The Power of Three in Cannabis Shotgun Proteomics: Proteases, Databases and Search Engines



Abstract
1. Introduction
2. Materials and Methods
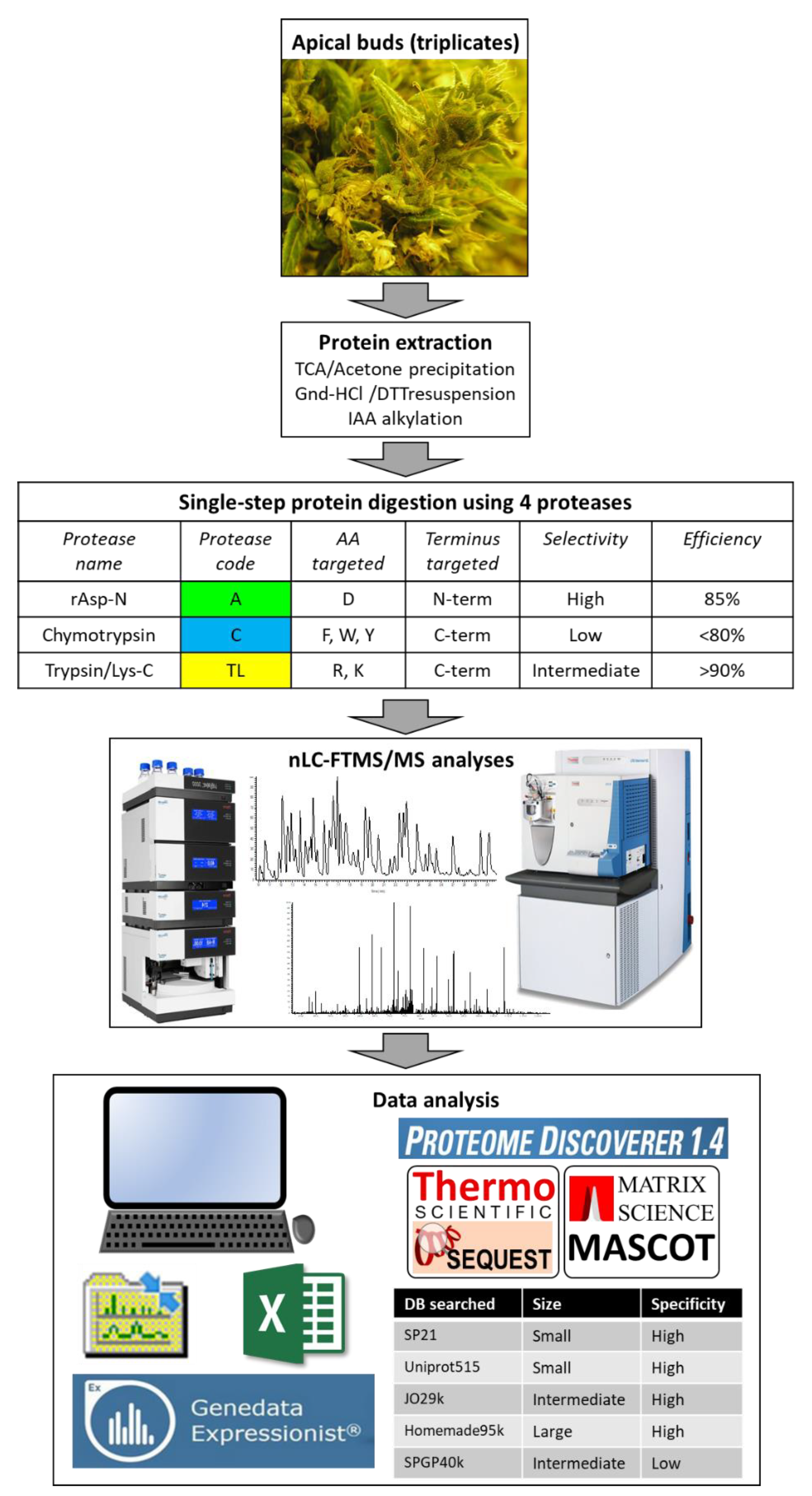
2.1. Protein Extraction, Digestion, and Analysis Using Nano Liquid Chromatography-Tandem Mass Spectrometry (nLC-MS/MS)
2.2. Protein Identification Using Five Databases and Statistical Analyses
3. Results and Discussion
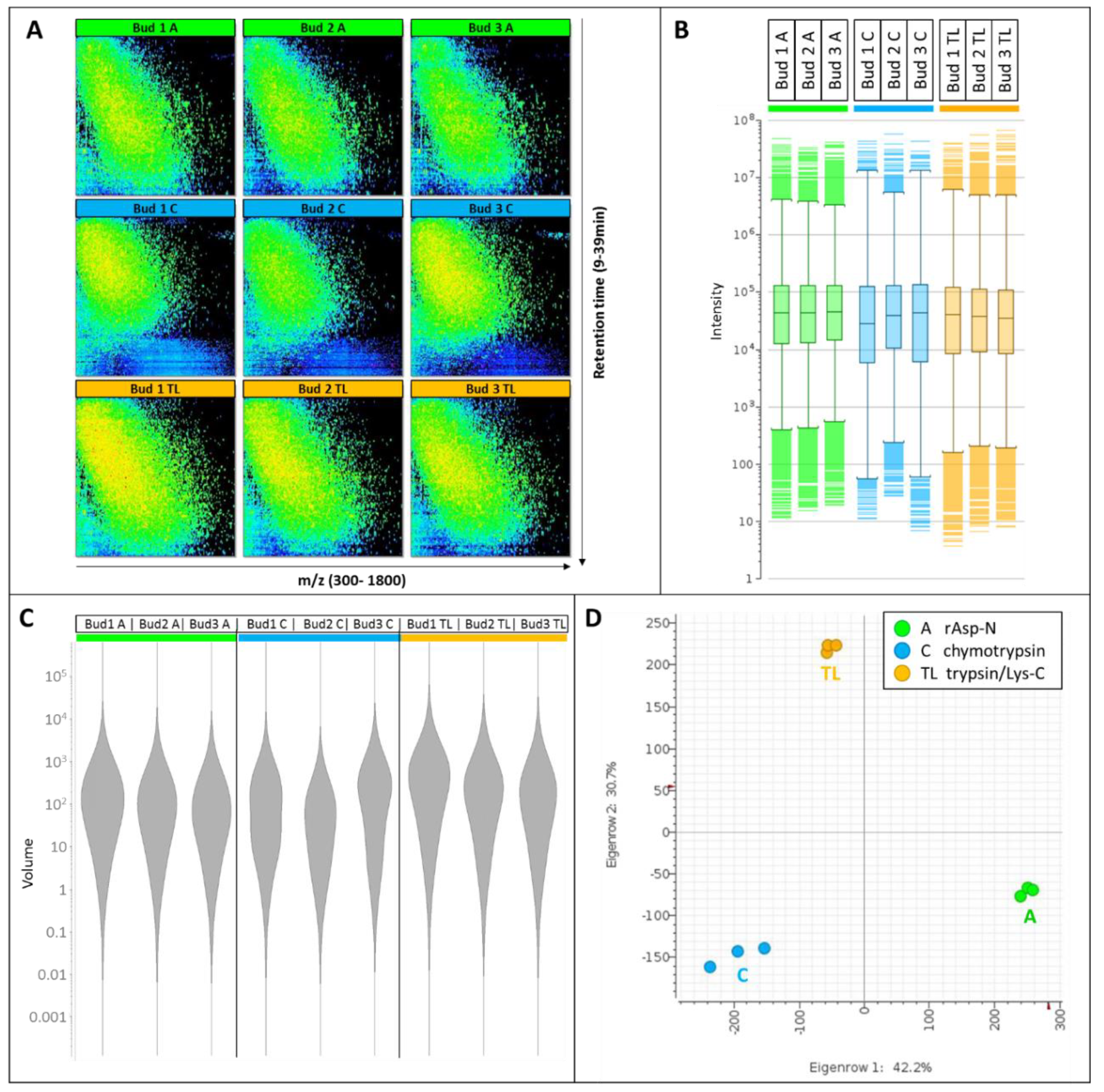
3.1. Comparison of the nLC-MS Files
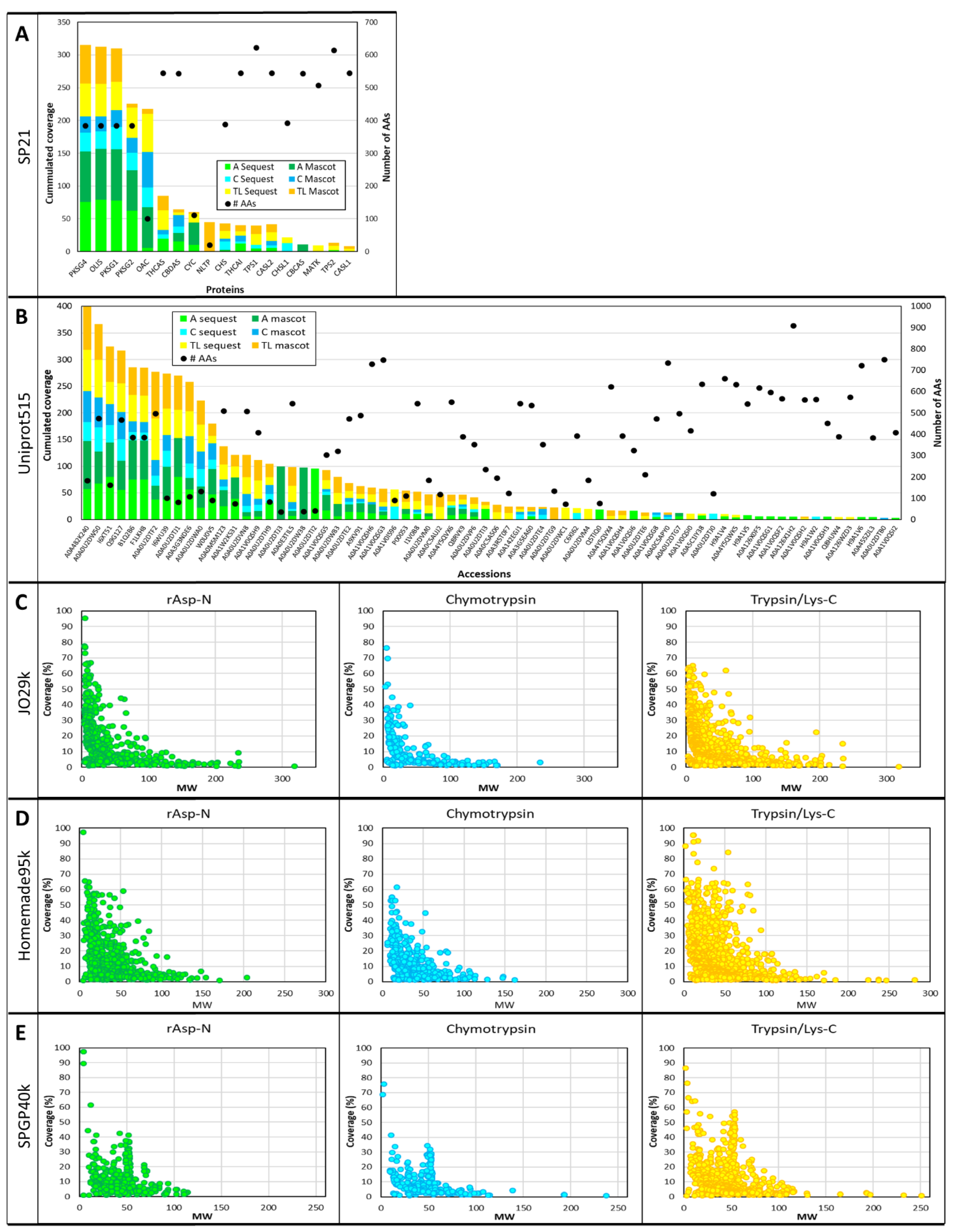
3.2. Database Search Yield and Duration
3.3. Comparison of Proteases and Their Proteolytic Efficiencies
3.4. Comparison of the Search Algorithms
3.5. Sequence Coverage and Post-Translational Modifications (PTMs)
3.6. Database Specificity and Gene Ontology (GO)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vincent, D.; Rochfort, S.; Spangenberg, G. Optimisation of Protein Extraction from Medicinal Cannabis Mature Buds for Bottom-Up Proteomics. Molecules 2019, 24, 659. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Ezernieks, V.; Rochfort, S.; Spangenberg, G. A Multiple Protease Strategy to Optimise the Shotgun Proteomics of Mature Medicinal Cannabis Buds. Int. J. Mol. Sci. 2019, 20, 5630. [Google Scholar] [CrossRef]
- Vincent, D.; Binos, S.; Rochfort, S.; Spangenberg, G. Top-down proteomics of medicinal cannabis. Proteomes 2019, 7, 33. [Google Scholar] [CrossRef]
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R., 3rd. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Aslanian, A.; Yates, J.R., 3rd. Mass spectrometry for proteomics. Curr. Opin. Chem. Biol. 2008, 12, 483–490. [Google Scholar] [CrossRef]
- Garcia, B.A.; Siuti, N.; Thomas, C.E.; Mizzen, C.A.; Kelleher, N.L. Characterization of neurohistone variants and post-translational modifications by electron capture dissociation mass spectrometry. Int. J. Mass Spectrom. 2007, 259, 184–196. [Google Scholar] [CrossRef]
- Kelleher, N.L. Top-down proteomics. Anal. Chem. 2004, 76, 197A–203A. [Google Scholar] [CrossRef]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., 3rd. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef]
- Biringer, R.G.; Amato, H.; Harrington, M.G.; Fonteh, A.N.; Riggins, J.N.; Hühmer, A.F. Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC-MS/MS. Brief. Funct. Genom. Proteom. 2006, 5, 144–153. [Google Scholar] [CrossRef]
- Choudhary, G.; Wu, S.L.; Shieh, P.; Hancock, W.S. Multiple enzymatic digestion for enhanced sequence coverage of proteins in complex proteomic mixtures using capillary LC with ion trap MS/MS. J. Proteome Res. 2003, 2, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Poetsch, A. Protein cleavage strategies for an improved analysis of the membrane proteome. Proteome. Sci. 2006, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Fischer, F.; Wolters, D.; Rogner, M.; Poetsch, A. Toward the complete membrane proteome: High coverage of integral membrane proteins through transmembrane peptide detection. Mol. Cell Proteom. 2006, 5, 444–453. [Google Scholar] [CrossRef] [PubMed]
