Is It Possible to Find Needles in a Haystack? Meta-Analysis of 1000+ MS/MS Files Provided by the Russian Proteomic Consortium for Mining Missing Proteins

 , , ,
, , ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. neXtProt Data Analysis
2.2. Expression Data Analysis
2.3. Virtual Proteolysis
2.4. Re-Analysis of MS Data
3. Results and Discussion
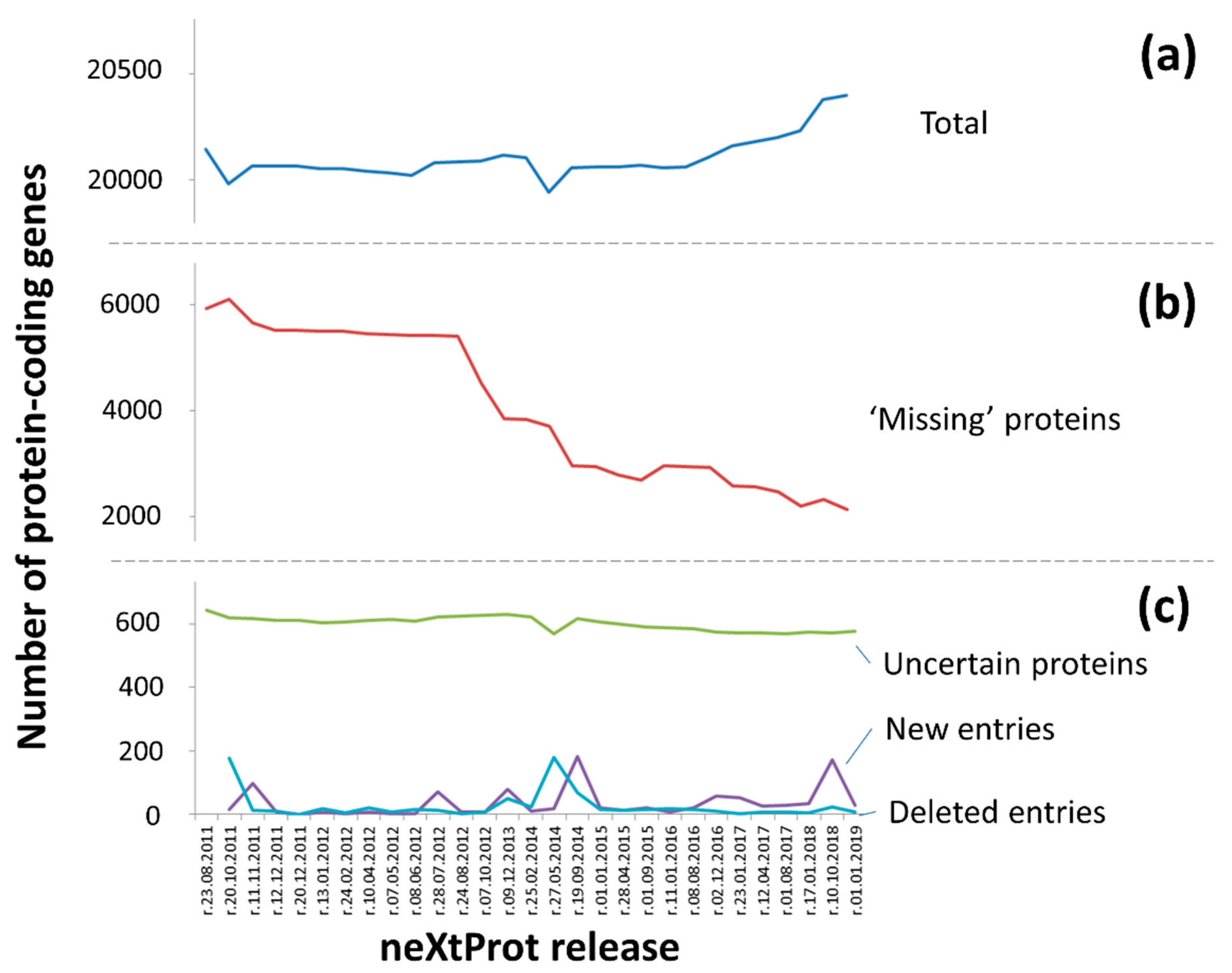
3.1. “Protein Existence” Features for Human Protein-Coding Genes
3.2. Is the mRNA a Good Helper in Searching for the Missing Proteins?
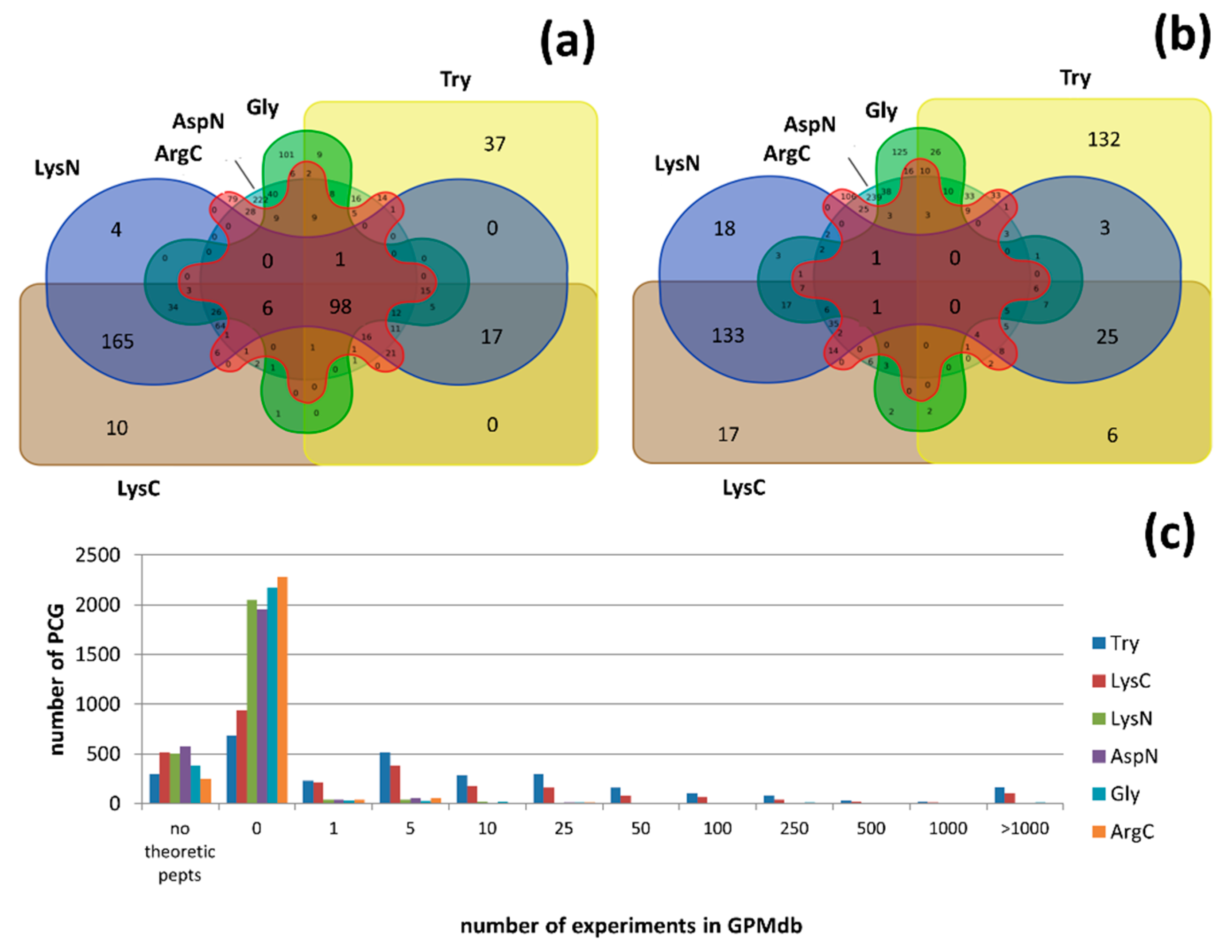
3.3. MS Detectable or Not?
3.4. Unique Cases—beyond the C-HPP Scope
3.5. One Hit Wonder!
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paik, Y.-K.; Jeong, S.-K.; Omenn, G.S.; Uhlen, M.; Hanash, S.; Cho, S.Y.; Lee, H.-J.; Na, K.; Choi, E.-Y.; Yan, F.; et al. The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat. Biotechnol. 2012, 30, 221–223. [Google Scholar] [CrossRef]
- Horvatovich, P.; Lundberg, E.K.; Chen, Y.-J.; Sung, T.-Y.; He, F.; Nice, E.C.; Goode, R.J.; Yu, S.; Ranganathan, S.; Baker, M.S.; et al. Quest for Missing Proteins: Update 2015 on Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 14, 3415–3431. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 1–6. [Google Scholar] [CrossRef]
- Poverennaya, E.V.; Ilgisonis, E.V.; Ponomarenko, E.A.; Kopylov, A.T.; Zgoda, V.G.; Radko, S.P.; Lisitsa, A.V.; Archakov, A.I. Why Are the Correlations between mRNA and Protein Levels so Low among the 275 Predicted Protein-Coding Genes on Human Chromosome 18? J. Proteome Res. 2017, 16, 4311–4318. [Google Scholar] [CrossRef] [PubMed]
- Ilgisonis, E.V.; Kopylov, A.T.; Ponomarenko, E.A.; Poverennaya, E.V.; Tikhonova, O.V.; Farafonova, T.E.; Novikova, S.; Lisitsa, A.V.; Zgoda, V.G.; Archakov, A.I. Increased Sensitivity of Mass Spectrometry by Alkaline Two-Dimensional Liquid Chromatography: Deep Cover of the Human Proteome in Gene-Centric Mode. J. Proteome Res. 2018, 17, 4258–4266. [Google Scholar] [CrossRef]
