B.; Henkel, C.; Poschmann, G.; Sipos, B.; Wiese, S.; Warscheid, B.; Broelsch, C.; Reiser, M.; Friedman, S.L.; et al. Detection of novel biomarkers of liver cirrhosis by proteomic analysis. Hepatology 2009, 49, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Bracht, T.; Mölleken, C.; Ahrens, M.; Poschmann, G.; Schlosser, A.; Eisenacher, M.; Stühler, K.; Meyer, H.E.; Schmiegel, W.H.; Holmskov, U.; et al. Evaluation of the biomarker candidate MFAP4 for non-invasive assessment of hepatic fibrosis in hepatitis C patients. J. Transl. Med. 2016, 14, 201. [Google Scholar] [CrossRef]
- Lottspeich, F.; Kellner, R. Microcharacterrization of Proteins, 1st ed.; Kellner, R., Lottspeich, F., Meyer, H.E., Eds.; Wiley: Hoboken, NJ, USA, 1999; ISBN 978-3-527-30084-6. [Google Scholar]
- Tran, J.C.; Zamdborg, L.; Ahlf, D.R.; Lee, J.E.; Catherman, A.D.; Durbin, K.R.; Tipton, J.D.; Vellaichamy, A.; Kellie, J.F.; Li, M.; et al. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 2011, 480, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, N.L. Top-down proteomics. Anal. Chem. 2004, 76, 197A–203A. [Google Scholar] [CrossRef]
- Claverol, S.; Burlet-Schiltz, O.; Girbal-Neuhauser, E.; Gairin, J.E.; Monsarrat, B. Mapping and structural dissection of human 20 S proteasome using proteomic approaches. Mol. Cell. Proteom. MCP 2002, 1, 567–578. [Google Scholar] [CrossRef]
- Claverol, S.; Burlet-Schiltz, O.; Gairin, J.E.; Monsarrat, B. Characterization of Protein Variants and Post-Translational Modifications: ESI-MSn Analyses of Intact Proteins Eluted from Polyacrylamide Gels. Mol. Cell. Proteom. 2003, 2, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Kachuk, C.; Stephen, K.; Doucette, A. Comparison of sodium dodecyl sulfate depletion techniques for proteome analysis by mass spectrometry. J. Chromatogr. A 2015, 1418, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Anderson, V.E. Prevention of artifactual protein oxidation generated during sodium dodecyl sulfate-gel electrophoresis. Electrophoresis 2004, 25, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T. Variations on a theme: Changes to electrophoretic separations that can make a difference. J. Proteom. 2010, 73, 1562–1572. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| 2D Gel-based Proteomics | Shotgun Proteomics | |
|---|---|---|
| Sample consuming | ++(+) * | + |
| Time consuming | +++ | ++ |
| Analysis depth | ++ | +++ |
| Separation/identification | ||
| Separation/detection of proteoforms | +++ | + |
| Identification on protein level | Multiple identifications | Only by inference |
| from peptides | ||
| Detection of proteoforms | +++ | - |
| Details at peptide level (e.g., sequence coverage) | +++ | + |
| Number of modulated proteins identified | + | +++ |
| Coupling with biochemical methods | ||
| Antibodies | +++ | + |
| Enzymes (zymography) | + | - |
| Robustness of quantification | ||
| Sensitivity | ++ | +++ |
| Linearity | +++ | + |
| Need of validation | +++ | +++ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcus, K.; Lelong, C.; Rabilloud, T. What Room for Two-Dimensional Gel-Based Proteomics in a Shotgun Proteomics World? Proteomes 2020, 8, 17. https://doi.org/10.3390/proteomes8030017
Marcus K, Lelong C, Rabilloud T. What Room for Two-Dimensional Gel-Based Proteomics in a Shotgun Proteomics World? Proteomes. 2020; 8(3):17. https://doi.org/10.3390/proteomes8030017
Chicago/Turabian StyleMarcus, Katrin, Cécile Lelong, and Thierry Rabilloud. 2020. "What Room for Two-Dimensional Gel-Based Proteomics in a Shotgun Proteomics World?" Proteomes 8, no. 3: 17. https://doi.org/10.3390/proteomes8030017
APA StyleMarcus, K., Lelong, C., & Rabilloud, T. (2020). What Room for Two-Dimensional Gel-Based Proteomics in a Shotgun Proteomics World? Proteomes, 8(3), 17. https://doi.org/10.3390/proteomes8030017