- MacCoss, M.J.; McDonald, W.H.; Saraf, A.; Sadygov, R.; Clark, J.M.; Tasto, J.J.; Gould, K.L.; Wolters, D.; Washburn, M.; Weiss, A.; et al. Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. USA 2002, 99, 7900–7905. [Google Scholar] [CrossRef]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Paabo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst.Biol. 2011, 7, 548. [Google Scholar] [CrossRef]
- Schlosser, A.; Vanselow, J.T.; Kramer, A. Mapping of phosphorylation sites by a multi-protease approach with specific phosphopeptide enrichment and NanoLC-MS/MS analysis. Anal. Chem. 2005, 77, 5243–5250. [Google Scholar] [CrossRef]
- Trevisan-Silva, D.; Bednaski, A.V.; Fischer, J.S.G.; Veiga, S.S.; Bandeira, N.; Guthals, A.; Marchini, F.K.; Leprevost, F.V.; Barbosa, V.C.; Senff-Ribeiro, A.; et al. A multi-protease, multi-dissociation, bottom-up-to-top-down proteomic view of the Loxosceles intermedia venom. Sci. Data 2017, 4, 170090. [Google Scholar] [CrossRef]
- Zhang, X. Less is More: Membrane Protein Digestion Beyond Urea-Trypsin Solution for Next-level Proteomics. Mol. Cell Proteom. 2015, 14, 2441–2453. [Google Scholar] [CrossRef]
- Trevisiol, S.; Ayoub, D.; Lesur, A.; Ancheva, L.; Gallien, S.; Domon, B. The use of proteases complementary to trypsin to probe isoforms and modifications. Proteomics 2016, 16, 715–728. [Google Scholar] [CrossRef]
- Tsiatsiani, L.; Heck, A.J. Proteomics beyond trypsin. FEBS J. 2015, 282, 2612–2626. [Google Scholar] [CrossRef]
- Vandermarliere, E.; Mueller, M.; Martens, L. Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom. Rev. 2013, 32, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Sadygov, R.G.; Cociorva, D.; Yates, J.R., 3rd. Large-scale database searching using tandem mass spectra: Looking up the answer in the back of the book. Nat. Methods 2004, 1, 195–202. [Google Scholar] [CrossRef]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Hoopmann, M.R.; Moritz, R.L. Current algorithmic solutions for peptide-based proteomics data generation and identification. Curr. Opin. Biotechnol. 2013, 24, 31–38. [Google Scholar] [CrossRef]
- Misra, B.B. Updates on resources, software tools, and databases for plant proteomics in 2016–2017. Electrophoresis 2018, 39, 1543–1557. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I. Protein Identification by Tandem Mass Spectrometry and Sequence Database Searching. In Mass Spectrometry Data Analysis in Proteomics; Matthiesen, R., Ed.; Humana Press Inc.: Totowa, NJ, USA, 2007; Volume 367, pp. 87–119. [Google Scholar]
- Shteynberg, D.; Nesvizhskii, A.I.; Moritz, R.L.; Deutsch, E.W. Combining results of multiple search engines in proteomics. Mol. Cell Proteom. 2013, 12, 2383–2393. [Google Scholar] [CrossRef]
- Tabb, D.L. The SEQUEST family tree. J. Am. Soc. Mass Spectrom. 2015, 26, 1814–1819. [Google Scholar] [CrossRef] [PubMed]
- Andre, C.M.; Hausman, J.-F.; Guerriero, G. Cannabis sativa: The Plant of the Thousand and One Molecules. Front. Plant Sci. 2016, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Bonini, S.A.; Premoli, M.; Tambaro, S.; Kumar, A.; Maccarinelli, G.; Memo, M.; Mastinu, A. Cannabis sativa: A comprehensive ethnopharmacological review of a medicinal plant with a long history. J. Ethnopharmacol. 2018, 227, 300–315. [Google Scholar] [CrossRef] [PubMed]