- Ezkurdia, I.; Juan, D.; Rodriguez, J.M.; Frankish, A.; Diekhans, M.; Harrow, J.; Vazquez, J.; Valencia, A.; Tress, M.L. Multiple evidence strands suggest that there may be as few as 19 000 human protein-coding genes. Hum. Mol. Genet. 2014, 23, 5866–5878. [Google Scholar] [CrossRef] [PubMed]
- NCBI Resource Coordinators Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2014, 42, D7–D17. [CrossRef]
- Carr, S.A.; Abbatiello, S.E.; Ackermann, B.L.; Borchers, C.; Domon, B.; Deutsch, E.W.; Grant, R.P.; Hoofnagle, A.N.; Huttenhain, R.; Koomen, J.M.; et al. Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-based Assay Development Using a Fit-for-Purpose Approach. Mol. Cell. Proteom. 2014, 13, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.K.; Omenn, G.S.; Uhlen, M.; Hanash, S.; Marko-Varga, G.; Aebersold, R.; Bairoch, A.; Yamamoto, T.; Legrain, P.; Lee, H.J.; et al. Standard guidelines for the chromosome-centric human proteome project. J. Proteome Res. 2012, 11, 2005–2013. [Google Scholar] [CrossRef]
- Omenn, G.S.; Lane, L.; Lundberg, E.K.; Beavis, R.C.; Overall, C.M.; Deutsch, E.W. Metrics for the Human Proteome Project 2016: Progress on Identifying and Characterizing the Human Proteome, Including Post-Translational Modifications. J. Proteome Res. 2016, 15, 3951–3960. [Google Scholar] [CrossRef]
- Omenn, G.S.; Lane, L.; Overall, C.M.; Corrales, F.J.; Schwenk, J.M.; Paik, Y.-K.; Van Eyk, J.E.; Liu, S.; Snyder, M.; Baker, M.S.; et al. Progress on Identifying and Characterizing the Human Proteome: 2018 Metrics from the HUPO Human Proteome Project. J. Proteome Res. 2018, 18. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Overall, C.M.; Van Eyk, J.E.; Baker, M.S.; Paik, Y.-K.; Weintraub, S.T.; Lane, L.; Martens, L.; Vandenbrouck, Y.; Kusebauch, U.; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2.1. J. Proteome Res. 2016, 15, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef]
- Giansanti, P.; Tsiatsiani, L.; Low, T.Y.; Heck, A.J.R. Six alternative proteases for mass spectrometry–based proteomics beyond trypsin. Nat. Protoc. 2016, 11, 993–1006. [Google Scholar] [CrossRef]
- Fenyö, D.; Beavis, R.C. The GPMDB REST interface. Bioinformatics 2015, 31, 2056–2058. [Google Scholar] [CrossRef]
