Exploring Morphine-Triggered PKC-Targets and Their Interaction with Signaling Pathways Leading to Pain via TrkA

 , and
, and 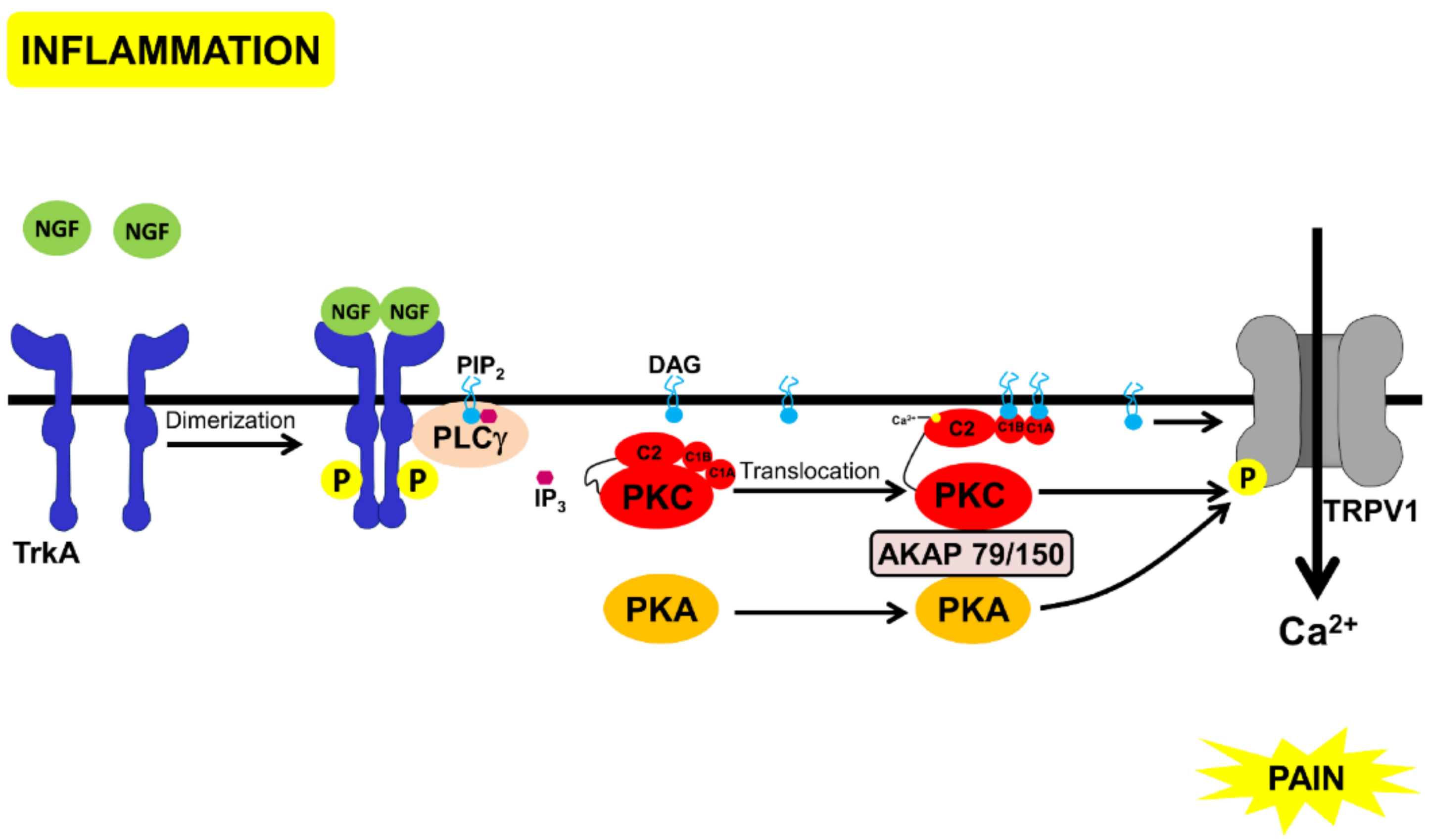{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Morphine-Mediated Signal Transduction Pathways and Receptor Desensitization
- (i)
- MOR activation enables the Gβγ subunit to activate PLC which then would lead to PKC activation [27],
- (ii)
- MOR activation leads to an activation of a Gq-coupled receptor that, in turn, leads to PLCβ activation, as seen in the case of M3 muscarinic receptor activation-mediated increase in the MOR desensitization [28].