- ElSohly, M.A.; Radwan, M.M.; Gul, W.; Chandra, S.; Galal, A. Phytochemistry of Cannabis sativa L. Prog. Chem. Org. Nat. Prod. 2017, 103, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Kovalchuk, I.; Pellino, M.; Rigault, P.; van Velzen, R.; Ebersbach, J.; R. Ashnest, J.; Mau, M.; Schranz, M.E.; Alcorn, J.; Laprairie, R.B.; et al. The Genomics of Cannabis and Its Close Relatives. Annu. Rev. Plant. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B.; Marcu, J. Cannabis Pharmacology: The Usual Suspects and a Few Promising Leads. Adv. Pharmacol. 2017, 80, 67–134. [Google Scholar] [CrossRef] [PubMed]
- Grassa, C.J.; Wenger, J.P.; Dabney, C.; Poplawski, S.G.; Motley, S.T.; Michael, T.P.; Schwartz, C.J.; Weiblen, G.D. A complete Cannabis chromosome assembly and adaptive admixture for elevated cannabidiol (CBD) content. bioRxiv 2018. [Google Scholar] [CrossRef]
- Laverty, K.U.; Stout, J.M.; Sullivan, M.J.; Shah, H.; Gill, N.; Holbrook, L.; Deikus, G.; Sebra, R.; Hughes, T.R.; Page, J.E.; et al. A physical and genetic map of Cannabis sativa identifies extensive rearrangement at the THC/CBD acid synthase locus. Genome Res. 2018. [Google Scholar] [CrossRef]
- Oh, H.; Seo, B.; Lee, S.; Ahn, D.H.; Jo, E.; Park, J.K.; Min, G.S. Two complete chloroplast genome sequences of Cannabis sativa varieties. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 2835–2837. [Google Scholar] [CrossRef]
- Van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12, R102. [Google Scholar] [CrossRef]
- Jenkins, C.; Orsburn, B. The Cannabis Proteome Draft Map Project. Int. J. Mol. Sci. 2020, 21, 965. [Google Scholar] [CrossRef]
- Page, J.; Boubakir, Z. Aromatic Prenyltransferase from Cannabis. Patent Application No. WO 2011/017798 Al, 17 February 2011. [Google Scholar]
- Page, J.; Stout, J. Cannabichromenic Acid Synthase from Cannabis Sativa. Patent Application No. WO 2015/196275 Al, 30 December 2015. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Creasy, D.M.; Cottrell, J.S. Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2002, 2, 1426–1434. [Google Scholar] [CrossRef]
- Verheggen, K.; Martens, L.; Berven, F.S.; Barsnes, H.; Vaudel, M. Database Search Engines: Paradigms, Challenges and Solutions. In Modern Proteomics—Sample Preparation, Analysis and Practical Applications; Carrasco, H.M.a.M., Ed.; Springer International Publishing: Kamm, Switzerland, 2016; Volume 919, pp. 147–156. [Google Scholar]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2014, 42, D32–D37. [Google Scholar] [CrossRef]
- McPartland, J.M. Cannabis Systematics at the Levels of Family, Genus, and Species. Cannabis Cannabinoid Res. 2018, 3, 203–212. [Google Scholar] [CrossRef]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J. Six alternative proteases for mass spectrometry-based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef]