- Holman, J.D.; Tabb, D.L.; Mallick, P. Employing ProteoWizard to Convert Raw Mass Spectrometry Data. Curr. Protoc. Bioinforma. 2014, 46, 13–24. [Google Scholar] [CrossRef]
- Fenyö, D.; Beavis, R.C. A method for assessing the statistical significance of mass spectrometry-based protein identifications using general scoring schemes. Anal. Chem. 2003, 75, 768–774. [Google Scholar] [CrossRef]
- Kim, S.; Pevzner, P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 2014, 5, 5277. [Google Scholar] [CrossRef]
- Geer, L.Y.; Markey, S.P.; Kowalak, J.A.; Wagner, L.; Xu, M.; Maynard, D.M.; Yang, X.; Shi, W.; Bryant, S.H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 3, 958–964. [Google Scholar] [CrossRef]
- Kiseleva, O.; Poverennaya, E.; Shargunov, A.; Lisitsa, A. Proteomic Cinderella: Customized analysis of bulky MS/MS data in one night. J. Bioinform. Comput. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Barsnes, H.; Vaudel, M. SearchGUI: A Highly Adaptable Common Interface for Proteomics Search and de Novo Engines. J. Proteome Res. 2018, 17, 2552–2555. [Google Scholar] [CrossRef] [PubMed]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Levitsky, L.I.; Ivanov, M.V.; Lobas, A.A.; Bubis, J.A.; Tarasova, I.A.; Solovyeva, E.M.; Pridatchenko, M.L.; Gorshkov, M.V. IdentiPy: An Extensible Search Engine for Protein Identification in Shotgun Proteomics. J. Proteome Res. 2018, 17, 2249–2255. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Gaudet, P.; Argoud-Puy, G.; Cusin, I.; Duek, P.; Evalet, O.; Gateau, A.; Gleizes, A.; Pereira, M.; Zahn-Zabal, M.; Zwahlen, C.; et al. neXtProt: Organizing protein knowledge in the context of human proteome projects. J. Proteome Res. 2013, 12, 293–298. [Google Scholar] [CrossRef]
- Zgoda, V.G.; Kopylov, A.T.; Tikhonova, O.V.; Moisa, A.a.; Pyndyk, N.V.; Farafonova, T.E.; Novikova, S.E.; Lisitsa, A.V.; Ponomarenko, E.a.; Poverennaya, E.V.; et al. Chromosome 18 transcriptome profiling and targeted proteome mapping in depleted plasma, liver tissue and HepG2 cells. J. Proteome Res. 2013, 12, 123–134. