Challenges in Clinical Metaproteomics Highlighted by the Analysis of Acute Leukemia Patients with Gut Colonization by Multidrug-Resistant Enterobacteriaceae

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
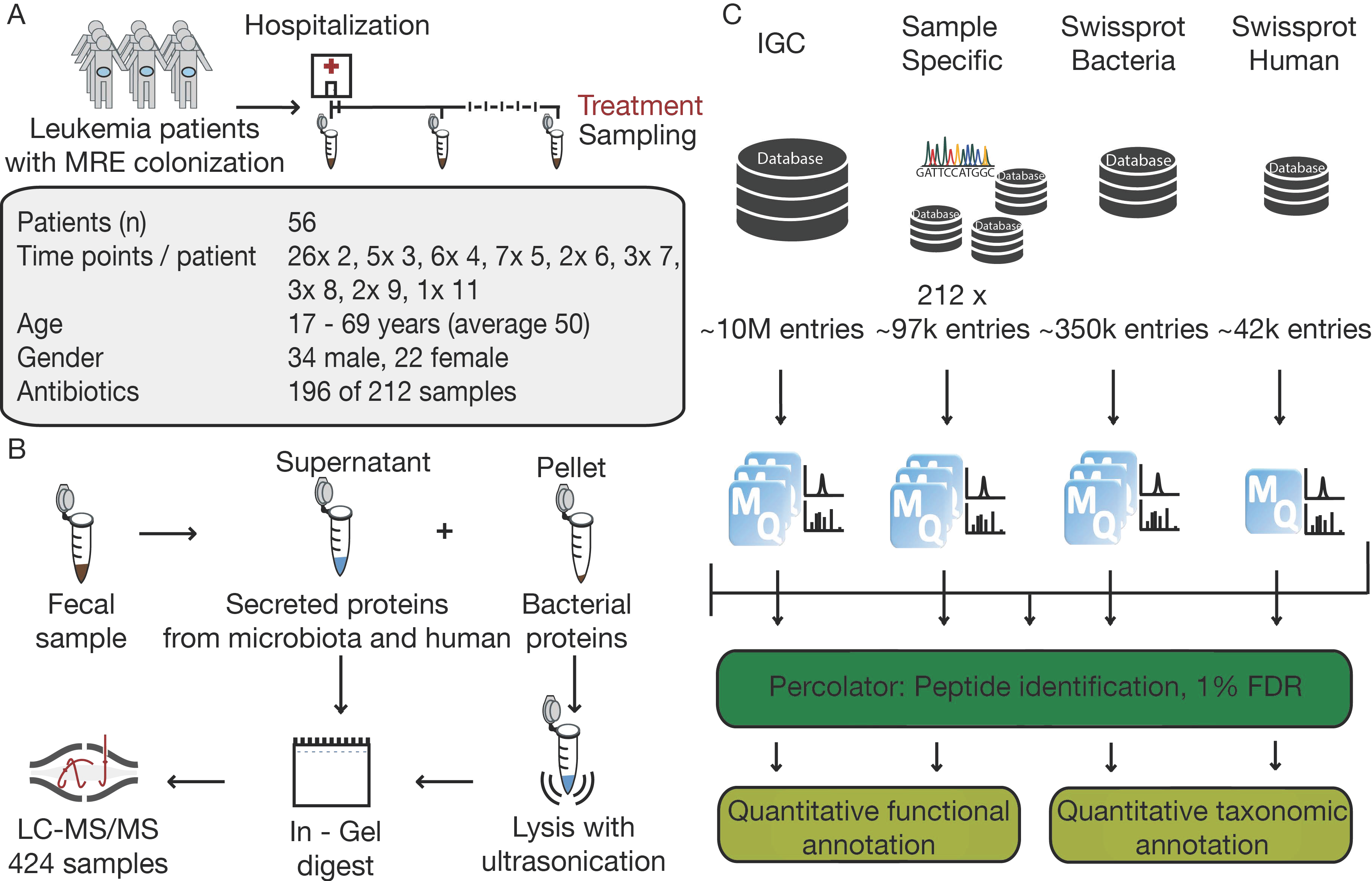
2.1. Sampling Process
2.2. Sample Preparation
2.3. LC-MS/MS Analysis
2.4. Data Processing and Analysis
2.5. Metagenomic Sequencing and Data Processing
2.6. 16S rRNA Sequencing and Data Processing
2.7. Determination of 16S rRNA Counts with qPCR
3. Results and Discussion
3.1. The Challenge of Experimental Design in Clinical Metaproteomics
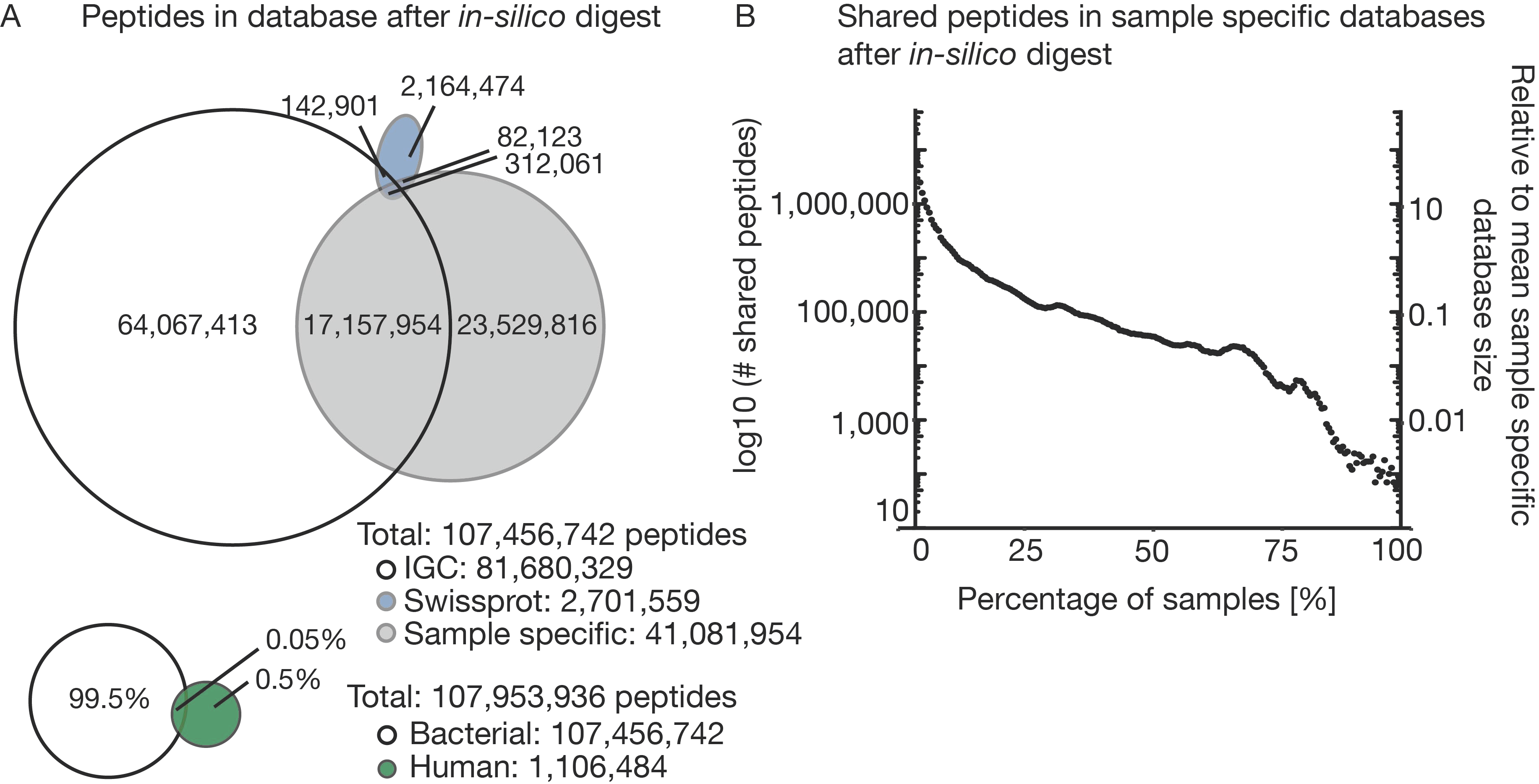
3.2. The Challenge of (the Lack of) a Comprehensive Sequence Search Space
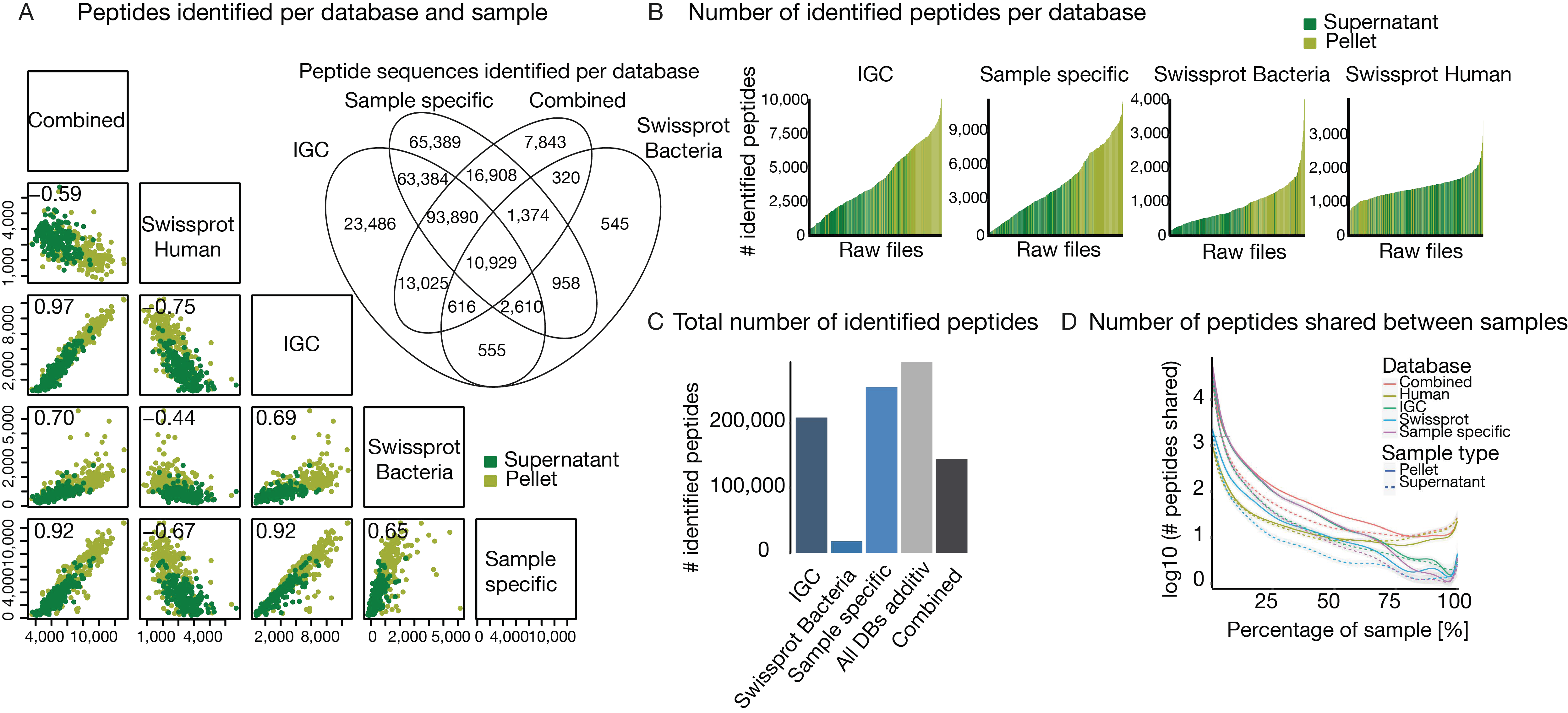
3.3. The Challenge of Extensive Demand for Computational Power and Storage Capacity
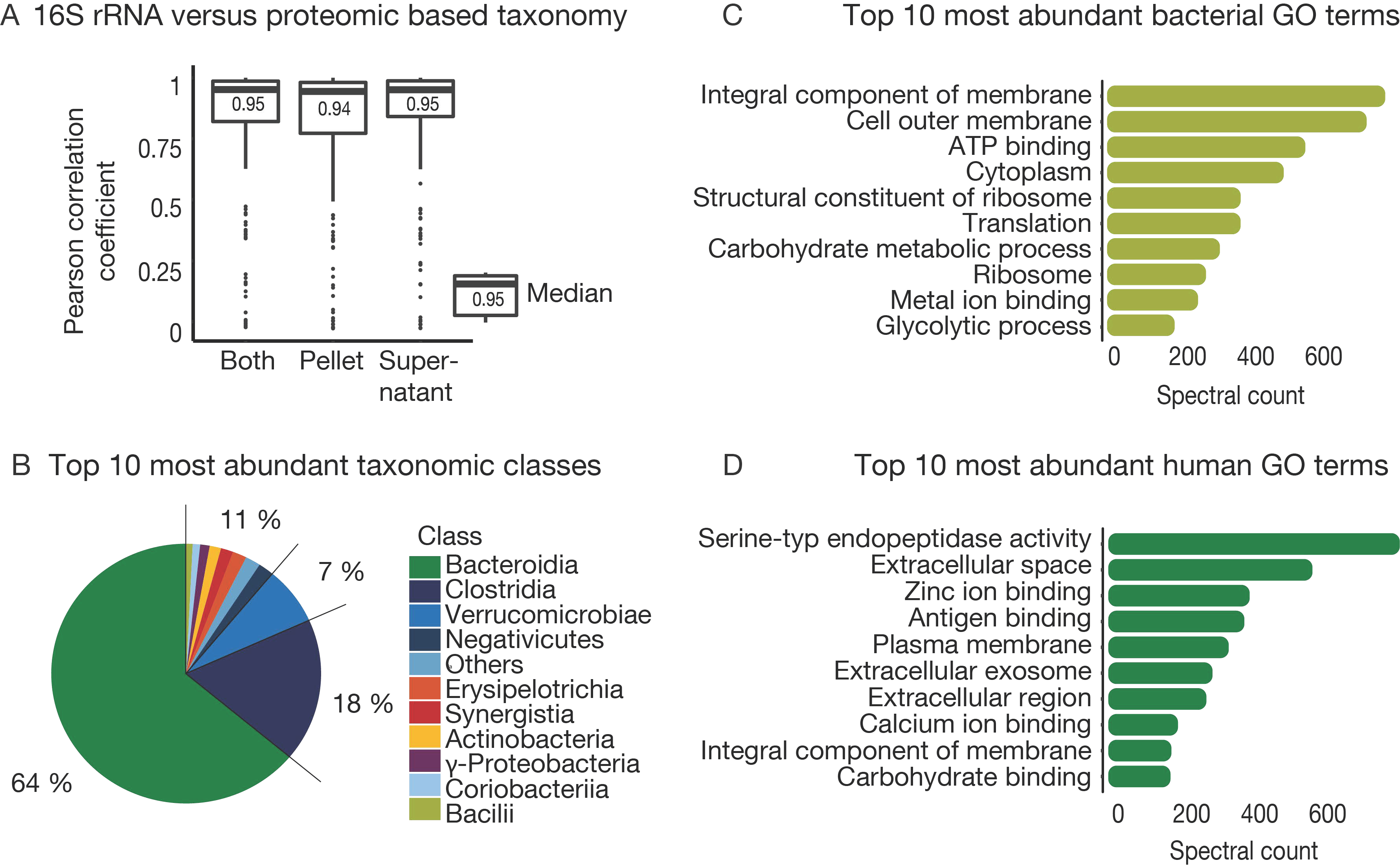
3.4. The Challenge of Functional and Taxonomic Annotation
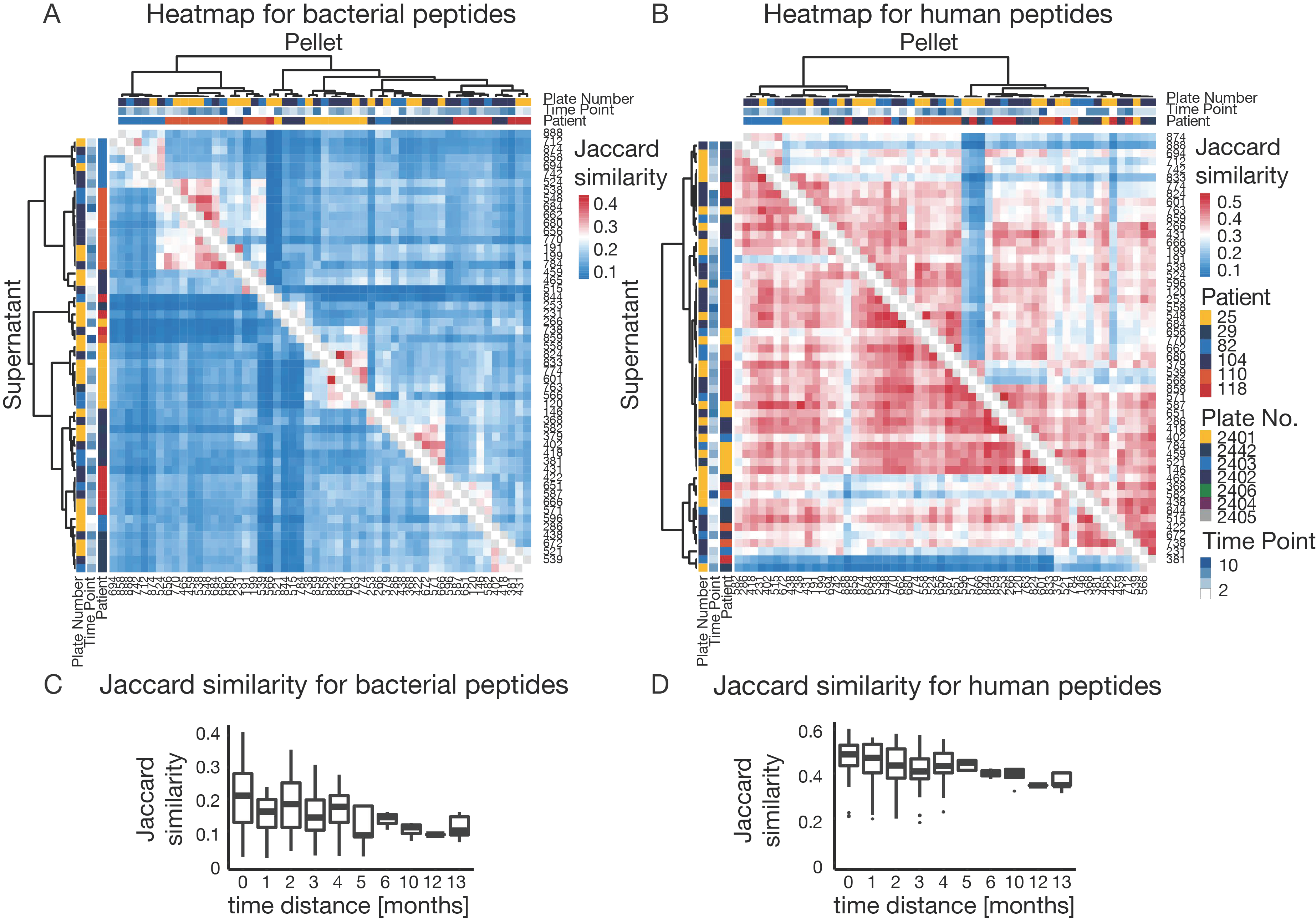
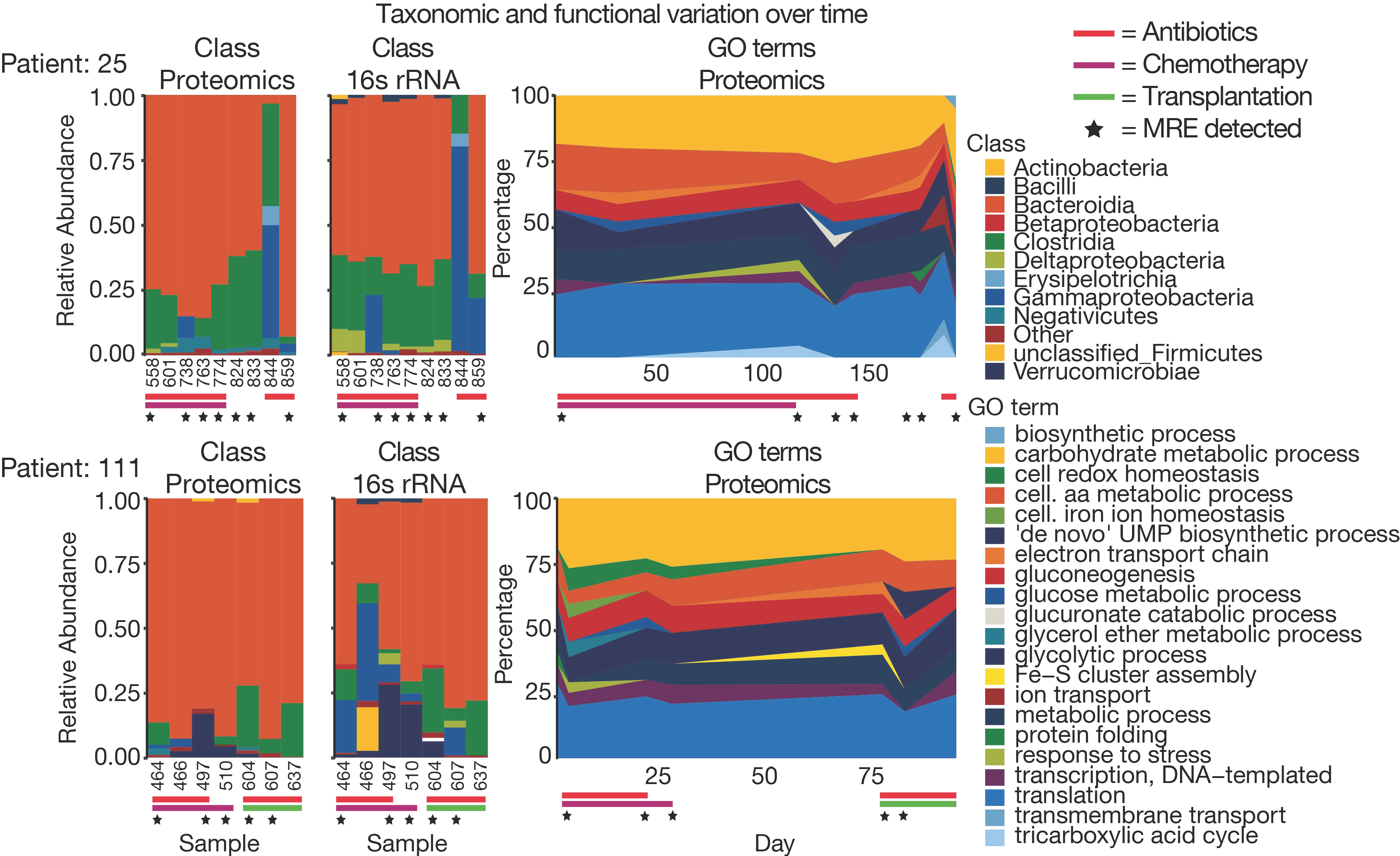
3.5. The Challenge of High Microbiome Variability in Clinical Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.S.; Kim, E.K.; Lee, W.J. Functional genomic and metagenomic approaches to understanding gut microbiota-animal mutualism. Curr. Opin. Microbiol. 2015, 24, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.J.; Graham, C.; McMullan, G. Microbial proteomics: A mass spectrometry primer for biologists. Microb. Cell Fact. 2007, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, P.; Bond, P.L. Metaproteomics: Studying functional gene expression in microbial ecosystems. Trends Microbiol. 2006, 14, 92–97. [Google Scholar] [CrossRef]