- Swaney, D.L.; Wenger, C.D.; Coon, J.J. Value of using multiple proteases for large-scale mass spectrometry-based proteomics. J. Proteome. Res. 2010, 9, 1323–1329. [Google Scholar] [CrossRef]
- Stead, D.A.; Preece, A.; Brown, A.J. Universal metrics for quality assessment of protein identifications by mass spectrometry. Mol. Cell Proteom. 2006, 5, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Cristobal, A.; Marino, F.; Post, H.; van den Toorn, H.W.; Mohammed, S.; Heck, A.J. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89, 3318–3325. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R., 3rd; Eng, J.K.; McCormack, A.L.; Schieltz, D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 1995, 67, 1426–1436. [Google Scholar] [CrossRef]
- Yates, J.R., 3rd; Eng, J.K.; McCormack, A.L. Mining genomes: Correlating tandem mass spectra of modified and unmodified peptides to sequences in nucleotide databases. Anal. Chem. 1995, 67, 3202–3210. [Google Scholar] [CrossRef]
- Agten, A.; Van Houtven, J.; Askenazi, M.; Burzykowski, T.; Laukens, K.; Valkenborg, D. Visualizing the agreement of peptide assignments between different search engines. J. Mass Spectrom. 2019. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Tolic, N.; Xie, F.; Zhao, R.; Purvine, S.O.; Schepmoes, A.A.; Moore, R.J.; Anderson, G.A.; Smith, R.D. Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: Comparison of peptide identification methods. J. Proteome. Res. 2011, 10, 3929–3943. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Li, J.; Shen, S.; Sheng, Q.; Shyr, Y.; Qu, J. Performance Investigation of Proteomic Identification by HCD/CID Fragmentations in Combination with High/Low-Resolution Detectors on a Tribrid, High-Field Orbitrap Instrument. PLoS ONE 2016, 11, e0160160. [Google Scholar] [CrossRef] [PubMed]
- Kapp, E.A.; Schutz, F.; Connolly, L.M.; Chakel, J.A.; Meza, J.E.; Miller, C.A.; Fenyo, D.; Eng, J.K.; Adkins, J.N.; Omenn, G.S.; et al. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: Sensitivity and specificity analysis. Proteomics 2005, 5, 3475–3490. [Google Scholar] [CrossRef]
- Balgley, B.M.; Laudeman, T.; Yang, L.; Song, T.; Lee, C.S. Comparative evaluation of tandem MS search algorithms using a target-decoy search strategy. Mol. Cell Proteom. 2007, 6, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A. Practical and Efficient Searching in Proteomics: A Cross Engine Comparison. Webmedcentral 2013, 4. [Google Scholar] [CrossRef]
- Suni, V.; Imanishi, S.Y.; Maiolica, A.; Aebersold, R.; Corthals, G.L. Confident site localization using a simulated phosphopeptide spectral library. J. Proteome. Res. 2015, 14, 2348–2359. [Google Scholar] [CrossRef] [PubMed]
- Shoyama, Y.; Tamada, T.; Kurihara, K.; Takeuchi, A.; Taura, F.; Arai, S.; Blaber, M.; Shoyama, Y.; Morimoto, S.; Kuroki, R. Structure and function of 1-tetrahydrocannabinolic acid (THCA) synthase, the enzyme controlling the psychoactivity of Cannabis sativa. J. Mol. Biol. 2012, 423, 96–105. [Google Scholar] [CrossRef]