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Kopylov, A.T.; Lisitsa, A.V.; Radko, S.P.; Kiseleva, Y.Y.; Kurbatov, L.K.; Ptitsyn, K.G.; Tikhonova, O.V.; Moisa, A.A.; Novikova, S.E.; et al. Chromosome 18 Transcriptoproteome of Liver Tissue and HepG2 Cells and Targeted Proteome Mapping in Depleted Plasma: Update 2013. J. Proteome Res. 2014, 13, 183–190. [Google Scholar] [CrossRef]
- Gaudet, P.; Michel, P.-A.; Zahn-Zabal, M.; Cusin, I.; Duek, P.D.; Evalet, O.; Gateau, A.; Gleizes, A.; Pereira, M.; Teixeira, D.; et al. The neXtProt knowledgebase on human proteins: Current status. Nucleic Acids Res. 2015, 43, D764–D770. [Google Scholar] [CrossRef]
- Brückner, A.; Polge, C.; Lentze, N.; Auerbach, D.; Schlattner, U. Yeast Two-Hybrid, a Powerful Tool for Systems Biology. Int. J. Mol. Sci. 2009, 10, 2763–2788. [Google Scholar] [CrossRef]
- Siddiqui, O.; Zhang, H.; Guan, Y.; Omenn, G.S. Chromosome 17 Missing Proteins: Recent Progress and Future Directions as Part of the neXt-MP50 Challenge. J. Proteome Res. 2018, 17, 4061–4071. [Google Scholar] [CrossRef] [PubMed]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium, K.G.; Deluca, D.S.; Segre, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; Lek, M.; et al. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Krupp, M.; Marquardt, J.U.; Sahin, U.; Galle, P.R.; Castle, J.; Teufel, A. RNA-Seq Atlas--a reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics 2012, 28, 1184–1185. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Poverennaya, E.V.; Kopylov, A.T.; Ponomarenko, E.A.; Ilgisonis, E.V.; Zgoda, V.G.; Tikhonova, O.V.; Novikova, S.E.; Farafonova, T.E.; Kiseleva, Y.Y.; Radko, S.P.; et al. State of the Art of Chromosome 18-Centric HPP in 2016: Transcriptome and Proteome Profiling of Liver Tissue and HepG2 Cells. J. Proteome Res. 2016, 15. [Google Scholar] [CrossRef]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Kumar, D.; Bansal, G.; Narang, A.; Basak, T.; Abbas, T.; Dash, D. Integrating transcriptome and proteome profiling: Strategies and applications. Proteomics 2016, 16, 2533–2544. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Kahles, A.; Lehmann, K.-V.; Toussaint, N.C.; Hüser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network, R.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef]