- Rodríguez-Valera, F. Environmental genomics, the big picture? FEMS Microbiol. Lett. 2004, 231, 153–158. [Google Scholar] [CrossRef]
- Lichtman, J.S.; Marcobal, A.; Sonnenburg, J.L.; Elias, J.E. Host-centric proteomics of stool: A novel strategy focused on intestinal responses to the gut microbiota. Mol. Cell Proteom. 2013, 12, 3310–3318. [Google Scholar] [CrossRef]
- Kolmeder, C.A.; de Been, M.; Nikkilä, J.; Ritamo, I.; Mättö, J.; Valmu, L.; Salojärvi, J.; Palva, A.; Salonen, A.; de Vos, W.M. Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS ONE 2012, 7, e29913. [Google Scholar] [CrossRef]
- Verberkmoes, N.C.; Russell, A.L.; Shah, M.; Godzik, A.; Rosenquist, M.; Halfvarson, J.; Lefsrud, M.G.; Apajalahti, J.; Tysk, C.; Hettich, R.L.; et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009, 3, 179–189. [Google Scholar] [CrossRef]
- Pérez-Cobas, A.E.; Gosalbes, M.J.; Friedrichs, A.; Knecht, H.; Artacho, A.; Eismann, K.; Otto, W.; Rojo, D.; Bargiell, R.; von Bergen, M.; et al. Gut microbiota disturbance during antibiotic therapy: A multi-omic approach. Gut 2012, 62, 1591–1601. [Google Scholar] [CrossRef]
- Chuong, K.H.; Mack, D.R.; Stintzi, A.; O’Doeherty, K.C. Human Microbiome and Learning Healthcare Systems: Integrating Research and Precision Medicine for Inflammatory Bowel Disease. OMICS 2018, 22, 119–126. [Google Scholar] [CrossRef]
- Juste, C.; Kreil, D.P.; Beauvallet, C.; Guillot, A.; Vaca, S.; Carapito, C.; Mondot, S.; Sykacek, P.; Sokol, H.; Blon, F.; et al. Bacterial protein signals are associated with Crohn’s disease. Gut 2014, 63, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Riuz, A.; Lanza, F.; Haange, S.B.; Oberbach, A.; Till, H.; Bargiella, R.; Campoy, C.; Segura, M.T.; Richter, M.; et al. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ. Microbiol. 2013, 15, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Kolmeder, C.A.; Ritari, J.; Verdam, F.J.; Muth, T.; Keskitalo, S.; Varosalo, M.; Fuentes, S.; Greve, J.W.; Buurman, W.A.; Reichl, U.; et al. Colonic metaproteomic signatures of active bacteria and the host in obesity. Proteomics 2015, 15, 3544–3552. [Google Scholar] [CrossRef] [PubMed]
- Gavin, P.G.; Mullaney, J.A.; Loo, D.; Gottlieb, P.A.; Hill, M.M.; Zipris, D.; Hamilton-Williams, E.E. Intestinal Metaproteomics Reveals Host-Microbiota Interactions in Subjects at Risk for Type 1 Diabetes. Diabetes Care 2018, 41, 2178–2186. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.P.; Baden, L.R. Antibiotic prophylaxis for patients with acute leukemia. Leuk Lymphoma 2008, 49, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Heyer, R.; Schallert, K.; Zoun, R.; Becher, B.; Saake, G.; Benndorf, D. Challenges and perspectives of metaproteomic data analysis. J. Biotechnol. 2017, 261, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Timmins-Schiffman, E.; May, D.H.; Mikan, M.; Riffle, M.; Frazar, C.; Harvey, H.R.; Noble, W.S.; Nunn, B.L. Critical decisions in metaproteomics: Achieving high confidence protein annotations in a sea of unknowns. ISME J. 2017, 11, 309–314. [Google Scholar] [CrossRef]