- (iii)
- MOR activation leads to activation of the receptor-coupled and non-coupled tyrosine kinases, which in turn lead to PLCγ activation.
- (iv)
- MOR activation leads to the activation of a small G protein which would then activate PLCε and subtypes of PLCβ and γ [29].
- (v)
- MOR activation leads to activation of PI3K which then activates PKC.
2. Methods
2.1. Cell Culture
2.2. Immunofluorescence
2.3. Western Blot
2.4. Real-Time PCR
2.5. Cross-Linking Antibodies to Beads
2.6. Immunoprecipitation Assays and iTRAQ®
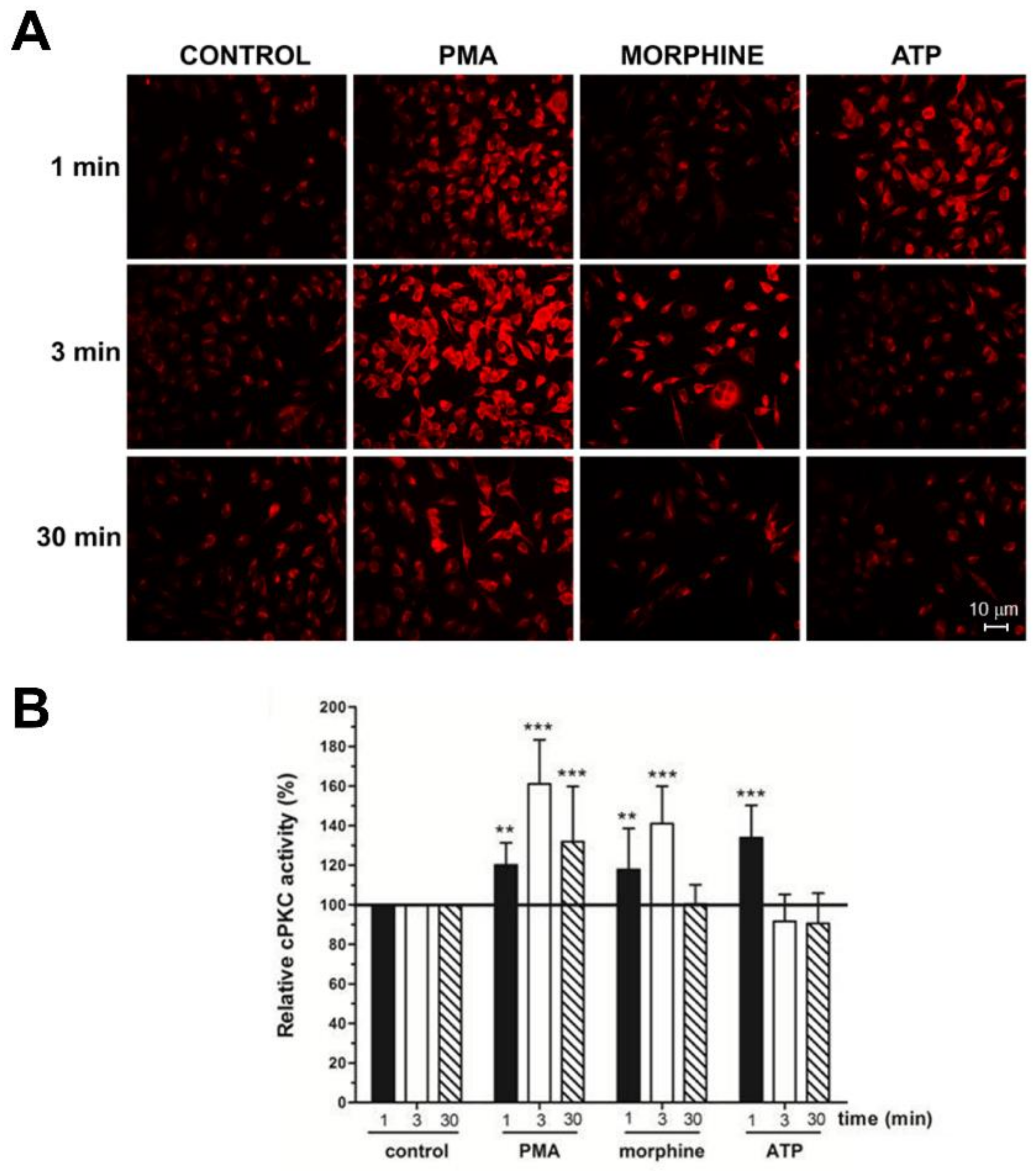
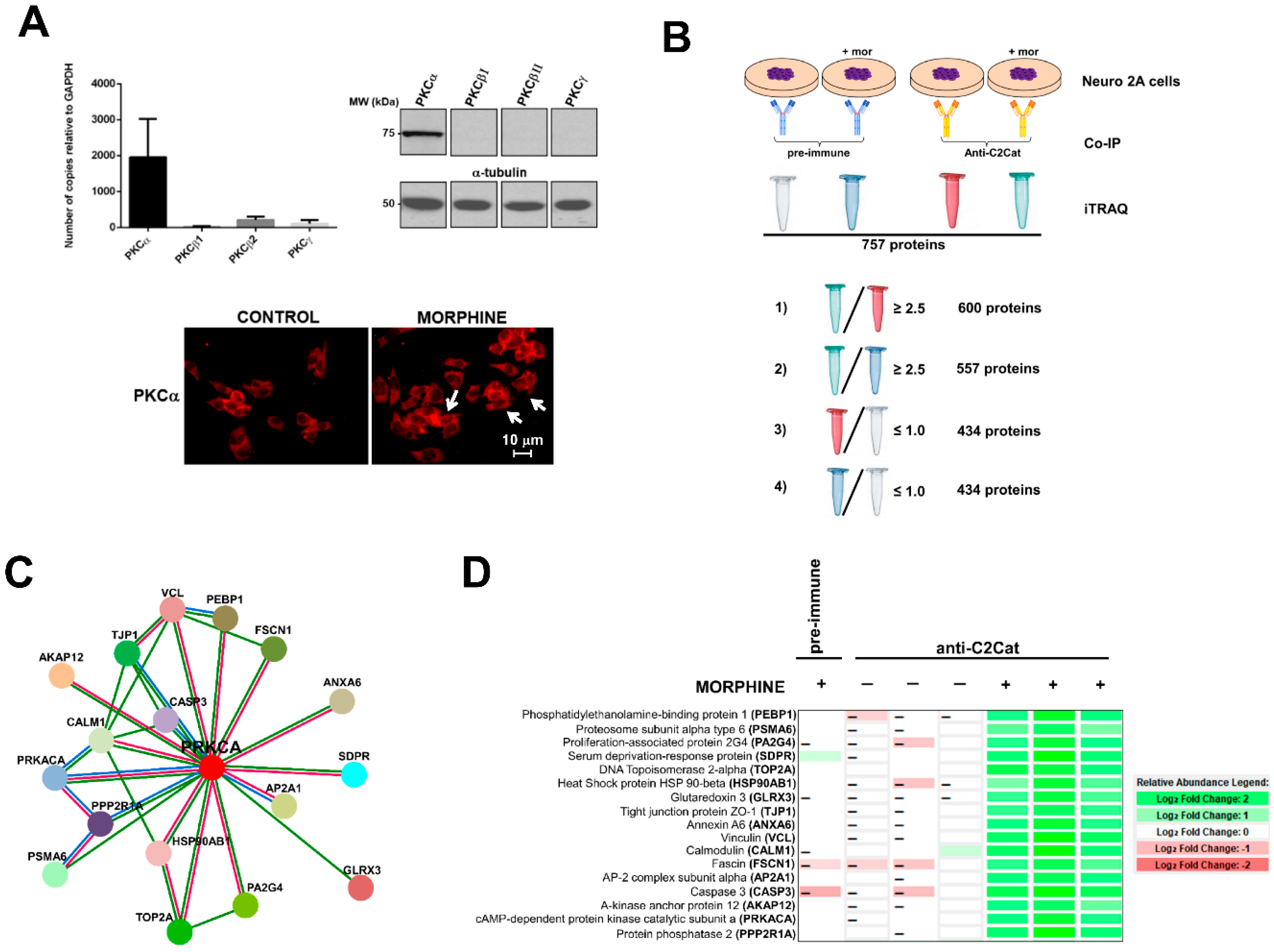
3. Results
- (i)
- To detect the proteins that interacted with active PKC upon morphine treatment, we eliminated proteins that immunoprecipitated from cells not treated with morphine. Six hundred proteins met this criterion (p ≤ 0.05, per t-test analysis) and these proteins exhibited a ratio of ≥2.5, when proteins immunoprecipitated from morphine-treated cells were compared with vehicle-treated cells.