- Choong, W.-K.; Chang, H.-Y.; Chen, C.-T.; Tsai, C.-F.; Hsu, W.-L.; Chen, Y.-J.; Sung, T.-Y. Informatics View on the Challenges of Identifying Missing Proteins from Shotgun Proteomics. J. Proteome Res. 2015, 14, 5396–5407. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Lane, L.; Overall, C.M.; Bandeira, N.; Baker, M.S.; Pineau, C.; Moritz, R.L.; Corrales, F.; Orchard, S.; Van Eyk, J.E.; et al. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 3.0. J. Proteome Res. 2019, 18, 4108–4116. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.; Zhang, Y.; Wei, W.; Li, Y.; Zhang, T.; He, F.; Gao, Y.; Xu, P. Multi-Protease Strategy Identifies Three PE2 Missing Proteins in Human Testis Tissue. J. Proteome Res. 2017, 16, 4352–4363. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shi, J.; Wang, Y.; Chen, Y.; Li, Y.; Kong, D.; Chang, L.; Liu, F.; Lv, Z.; Zhou, Y.; et al. Multiproteases Combined with High-pH Reverse-Phase Separation Strategy Verified Fourteen Missing Proteins in Human Testis Tissue. J. Proteome Res. 2018, 17, 4171–4177. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Claassen, M.; Schmidt, A.; Aebersold, R. Estimation of Absolute Protein Quantities of Unlabeled Samples by Selected Reaction Monitoring Mass Spectrometry. Mol. Cell. Proteom. 2012, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.; Wasinger, V.C. Protein and peptide fractionation, enrichment and depletion: Tools for the complex proteome. Proteomics 2011, 11, 513–534. [Google Scholar] [CrossRef]
- Kopylov, A.; Zgoda, V.; Lisitsa, A.; Archakov, A. Combined use of irreversible binding and MRM technology for low- and ultralow copy-number protein detection and quantitation. Proteomics 2013, 13, 727–742. [Google Scholar] [CrossRef]
- Omenn, G.S. The HUPO Human Proteome Project (HPP), a Global Health Research Collaboration. Cent. Asian J. Glob. Heal. 2012, 1. [Google Scholar] [CrossRef]