- Muth, T.; Renard, B.Y.; Martens, L. Metaproteomic data analysis at a glance: Advances in computational microbial community proteomics. Expert Rev. Proteom. 2016, 13, 757–769. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017, 550, 61–66. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kleiner, M.; Thorson, E.; Sharp, C.E.; Dong, X.; Liu, D.; Li, C.; Strous, M. Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 2017, 8, 1558. [Google Scholar] [CrossRef] [PubMed]
- Haange, S.B.; Jehmlich, N. Proteomic interrogation of the gut microbiota: Potential clinical impact. Expert Rev. Proteom. 2016, 13, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbath, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2014, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Tomas, H.; Halvis, J.; Olsen, B.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Hahne, H.; Pachl, F.; Ruprecht, B.; Maier, S.K.; Klaeger, S.; Helm, D.; Médard, G.; Wilm, M.; Lemeer, S.; Kuster, B. DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat. Methods 2013, 10, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; Nielsen, T.; et al. MetaHIT Consortium An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef]
- Mesuere, B.; Devreese, B.; Debyser, G.; Aerts, M.; Vandamme, P.; Dawyndt, P. Unipept: Tryptic peptide-based biodiversity analysis of metaproteome samples. J. Proteome Res. 2012, 11, 5773–5780. [Google Scholar] [CrossRef]
- Gurdeep, S.R.; Tanca, A.; Palomba, A.; Van der Jeugt, F.; Verschaffelt, P.; Uzzau, S.; Martens, L.; Dawyndt, P.; Mesuere, B. Unipept 4.0: Functional analysis of metaproteome data. J Proteome Res. 2018. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Côté, R.G.; Csordas, A.; Dianes, J.A.; Fabregat, A.; Foster, J.M.; Griss, J.; Alpi, E.; Birim, M.; Contell, J.; et al. The Proteomics Identifications (PRIDE) database and associated tools: Status in 2013. Nucleic Acids Res. 2013, 41, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 18, e2584. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W. The mathematical theory of communication. Bell Syst. Tech. J. 1948, 34, 312–313. [Google Scholar] [CrossRef]
- Wang, O.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Deligios, M.; Cubeddu, T.; Fraumene, C.; Biossa, G.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Evaluating the impact of different sequence databases on metaproteome analysis: Insights from a lab-assembled microbial mixture. PLoS ONE 2013, 8, e82981. [Google Scholar] [CrossRef]
- Muth, T.; Kolmeder, C.A.; Salojärvi, J.; Keskitalo, S.; Varjosalo, S.; Verdam, F.J.; Rensen, S.S.; Reichl, U.; de Vos, W.M.; Rapp, E.; et al. Navigating through metaproteomics data: A logbook of database searching. Proteomics 2015, 15, 3439–3453. [Google Scholar] [CrossRef]
- Hershberg, R. Mutation—The Engine of Evolution: Studying Mutation and Its Role in the Evolution of Bacteria. Cold Spring Harb. Perspect. Biol. 2015, 7, a018077. [Google Scholar] [CrossRef]
- Troung, D.T.; Tett, A.; Pasolli, E.; Huttenhower, C.; Segata, N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017, 27, 626–638. [Google Scholar] [CrossRef]
- Savitski, M.M.; Wilhelm, M.; Hahne, H.; Kuster, B.; Bantscheff, M. A Scalable Approach for Protein False Discovery Rate Estimation in Large Proteomic Data Sets. Mol. Cell. Proteom. 2015, 14, 2394–2404. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic Analysis of the Human Distal Gut Microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Deeke, S.A.; Ning, Z.; Starr, A.E.; Butcher, J.; Li, J.; Mayne, J.; Cheng, K.; Liao, B.; Li, L.; et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun. 2018, 9, 2873. [Google Scholar] [CrossRef] [PubMed]