- (ii)
- To exclude proteins that bound non-specifically to the anti-C2Cat antibody, we eliminated proteins that interacted with pre-immune serum in the presence of morphine. This reduced the list to 557 proteins and these proteins exhibited a ratio ≥2.5, when proteins that were immunoprecipitated by anti-C2Cat antibody were compared to proteins immunoprecipitated with pre-immune serum (from cells treated with morphine).
- (iii)
- To exclude proteins that interacted with PKC under basal conditions (absence of morphine), we only considered proteins that had a ratio ≤1.0, when comparing proteins immunoprecipitated with anti-C2Cat, to those with pre-immune serum, in vehicle-treated cells. This further reduced the list to 434 proteins.
3.1. Phosphatidylethanolamine Binding Protein 1 (PEBP1)
3.2. Scaffolds (Annexin 6 and AKAP12) and PKA
3.3. Neurogranin and Calmodulin
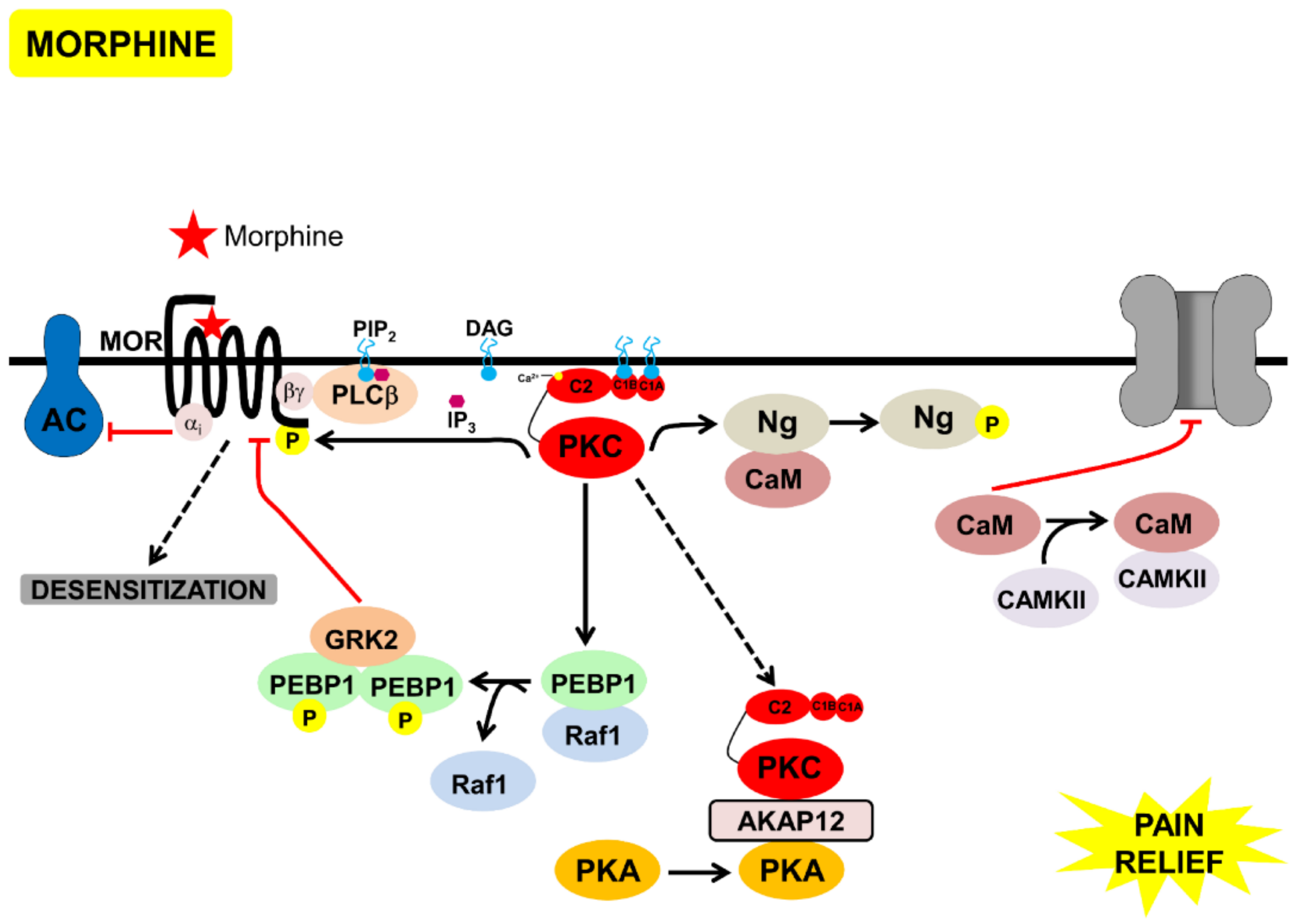
3.4. Morphine Inhibition of TrKA-Signaling Pathways via PKC
- (a)
- Inhibition of PKA by Gαi.