- Tarasova, I.A.; Tereshkova, A.V.; Lobas, A.A.; Solovyeva, E.M.; Sidorenko, A.S.; Gorshkov, V.; Kjeldsen, F.; Bubis, J.A.; Ivanov, M.V.; Ilina, I.Y.; et al. Comparative proteomics as a tool for identifying specific alterations within interferon response pathways in human glioblastoma multiforme cells. Oncotarget 2018, 9, 1785–1802. [Google Scholar] [CrossRef]
- Lobas, A.A.; Pyatnitskiy, M.A.; Chernobrovkin, A.L.; Ilina, I.Y.; Karpov, D.S.; Solovyeva, E.M.; Kuznetsova, K.G.; Ivanov, M.V.; Lyssuk, E.Y.; Kliuchnikova, A.A.; et al. Proteogenomics of Malignant Melanoma Cell Lines: The Effect of Stringency of Exome Data Filtering on Variant Peptide Identification in Shotgun Proteomics. J. Proteome Res. 2018, 17, 1801–1811. [Google Scholar] [CrossRef]
- Naryzhny, S.; Maynskova, M.; Zgoda, V.; Archakov, A. Dataset of protein species from human liver. Data Br. 2017, 12, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.N.; Zgoda, V.G.; Maynskova, M.A.; Novikova, S.E.; Ronzhina, N.L.; Vakhrushev, I.V.; Khryapova, E.V.; Lisitsa, A.V.; Tikhonova, O.V.; Ponomarenko, E.A.; et al. Combination of virtual and experimental 2DE together with ESI LC-MS/MS gives a clearer view about proteomes of human cells and plasma. Electrophoresis 2016, 37, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Starodubtseva, N.L.; Brzhozovskiy, A.G.; Bugrova, A.E.; Kononikhin, A.S.; Indeykina, M.I.; Gusakov, K.I.; Chagovets, V.V.; Nazarova, N.M.; Frankevich, V.E.; Sukhikh, G.T.; et al. Label-free cervicovaginal fluid proteome profiling reflects the cervix neoplastic transformation. J. Mass Spectrom. 2019, 54, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Kaysheva, A.L.; Kopylov, A.T.; Ponomarenko, E.A.; Kiseleva, O.I.; Teryaeva, N.B.; Potapov, A.A.; Izotov, A.A.; Morozov, S.G.; Kudryavtseva, V.Y.; Archakov, A.I. Relative Abundance of Proteins in Blood Plasma Samples from Patients with Chronic Cerebral Ischemia. J. Mol. Neurosci. 