- Taura, F.; Sirikantaramas, S.; Shoyama, Y.; Yoshikai, K.; Shoyama, Y.; Morimoto, S. Cannabidiolic-acid synthase, the chemotype-determining enzyme in the fiber-type Cannabis sativa. FEBS Lett. 2007, 581, 2929–2934. [Google Scholar] [CrossRef]
- Zirpel, B.; Kayser, O.; Stehle, F. Elucidation of structure-function relationship of THCA and CBDA synthase from Cannabis sativaL. J. Biotechnol. 2018, 284, 17–26. [Google Scholar] [CrossRef]
- Sevier, C.S.; Kaiser, C.A. Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol. 2002, 3, 836–847. [Google Scholar] [CrossRef]
- Taura, F.; Dono, E.; Sirikantaramas, S.; Yoshimura, K.; Shoyama, Y.; Morimoto, S. Production of Delta(1)-tetrahydrocannabinolic acid by the biosynthetic enzyme secreted from transgenic Pichia pastoris. Biochem. Biophys. Res. Commun. 2007, 361, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Venne, A.S.; Kollipara, L.; Zahedi, R.P. The next level of complexity: Crosstalk of posttranslational modifications. Proteomics 2014, 14, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J.; Trapp, J.; Pible, O.; Geffard, O.; Chaumot, A.; Hartmann, E.M. Non-model organisms, a species endangered by proteogenomics. J. Proteom. 2014, 105, 5–18. [Google Scholar] [CrossRef]
- Bryant, L.; Flatley, B.; Patole, C.; Brown, G.D.; Cramer, R. Proteomic analysis of Artemisia annua--towards elucidating the biosynthetic pathways of the antimalarial pro-drug artemisinin. BMC Plant Biol. 2015, 15, 175. [Google Scholar] [CrossRef] [PubMed]
- Scollo, E.; Neville, D.; Oruna-Concha, M.J.; Trotin, M.; Cramer, R. Characterization of the Proteome of Theobroma cacao Beans by Nano-UHPLC-ESI MS/MS. Proteomics 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, A.L.; Caruso, G.; Cavaliere, C.; Foglia, P.; Piovesana, S.; Samperi, R.; Lagana, A. Proteome investigation of the non-model plant pomegranate (Punica granatum L.). Anal. Bioanal. Chem. 2013, 405, 9301–9309. [Google Scholar] [CrossRef] [PubMed]
- Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Stampachiacchiere, S.; Ventura, S.; Zenezini Chiozzi, R.; Lagana, A. Characterization of quinoa seed proteome combining different protein precipitation techniques: Improvement of knowledge of nonmodel plant proteomics. J. Sep. Sci. 2015, 38, 1017–1025. [Google Scholar] [CrossRef]
- Rodriguez de Francisco, L.; Romero-Rodriguez, M.C.; Navarro-Cerrillo, R.M.; Minino, V.; Perdomo, O.; Jorrin-Novo, J.V. Characterization of the orthodox Pinus occidentalis seed and pollen proteomes by using complementary gel-based and gel-free approaches. J. Proteom. 2016, 143, 382–389. [Google Scholar] [CrossRef]
- Zaman, U.; Urlaub, H.; Abbasi, A. Protein Profiling of Non-model Plant Cuminum cyminum by Gel-Based Proteomic Approach. Phytochem. Anal. 2018, 29, 242–249. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample | MS Scans | MS/MS Scans | MS Clusters |
|---|---|---|---|
| bud1_A | 12,582 | 10,990 | 91,784 |
| bud2_A | 11,820 | 10,174 | 85,566 |
| bud3_A | 11,686 | 10,079 | 85,388 |
| bud1_C | 11,345 | 9532 | 89,030 |
| bud2_C | 10,391 | 8458 | 82,091 |
| bud3_C | 11,562 | 9597 | 83,440 |
| bud1_TL | 13,423 | 11,828 | 91,320 |
