Identification of Potential Plasma Biomarkers for Abdominal Aortic Aneurysm Using Tandem Mass Tag Quantitative Proteomics

 ,
,
Abstract
1. Introduction
2. Methods
2.1. Subject Sample
2.2. Discovery Study
2.2.1. Sample Preparation
2.2.2. Nano LC-MS Analysis
2.2.3. Database Search and TMT Quantification
2.2.4. Statistical Analysis
2.3. Validation Study
2.3.1. ELISA
2.3.2. Statistical Analysis
3. Results
3.1. Discovery Study
3.2. Validation Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sakalihasan, N.; Limet, R.; Defawe, O.D. Abdominal aortic aneurysm. Lancet 2005, 365, 1577–1589. [Google Scholar] [CrossRef]
- Choke, E.; Cockerill, G.; Wilson, W.R.; Sayed, S.; Dawson, J.; Loftus, I.; Thompson, M.M. A review of biological factors implicated in abdominal aortic aneurysm rupture. Eur. J. Vasc. Endovasc. Surg. 2005, 30, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 11, 845–867. [Google Scholar] [CrossRef]
- Urbonavicius, S.; Urbonaviciene, G.; Honoré, B.; Henneberg, E.W.; Vorum, H.; Lindholt, J.S. Potential circulating biomarkers for abdominal aortic aneurysm expansion and rupture—A systematic review. Eur. J. Vasc. Endovasc. Surg. 2008, 36, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Tsao, P.S.; Dalman, R.L.; Norman, P.E. Circulating markers of abdominal aortic aneurysm presence and progression. Circulation 2008, 118, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Nordon, I.; Brar, R.; Hinchliffe, R.; Cockerill, G.; Loftus, I.; Thompson, M. The role of proteomic research in vascular disease. J. Vasc. Surg. 2009, 49, 1602–1612. [Google Scholar] [CrossRef] [PubMed]
- Nordon, I.M.; Brar, R.; Hinchliffe, R.J.; Cockerill, G.; Thompson, M.M. Proteomics and pitfalls in the search for potential biomarkers of abdominal aortic aneurysms. Vascular 2010, 18, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Gamberi, T.; Puglia, M.; Guidi, F.; Magherini, F.; Bini, L.; Marzocchini, R.; Modesti, A.; Modesti, P.A. A proteomic approach to identify plasma proteins in patients with abdominal aortic aneurysm. Mol. Biosyst. 2011, 7, 2855–2862. [Google Scholar] [CrossRef] [PubMed]
- Pulinx, B.; Hellenthal, F.A.; Hamulyák, K.; van Dieijen-Visser, M.P.; Schurink, G.W.; Wodzig, W.K. Differential protein expression in serum of abdominal aortic aneurysm patients—A proteomic approach. Eur. J. Vasc. Endovasc. Surg. 2011, 42, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Martin, A.E.; Panchaud, A.; Chwastyniak, M.; Dupont, A.; Juthier, F.; Gautier, C.; Jude, B.; Amouyel, P.; Goodlett, D.R.; Pinet, F. Quantitative mass spectrometry analysis using PAcIFIC for the identification of plasma diagnosic biomarkers for abdominal aortic aneurysm. PLoS ONE 2011, 6, e28698. [Google Scholar] [CrossRef] [PubMed]