2018, 64, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Naryzhny, S.; Zgoda, V.; Kopylov, A.; Petrenko, E.; Kleist, O.; Archakov, A. Variety and Dynamics of Proteoforms in the Human Proteome: Aspects of Markers for Hepatocellular Carcinoma. Proteomes 2017, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, I.A.; Chumakov, P.M.; Moshkovskii, S.A.; Gorshkov, M.V. Profiling modifications for glioblastoma proteome using ultra-tolerant database search: Are the peptide mass shifts biologically relevant or chemically induced? J. Proteomics 2019, 191, 16–21. [Google Scholar] [CrossRef]
- Yampolsky, L.Y.; Stoltzfus, A. The exchangeability of amino acids in proteins. Genetics 2005, 170, 1459–1472. [Google Scholar] [CrossRef]
- Ivanov, M.V.; Levitsky, L.I.; Bubis, J.A.; Gorshkov, M. V Scavager: A Versatile Postsearch Validation Algorithm for Shotgun Proteomics Based on Gradient Boosting. Proteomics 2019, 19, e1800280. [Google Scholar] [CrossRef]
- Amaral, A.; Castillo, J.; Ramalho-Santos, J.; Oliva, R. The Combined Human Sperm Proteome: Cellular Pathways and Implications for Basic and Clinical Science. Hum. Reprod. Update 2014, 20. [Google Scholar] [CrossRef]
- Vandenbrouck, Y.; Lane, L.; Carapito, C.; Duek, P.; Rondel, K.; Bruley, C.; Macron, C.; Gonzalez de Peredo, A.; Couté, Y.; Chaoui, K.; et al. Looking for Missing Proteins in the Proteome of Human Spermatozoa: An Update. J. Proteome Res. 2016, 15. [Google Scholar] [CrossRef]
- Poverennaya, E.; Lisitsa, A. Gene Editing—A path forward for annotating the uPE1s for the C-HPP. Newsl. C-HPP 2018, 7, 1–2. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
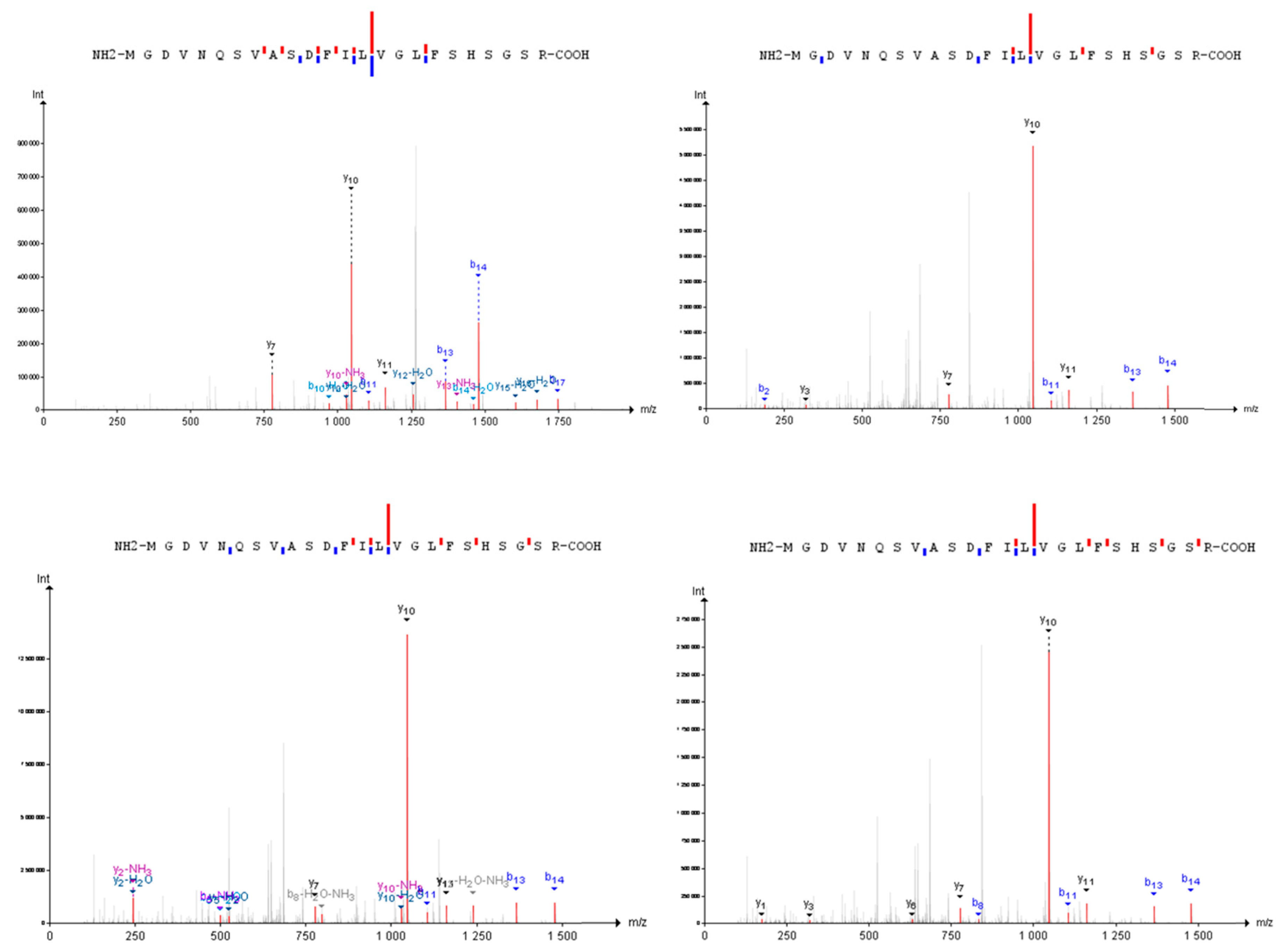{kind=link}
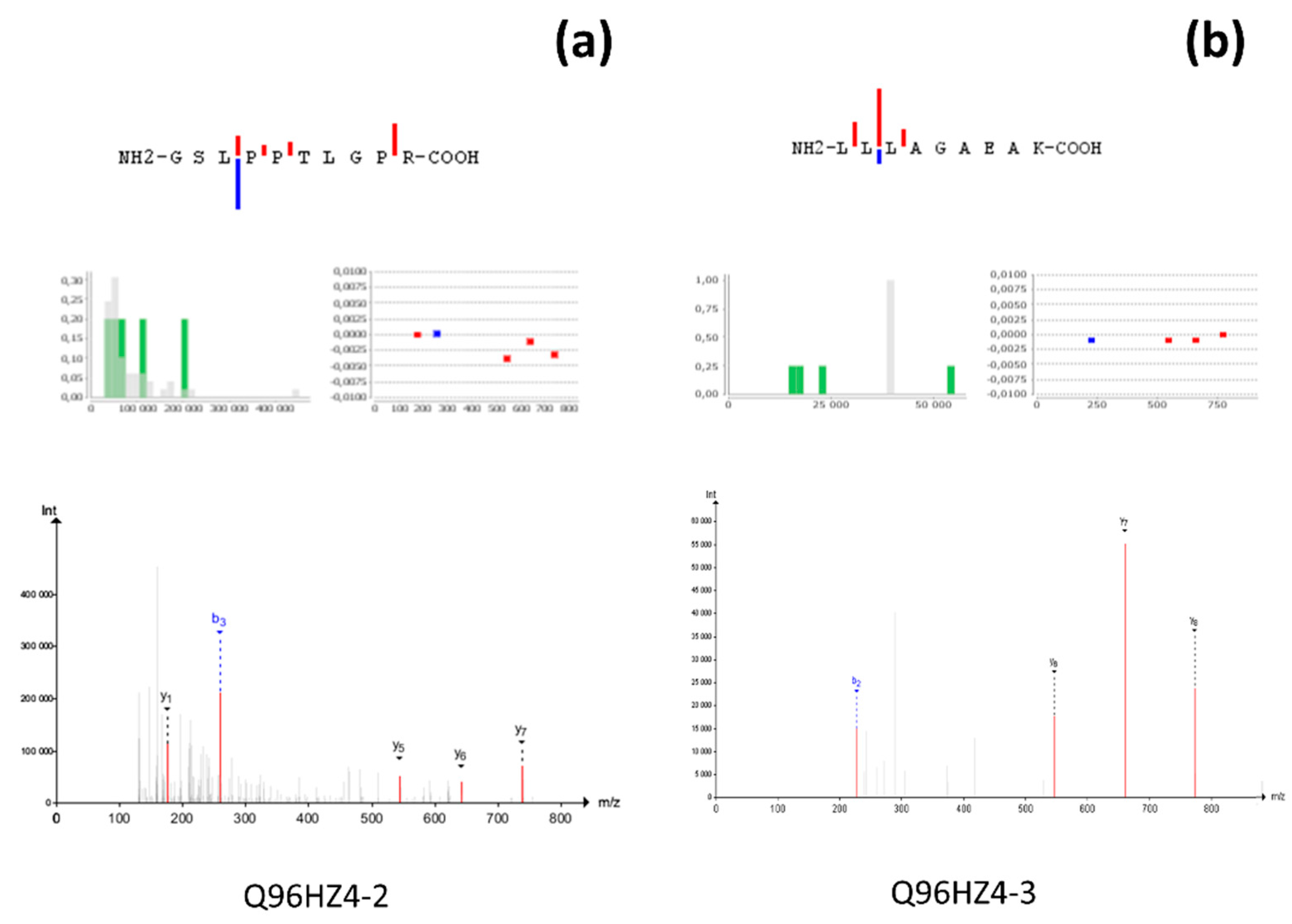{kind=link}
| Category of Biomaterial, Where Gene of Interest Was Observed | Total Number of Genes | Missing Proteins | Uncertain Proteins (PE5) | ||
|---|---|---|---|---|---|
| PE2 | PE3 | PE4 | |||
| All biomaterials | 9542 | 311 | 10 | 6 | 79 |
| Part of biomaterials | 3074 | 429 | 41 | 8 | 56 |
| Normal or tumor biomaterials * | 161 | 46 | 15 | 0 | 3 |
| Only normal | 274 | 55 | 36 | 3 | 4 |
| Only cancer | 58 | 12 | 8 | 0 | 4 |
| Total | 13,109 | 853 | 110 | 17 | 146 |
| # | AC | Gene | Number of Samples | Number of Unique Detectable Tryptic Peptides | ||