| bud2_TL | 12,858 | 11,242 | 87,335 |
| bud3_TL | 12,330 | 10,665 | 84,845 |
| mean A | 12,029 | 10,414 | 87,579 |
| SD A | 483 | 501 | 3642 |
| CV A | 4 | 5 | 4 |
| mean C | 11,099 | 9196 | 84,854 |
| SD C | 623 | 640 | 3679 |
| CV C | 6 | 7 | 4 |
| mean TL | 12,870 | 11,245 | 87,833 |
| SD TL | 547 | 582 | 3266 |
| CV TL | 4 | 5 | 4 |
| Database | # Proteins in Database | Sample | # Proteins with SEQUEST | # Proteins with Mascot | % Proteins with SEQUEST | % Proteins with Mascot |
|---|---|---|---|---|---|---|
| SP21 | 21 | bud1_A | 15 | 9 | 71.4 | 42.9 |
| SP21 | 21 | bud2_A | 15 | 9 | 71.4 | 42.9 |
| SP21 | 21 | bud3_A | 15 | 9 | 71.4 | 42.9 |
| SP21 | 21 | bud1_C | 15 | 12 | 71.4 | 57.1 |
| SP21 | 21 | bud2_C | 15 | 12 | 71.4 | 57.1 |
| SP21 | 21 | bud3_C | 15 | 11 | 71.4 | 52.4 |
| SP21 | 21 | bud1_TL | 16 | 15 | 76.2 | 71.4 |
| SP21 | 21 | bud2_TL | 15 | 14 | 71.4 | 66.7 |
| SP21 | 21 | bud3_TL | 16 | 16 | 76.2 | 76.2 |
| Uniprot515 | 515 | bud1_A | 65 | 40 | 12.6 | 7.8 |
| Uniprot515 | 515 | bud2_A | 63 | 35 | 12.2 | 6.8 |
| Uniprot515 | 515 | bud3_A | 67 | 36 | 13.0 | 7.0 |
| Uniprot515 | 515 | bud1_C | 67 | 46 | 13.0 | 8.9 |
| Uniprot515 | 515 | bud2_C | 70 | 39 | 13.6 | 7.6 |
| Uniprot515 | 515 | bud3_C | 70 | 38 | 13.6 | 7.4 |
| Uniprot515 | 515 | bud1_TL | 70 | 48 | 13.6 | 9.3 |
| Uniprot515 | 515 | bud2_TL | 69 | 39 | 13.4 | 7.6 |
| Uniprot515 | 515 | bud3_TL | 69 | 48 | 13.4 | 9.3 |
| JO29k | 29,057 | bud1_A | 1071 | n.a. | 3.7 | n.a. |
| JO29k | 29,057 | bud2_A | 1037 | n.a. | 3.6 | n.a. |
| JO29k | 29,057 | bud3_A | 1034 | n.a. | 3.6 | n.a. |
| JO29k | 29,057 | bud1_C | 748 | n.a. | 2.6 | n.a. |
| JO29k | 29,057 | bud2_C | 766 | n.a. | 2.6 | n.a. |
| JO29k | 29,057 | bud3_C | 807 | n.a. | 2.8 | n.a. |
| JO29k | 29,057 | bud1_TL | 1244 | n.a. | 4.3 | n.a. |
| JO29k | 29,057 | bud2_TL | 1162 | n.a. | 4.0 | n.a. |
| JO29k | 29,057 | bud3_TL | 1188 | n.a. | 4.1 | n.a. |
| Homenade95k | 95,069 | bud1_A | 1130 | 792 | 1.2 | 0.8 |
| Homenade95k | 95,069 | bud2_A | 1115 | 741 | 1.2 | 0.8 |
| Homenade95k | 95,069 | bud3_A | 1085 | 699 | 1.1 | 0.7 |
| Homenade95k | 95,069 | bud1_C | 981 | 552 | 1.0 | 0.6 |
| Homenade95k | 95,069 | bud2_C | 988 | 555 | 1.0 | 0.6 |
| Homenade95k | 95,069 | bud3_C | 1002 | 549 | 1.1 | 0.6 |
| Homenade95k | 95,069 | bud1_TL | 1322 | 1126 | 1.4 | 1.2 |
| Homenade95k | 95,069 | bud2_TL | 1192 | 922 | 1.3 | 1.0 |
| Homenade95k | 95,069 | bud3_TL | 1237 | 1009 | 1.3 | 1.1 |
| SPGP40k | 39,800 | bud1_A | 627 | 439 | 1.6 | 1.1 |
| SPGP40k | 39,800 | bud2_A | 620 | 415 | 1.6 | 1.0 |
| SPGP40k | 39,800 | bud3_A | 605 | 394 | 1.5 | 1.0 |
| SPGP40k | 39,800 | bud1_C | 604 | 443 | 1.5 | 1.1 |
| SPGP40k | 39,800 | bud2_C | 605 | 395 | 1.5 | 1.0 |
| SPGP40k | 39,800 | bud3_C | 621 | 416 | 1.6 | 1.0 |
| SPGP40k | 39,800 | bud1_TL | 756 | 688 | 1.9 | 1.7 |
| SPGP40k | 39,800 | bud2_TL | 706 | 562 | 1.8 | 1.4 |
| SPGP40k | 39,800 | bud3_TL | 730 | 624 | 1.8 | 1.6 |
| Database | Sample | Total Search Duration 1 | SEQUEST/Decoy 2 Search Duration | Mascot/Decoy 2 Search Duration |
|---|---|---|---|---|