- Wallinder, J.; Bergström, J.; Henriksson, A.E. Discovery of a novel circulating biomarker in patients with abdominal aortic aneurysm: A pilot study using a proteomic approach. Clin. Trans. Sci. 2012, 5, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Spadaccio, C.; di Domenico, F.; Perluigi, M.; Lusini, M.; Giorgi, A.; Schininà, M.E.; Blarzino, C.; Covino, E.; Chello, M.; Coccia, R. Serum proteomics in patients with diagnosis of abdominal aortic aneurysm. Cardiovasc. Pathol. 2012, 21, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Bylund, D.; Henriksson, A.E. Proteomic approaches to identify circulating biomarkers in patients with abdominal aortic aneurysm. Am. J. Cardiovasc. Dis. 2015, 5, 140–145. [Google Scholar] [PubMed]
- Grimes, D.A.; Schulz, K.F. Compared to what? Finding controls for case-control studies. Lancet 2005, 365, 1429–1433. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ventura, J.L.; Madrigal-Matute, J.; Martinez-Pinna, R.; Ramos-Mozo, P.; Blanco-Colio, L.M.; Moreno, J.A.; Tarin, C.; Burillo, E.; Fernandez-Garcia, C.E.; Egido, J.; et al. Erythrocytes, leukocytes and platelets as a source of oxidative stress in chronic vascular diseases: Detoxifying mechanisms and potential therapeutic options. Thromb. Haemost. 2012, 108, 435–442. [Google Scholar] [PubMed]
- Belcher, J.D.; Beckman, J.D.; Balla, G.; Balla, J.; Vercellotti, G. Heme degradation and vascular injury. Antioxid. Redox Signal 2010, 12, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, M.; Wallinder, J.; Henriksson, A.E. Soluble urokinase plasminogen activator receptor in patients with abdominal aortic aneurysm. Thromb. Res. 2012, 130, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Mayilyan, K.R. Complement genetics, deficiencies, and disease associations. Protein Cell 2012, 3, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.K.; Mitchell, A.; Endoh, Y.; Hampartzoumian, T.; Huynh, O.; Borges, L.; Geczy, C.; Bryant, K.; Tedla, N. LILRA2 selectively modulates LPS-mediated cytokine production and inhibits phagocytosis by monocytes. PLoS ONE 2012, 7, e33478. [Google Scholar] [CrossRef] [PubMed]
- Sidloff, D.A.; Stather, P.W.; Choke, E.; Bown, M.J.; Sayers, R.D. A systematic review and meta-analysis of the association between markers of hemostasis and abdominal aortic aneurysm presence and size. J. Vasc. Surg. 2014, 59, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Hobbs, R.P.; Coulombe, P.A. The expanding significance of keratin intermediate filaments in normal and diseased epithelia. Curr. Opin. Cell Biol. 2013, 25, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, F.; Lunardi, F.; Balestro, E.; Marulli, G.; Perissinotto, E.; Loy, M.; Nannini, N.; Valente, M.; Saetta, M.; Agostini, C.; et al. Serpin B4 isoform overexpression is associated with aberrant epithelial proliferation and lung cancer in idiopathic pulmonary fibrosis. Pathology 2012, 44, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, H.; Hori, S.; Sawa, T.; Yoshioka, T.; Takeuchi, T. A bleomycin-inactivating enzyme in mouse liver. J. Antibiot. 