- Cebula, T.A. Genetic and physiological modulation of anthracycline-induced mutagenesis in Salmonella typhimurium. Environ. Mutagen. 1986, 8, 675–692. [Google Scholar] [CrossRef] [PubMed]
- Riffle, M.; May, D.H.; Timmins-Schiffman, E.; Mikan, M.P.; Jaschib, D.; Noble, W.S.; Nunn, B.L. MetaGOmics: A Web-Based Tool for Peptide-Centric Functional and Taxonomic Analysis of Metaproteomics Data. Proteomes 2018, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Kolmeder, C.A.; de Vos, W.S. Metaproteomics of our microbiome—Developing insight in function and activity in man and model systems. J. Proteom. 2014, 97, 3–16. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 604, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Gonnelli, G.; Stock, M.; Verwaeren, J.; Maddelein, D.; De Baets, B.; Martens, L.; Degroeve, S. A decoy-free approach to the identification of peptides. J. Proteome Res. 2015, 14, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Marx, H.; Lemeer, S.; Klaeger, S.; Rattei, T.; Kuster, B. MScDB: A mass spectrometry-centric protein sequence database for proteomics. J. Proteome Res. 2013, 12, 2386–2398. [Google Scholar] [CrossRef] [PubMed]
- May, D.H.; Timmins-Schiffman, E.; Mikan, M.P.; Harvey, H.R.; Borenstein, E.; Nunn, B.L.; Noble, W.S. An alignment-free ‘metapeptide’ strategy for metaproteomic characterization of microbiome samples using shotgun metagenomic sequencing. J. Proteome Res. 2016, 15, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Goslinga, J.; Kooren, J.A.; McGowan, T.; Wroblewski, M.S.; Seymour, S.L.; Griffin, T.J. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics 2013, 13, 1352–1357. [Google Scholar] [CrossRef]