| SP21 | bud1_A | 11 min 0 s | 2 min 0 s | 6 min 43 s |
| SP21 | bud2_A | 10 min 0 s | 1 min 30 s | 6 min 44 s |
| SP21 | bud3_A | 10 min 0 s | 1 min 31 s | 6 min 25 s |
| SP21 | bud1_C | 10 min 0 s | 2 min 35 s | 4 min 52 s |
| SP21 | bud2_C | 8 min 0 s | 1 min 54 s | 4 min 4 s |
| SP21 | bud3_C | 10 min 0 s | 2 min 21 s | 5 min 12 s |
| SP21 | bud1_T | 12 min 0 s | 2 min 28 s | 6 min 42 s |
| SP21 | bud2_T | 11 min 0 s | 2 min 18 s | 6 min 28 s |
| SP21 | bud3_T | 11 min 0 s | 2 min 12 s | 6 min 1 s |
| Uniprot515 | bud1_A | 20 min 0 s | 5 min 30 s | 10 min 12 s |
| Uniprot515 | bud2_A | 19 min 0 s | 5 min 10 s | 10 min 53 s |
| Uniprot515 | bud3_A | 21 min 0 s | 5 min 15 s | 11 min 42 s |
| Uniprot515 | bud1_C | 18 min 0 s | 8 min 28 s | 5 min 12 s |
| Uniprot515 | bud2_C | 16 min 0 s | 7 min 1 s | 4 min 22 s |
| Uniprot515 | bud3_C | 19 min 0 s | 8 min 53 s | 5 min 4 s |
| Uniprot515 | bud1_T | 26 min 0 s | 11 min 33 s | 8 min 25 s |
| Uniprot515 | bud2_T | 20 min 0 s | 8 min 55 s | 6 min 4 s |
| Uniprot515 | bud3_T | 21 min 0 s | 8 min 49 s | 7 min 22 s |
| JO29k | bud1_A | 1 h 14 min 0 s | 1 h 9 min | n.a. |
| JO29k | bud2_A | 1 h 17 min 0 s | 1 h 13 min | n.a. |
| JO29k | bud3_A | 1 h 22 min 0 s | 1 h 18 min | n.a. |
| JO29k | bud1_C | 28 min 0 s | 24 min 3 s | n.a. |
| JO29k | bud2_C | 19 min 0 s | 16 min 14 s | n.a. |
| JO29k | bud3_C | 25 min 0 s | 21 min 4 s | n.a. |
| JO29k | bud1_T | 56 min 0 s | 51 min 50 s | n.a. |
| JO29k | bud2_T | 45 min 0 s | 40 min 29 s | n.a. |
| JO29k | bud3_T | 49 min 0 s | 44 min 30 s | n.a. |
| Homemade95k | bud1_A | 19 h 13 min 0 s | 4 h 47 min | 14 h 17 min |
| Homemade95k | bud2_A | 22 h 16 min 0 s | 5 h 14 min | 16 h 54 min |
| Homemade95k | bud3_A | 25 h 28 min 0 s | 5 h 56 min | 19 h 24 min |
| Homemade95k | bud1_C | 8 h 31 min 0 s | 2 h 53 min | 5 h 31 min |
| Homemade95k | bud2_C | 5 h 21 min 0 s | 1 h 31 min | 3 h 43 min |
| Homemade95k | bud3_C | 5 h 29 min 0 s | 1 h 57 min | 3 h 25 min |
| Homemade95k | bud1_T | 9 h 20 min 0 s | 2 h 50 min | 6 h 22 min |
| Homemade95k | bud2_T | 5 h 29 min 0 s | 1 h 49 min s | 3 h 30 min |
| Homemade95k | bud3_T | 8 h 10 min 0 s | 2 h 19 min s | 5 h 43 min |
| SPGP40k | bud1_A | 6 h 48 min 0 s | 3 h 33 min | 3 h 8 min |
| SPGP40k | bud2_A | 7 h 41 min 0 s | 3 h 50 min | 3 h 45 min |
| SPGP40k | bud3_A | 8 h 39 min 0 s | 4 h 17 min | 4 h 15 min |
| SPGP40k | bud1_C | 3 h 35 min 0 s | 2 h 3 min | 1 h 26 min |
| SPGP40k | bud2_C | 2 h 18 min 0 s | 1 h 14 min | 59 min 41 s |
| SPGP40k | bud3_C | 2 h 42 min 0 s | 1 h 39 min | 57 min 18 s |
| SPGP40k | bud1_T | 4 h 22 min 0 s | 2 h 27 min | 1 h 48 min |
| SPGP40k | bud2_T | 2 h 43 min 0 s | 1 h 34 min | 1 h 2 min |
| SPGP40k | bud3_T | 3 h 42 min 0 s | 1 h 59 min | 1 h 36 min |
| # Miscleavage | SP21 | Uniprot515 | JO29k | Homemade95k | SPGP40k |
|---|---|---|---|---|---|
| 0 | 116 | 433 | 2822 | 5818 | 2060 |
| 1 | 33 | 95 | 282 | 1091 | 403 |
| 2 | 20 | 51 | 32 | 339 | 140 |
| 3 | 7 | 16 | 13 | 158 | 60 |
| 4 | 8 | 9 | 5 | 54 | 28 |
| 5 | 1 | 1 | 6 | 22 | 7 |
| 6 | 4 | 3 | 4 | 8 | 5 |
| 7 | 2 | 3 | 1 | 8 | 4 |