1974, 27, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Zimny, J.; Sikora, M.; Guranowski, A.; Jakubowski, H. Protective mechanisms against homocysteine toxicity: The role of bleomycin hydrolase. J. Biol. Chem. 2006, 281, 22485–22492. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.Y.; Golledge, J.; Flicker, L.; McCaul, K.A.; Hankey, G.J.; van Bockxmeer, F.M.; Yeap, B.B.; Norman, P.E. Plasma total homocysteine is associated with abdominal aortic aneurysm and aortic diameter in older men. J. Vasc. Surg. 2013, 58, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
| Parameter | Controls (n = 41) | Non-Ruptured AAA (n = 78) | Ruptured AAA | p-Values (Non-Ruptured vs. Ruptured AAA) | |
|---|---|---|---|---|---|
| Small AAA (n = 38) | Large AAA (n = 40) | (n = 55) | |||
| Age, years | 72 (67–79) | 70 (66–76) NS | 71 (63–78) NS | 73 (70–79) NS | 0.509 |
| Sex, male | 33 (80%) | 27 (71%) NS | 35 (88%) NS | 44 (80%) NS | 0.942 |
| Current smoking | 18 (44%) | 16 (42%) NS | 19 (48%) NS | 22 (40%) NS | 0.576 |
| Aneurysm diameter, cm | No aneurysm | 4.0 (3.5–4.3) | 6.0 (5.2–7.1) | 7.3 (6.0–8.0) | <0.001 |
| Hemoglobin, g/L | 141 (130–147) | 142 (134–151) NS | 139 (131–145) NS | 118 (101–136) ** | <0.001 |
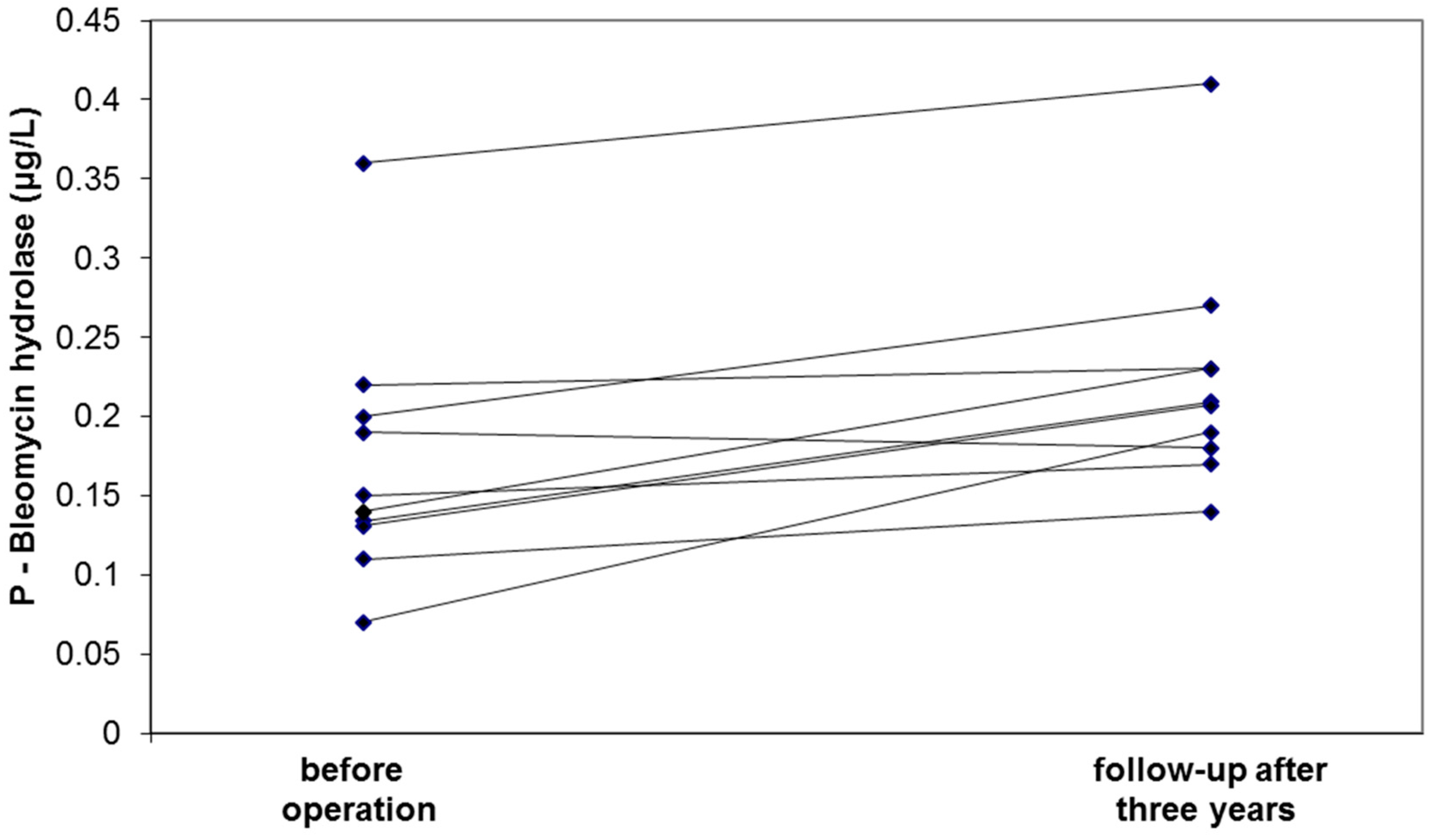
| Bleomycin hydrolase, μg/L | 0.19 (0.16–0.25) | 0.20 (0.16–0.24) NS | 0.14 (0.09–0.19) * | 0.12 (0.08–0.19) ** | 0.001 |
| Protein | UniProt Accession | Fold Change (AAA/C) | Welch Test p-Value |
|---|---|---|---|
| Complement component C9 | P02748 | 1.506 | 0.001 |
| Keratin 77 | Q0IIN1 | 0.440 | 0.004 |
| Bleomycin hydrolase | Q13867 | 0.565 | 0.008 |
| Hemoglobin subunit β | P68871 | 1.974 | 0.016 |
| Fibrinogen γ chain | P02679 | 1.558 | 0.017 |
| Leukocyte immunoglobulin-like receptor subfamily A member 2 | A8MZH0 | 2.496 | 0.021 |
| C-reactive protein | P02741 | 2.485 | 0.026 |
| Serpin B4 | F8W9L1 | 0.366 | 0.049 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henriksson, A.E.; Lindqvist, M.; Sihlbom, C.; Bergström, J.; Bylund, D. Identification of Potential Plasma Biomarkers for Abdominal Aortic Aneurysm Using Tandem Mass Tag Quantitative Proteomics. Proteomes 2018, 6, 43. https://doi.org/10.3390/proteomes6040043
Henriksson AE, Lindqvist M, Sihlbom C, Bergström J, Bylund D. Identification of Potential Plasma Biomarkers for Abdominal Aortic Aneurysm Using Tandem Mass Tag Quantitative Proteomics. Proteomes. 2018; 6(4):43. https://doi.org/10.3390/proteomes6040043
Chicago/Turabian StyleHenriksson, Anders E., Markus Lindqvist, Carina Sihlbom, Jörgen Bergström, and Dan Bylund. 2018. "Identification of Potential Plasma Biomarkers for Abdominal Aortic Aneurysm Using Tandem Mass Tag Quantitative Proteomics" Proteomes 6, no. 4: 43. https://doi.org/10.3390/proteomes6040043
APA StyleHenriksson, A. E., Lindqvist, M., Sihlbom, C., Bergström, J., & Bylund, D. (2018). Identification of Potential Plasma Biomarkers for Abdominal Aortic Aneurysm Using Tandem Mass Tag Quantitative Proteomics. Proteomes, 6(4), 43. https://doi.org/10.3390/proteomes6040043