- Zolg, D.P.; Wilhelm, M.; Schnatbaum, K.; Zerweck, J.; Knaute, T.; Delanghe, B.; Bailey, D.J.; Gessulat, S.; Ehrlich, H.C.; Weininger, M.; et al. Building ProteomeTools based on a complete synthetic human proteome. Nat. Methods 2017, 14, 259–262. [Google Scholar] [CrossRef]
- Papanicolas, L.E.; Gordon, D.L.; Wesselingh, S.L.; Rogers, G.B. Not Just Antibiotics: Is Cancer Chemotherapy Driving Antimicrobial Resistance? Trends Microbiol. 2018, 26, 393–400. [Google Scholar] [CrossRef]
- Xiong, W.; Giannone, R.J.; Morowitz, M.J.; Banfield, J.F.; Hettich, R.L. Development of an enhanced metaproteomic approach for deepening the microbiome characterization of the human infant gut. J. Proteome Res. 2014, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Chin, S.F.; Neoh, H.M.; Jamal, R. Metaproteomic analysis of human gut microbiota: Where are we heading? J. Biomed. Sci. 2017, 24, 36. [Google Scholar] [CrossRef] [PubMed]
- Griffin, N.M.; Yu, J.; Long, F.; Oh, P.; Shore, S.; Li, Y.; Koziol, J.A.; Schnitzer, J.E. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat. Biotechnol. 2010, 28, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Weber, N. Microbial community analysis using MEGAN. Methods Enzymol. 2013, 531, 465–485. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. EGGNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, 457–462. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rechenberger, J.; Samaras, P.; Jarzab, A.; Behr, J.; Frejno, M.; Djukovic, A.; Sanz, J.; González-Barberá, E.M.; Salavert, M.; López-Hontangas, J.L.; et al. Challenges in Clinical Metaproteomics Highlighted by the Analysis of Acute Leukemia Patients with Gut Colonization by Multidrug-Resistant Enterobacteriaceae. Proteomes 2019, 7, 2. https://doi.org/10.3390/proteomes7010002
Rechenberger J, Samaras P, Jarzab A, Behr J, Frejno M, Djukovic A, Sanz J, González-Barberá EM, Salavert M, López-Hontangas JL, et al. Challenges in Clinical Metaproteomics Highlighted by the Analysis of Acute Leukemia Patients with Gut Colonization by Multidrug-Resistant Enterobacteriaceae. Proteomes. 2019; 7(1):2. https://doi.org/10.3390/proteomes7010002
Chicago/Turabian StyleRechenberger, Julia, Patroklos Samaras, Anna Jarzab, Juergen Behr, Martin Frejno, Ana Djukovic, Jaime Sanz, Eva M. González-Barberá, Miguel Salavert, Jose Luis López-Hontangas, and et al. 2019. "Challenges in Clinical Metaproteomics Highlighted by the Analysis of Acute Leukemia Patients with Gut Colonization by Multidrug-Resistant Enterobacteriaceae" Proteomes 7, no. 1: 2. https://doi.org/10.3390/proteomes7010002
APA StyleRechenberger, J., Samaras, P., Jarzab, A., Behr, J., Frejno, M., Djukovic, A., Sanz, J., González-Barberá, E. M., Salavert, M., López-Hontangas, J. L., Xavier, K. B., Debrauwer, L., Rolain, J.-M., Sanz, M., Garcia-Garcera, M., Wilhelm, M., Ubeda, C., & Kuster, B. (2019). Challenges in Clinical Metaproteomics Highlighted by the Analysis of Acute Leukemia Patients with Gut Colonization by Multidrug-Resistant Enterobacteriaceae. Proteomes, 7(1), 2. https://doi.org/10.3390/proteomes7010002