| 8 | 1 | 0 | 3 | 5 | 1 |
| 10 | 1 | 0 | 1 | 1 | 1 |
| TOTAL | 193 | 611 | 3169 | 7504 | 2709 |
| TOTAL miscleavage = 0 | 116 | 433 | 2822 | 5818 | 2060 |
| TOTAL miscleavage > 0 | 77 | 178 | 347 | 1686 | 649 |
| % miscleavage > 0 | 39.9 | 29.1 | 10.9 | 22.5 | 24.0 |
| ELPD a | 39 | 255 | 2475 | 4132 | 1411 |
| A. Peptide Mass | SP21 | Uniprot515 | JO29k | Homemade95k | SPGP40k |
|---|---|---|---|---|---|
| min | 626.4 | 626.4 | 969.5 | 604.3 | 604.3 |
| max | 7600.9 | 6385.2 | 6724.5 | 6993.1 | 6448.6 |
| average | 2123.2 | 2023.2 | 2173.6 | 1975.8 | 1866.0 |
| SD | 1099.7 | 1048.9 | 791.1 | 830.3 | 776.8 |
| B. Protease | Database | min Mass | max mass | average Mass | SD Mass |
| A | SP21 | 1006.6 | 7600.9 | 2475.2 | 1166.7 |
| A | Uniprot515 | 631.3 | 5994.1 | 2363.4 | 1192.1 |
| A | JO29k | 969.5 | 6724.5 | 2280.9 | 905.8 |
| A | Homemade95k | 653.4 | 6375.2 | 2147.2 | 939.1 |
| A | SPGP40k | 653.4 | 6448.6 | 2028.9 | 929.2 |
| C | SP21 | 774.4 | 5520.9 | 1807.1 | 927.0 |
| C | Uniprot515 | 704.4 | 5520.9 | 1779.1 | 793.0 |
| C | JO29k | 1034.6 | 6061.9 | 2108.9 | 776.2 |
| C | Homemade95k | 789.5 | 6954.3 | 1901.9 | 724.2 |
| C | SPGP40k | 789.5 | 5121.4 | 1832.0 | 581.4 |
| TL | SP21 | 626.4 | 5303.5 | 2007.0 | 1058.9 |
| TL | Uniprot515 | 626.4 | 6385.2 | 1926.4 | 1015.7 |
| TL | JO29k | 1055.5 | 6369.2 | 2112.1 | 705.8 |
| TL | Homemade95k | 604.3 | 6369.2 | 1922.4 | 789.4 |
| TL | SPGP40k | 604.3 | 6369.2 | 1795.0 | 706.0 |
| PTM | SP21 | Uniprot515 | JO29k | Homemade95k | SPGP40k |
|---|---|---|---|---|---|
| Carbamidomethyl (C) | 34 | 94 | 493 | 602 | 226 |
| N-term acetyl (K) | 21 | 16 | 27 | 91 | 44 |
| Acetyl (K) | 47 | 32 | 47 | 132 | 71 |
| Methyl (K) | 61 | 49 | 114 | 163 | 158 |
| NAG (N) | 10 | 5 | 9 | 17 | 7 |
| Oxidation (M) | 18 | 24 | 43 | 66 | 90 |
| Phospho (STY) | 86 | 57 | 100 | 201 | 71 |
| TOTAL PTMs | 277 | 277 | 833 | 1272 | 667 |
| # identified peptides | 344 | 611 | 3169 | 7504 | 2709 |
| # unmodified peptides | 192 | 450 | 2255 | 5593 | 1834 |
| # modified peptides | 152 | 161 | 914 | 1911 | 875 |
| % modified peptides | 44.2 | 26.4 | 28.8 | 25.5 | 32.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, D.; Savin, K.; Rochfort, S.; Spangenberg, G. The Power of Three in Cannabis Shotgun Proteomics: Proteases, Databases and Search Engines. Proteomes 2020, 8, 13. https://doi.org/10.3390/proteomes8020013
Vincent D, Savin K, Rochfort S, Spangenberg G. The Power of Three in Cannabis Shotgun Proteomics: Proteases, Databases and Search Engines. Proteomes. 2020; 8(2):13. https://doi.org/10.3390/proteomes8020013
Chicago/Turabian StyleVincent, Delphine, Keith Savin, Simone Rochfort, and German Spangenberg. 2020. "The Power of Three in Cannabis Shotgun Proteomics: Proteases, Databases and Search Engines" Proteomes 8, no. 2: 13. https://doi.org/10.3390/proteomes8020013
APA StyleVincent, D., Savin, K., Rochfort, S., & Spangenberg, G. (2020). The Power of Three in Cannabis Shotgun Proteomics: Proteases, Databases and Search Engines. Proteomes, 8(2), 13. https://doi.org/10.3390/proteomes8020013