|---|---|---|---|---|---|---|
| Theoretically | Observed in GPMdb | Observed (SRM synt) in PeptideAtlas | ||||
| Missing proteins | ||||||
| 1 | P22532 | SPRR2D | 10 | 1 | 1 | 1/0 |
| 2 | A0A087WSY6 | IGKV3D-15 | 3 | 1 | 1 | 1/0 |
| Uncertain proteins | ||||||
| 3 | Q58FF3 | HSP90B2P | 1 | 10 | 3 | 1/2 |
| 4 | Q58FG1 | HSP90AA4P | 1 | 14 | 13 | 7/5 |
| 5 | Q9BYX7 | POTEKP | 3 | 8 | 8 | 5/5 |
| 6 | Q9BZK3 | NACA4P | 1 | 5 | 5 | 4/4 |
| 7 | Q9H853 | TUBA4B | 35 | 9 | 4 | 2/4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poverennaya, E.; Kiseleva, O.; Ilgisonis, E.; Novikova, S.; Kopylov, A.; Ivanov, Y.; Kononikhin, A.; Gorshkov, M.; Kushlinskii, N.; Archakov, A.; et al. Is It Possible to Find Needles in a Haystack? Meta-Analysis of 1000+ MS/MS Files Provided by the Russian Proteomic Consortium for Mining Missing Proteins. Proteomes 2020, 8, 12. https://doi.org/10.3390/proteomes8020012
Poverennaya E, Kiseleva O, Ilgisonis E, Novikova S, Kopylov A, Ivanov Y, Kononikhin A, Gorshkov M, Kushlinskii N, Archakov A, et al. Is It Possible to Find Needles in a Haystack? Meta-Analysis of 1000+ MS/MS Files Provided by the Russian Proteomic Consortium for Mining Missing Proteins. Proteomes. 2020; 8(2):12. https://doi.org/10.3390/proteomes8020012
Chicago/Turabian StylePoverennaya, Ekaterina, Olga Kiseleva, Ekaterina Ilgisonis, Svetlana Novikova, Arthur Kopylov, Yuri Ivanov, Alexei Kononikhin, Mikhail Gorshkov, Nikolay Kushlinskii, Alexander Archakov, and et al. 2020. "Is It Possible to Find Needles in a Haystack? Meta-Analysis of 1000+ MS/MS Files Provided by the Russian Proteomic Consortium for Mining Missing Proteins" Proteomes 8, no. 2: 12. https://doi.org/10.3390/proteomes8020012
APA StylePoverennaya, E., Kiseleva, O., Ilgisonis, E., Novikova, S., Kopylov, A., Ivanov, Y., Kononikhin, A., Gorshkov, M., Kushlinskii, N., Archakov, A., & Ponomarenko, E. (2020). Is It Possible to Find Needles in a Haystack? Meta-Analysis of 1000+ MS/MS Files Provided by the Russian Proteomic Consortium for Mining Missing Proteins. Proteomes, 8(2), 12. https://doi.org/10.3390/proteomes8020012