- (b)
- Transient activation of cPKC, initially via Gβγ. Sustained PKC activation, possibly of nPKCs, could be mediated through other mechanisms discussed above.
- (c)
- PKC-mediated phosphorylation of neurogranin, making CAM available to activate CAMKII and to bind to TRPV1, and inhibit the channel and nociception.
- (d)
- Displacement of PKC and PKA, through scaffold proteins, annexin 6, and AKAP 12, inhibiting these kinases from binding to AKAP79/150 and activating TRPV1 (Figure 4).
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koob, G.F.; Sanna, P.P.; Bloom, F.E. Neuroscience of addiction. Neuron 1998, 21, 467–476. [Google Scholar] [CrossRef]
- Williams, J.T.; Ingram, S.L.; Henderson, G.; Chavkin, C.; von Zastrow, M.; Schulz, S.; Koch, T.; Evans, C.J.; Christie, M.J. Regulation of mu-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev. 2013, 65, 223–254. [Google Scholar] [CrossRef] [PubMed]
- Koch, W.J.; Hawes, B.E.; Allen, L.F.; Lefkowitz, R.J. Direct evidence that Gi-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G beta gamma activation of p21ras. Proc. Natl. Acad. Sci. USA 1994, 91, 12706–12710. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys. Sin. (Shanghai) 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Guitart, X.; Nestler, E.J. Second messenger and protein phosphorylation mechanisms underlying opiate addiction: Studies in the rat locus coeruleus. Neurochem. Res. 1993, 18, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Tallman, J.F.; Nestler, E.J. Acute and chronic opiate-regulation of adenylate cyclase in brain: specific effects in locus coeruleus. J. Pharmacol. Exp. Ther. 1988, 246, 1033–1039. [Google Scholar] [PubMed]
- Terwilliger, R.Z.; Beitner-Johnson, D.; Sevarino, K.A.; Crain, S.M.; Nestler, E.J. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991, 548, 100–110. [Google Scholar] [CrossRef]
- Fukuda, K.; Kato, S.; Morikawa, H.; Shoda, T.; Mori, K. Functional coupling of the delta-, mu-, and kappa-opioid receptors to mitogen-activated protein kinase and arachidonate release in Chinese hamster ovary cells. J. Neurochem. 1996, 67, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Law, P.Y.; Wong, Y.H.; Loh, H.H. Molecular mechanisms and regulation of opioid receptor signaling. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 389–430. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Akai, H.; Nagumo, Y.; Sunagawa, N.; Hasebe, K.; Nagase, H.; Kita, T.; Hara, C.; Suzuki, T. Implications of protein kinase C in the nucleus accumbens in the development of sensitization to methamphetamine in rats. Neuroscience 2004, 127, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Polakiewicz, R.D.; Schieferl, S.M.; Dorner, L.F.; Kansra, V.; Comb, M.J. A mitogen-activated protein kinase pathway is required for mu-opioid receptor desensitization. J. Biol. Chem. 1998, 273, 12402–12406. [Google Scholar] [CrossRef] [PubMed]
- Pak, Y.; O’Dowd, B.F.; George, S.R. Agonist-induced desensitization of the mu opioid receptor is determined by threonine 394 preceded by acidic amino acids in the COOH-terminal tail. J. Biol. Chem. 1997, 272, 24961–24965. [Google Scholar] [CrossRef] [PubMed]
- Pei, G.; Kieffer, B.L.; Lefkowitz, R.J.; Freedman, N.J. Agonist-dependent phosphorylation of the mouse delta-opioid receptor: involvement of G protein-coupled receptor kinases but not protein kinase C. Mol. Pharmacol. 1995, 48, 173–177. [Google Scholar] [PubMed]
- Liu, J.G.; Anand, K.J. Protein kinases modulate the cellular adaptations associated with opioid tolerance and dependence. Brain Res. Brain. Res. Rev. 2001, 38, 1–19. [Google Scholar] [CrossRef]
- Ferrer-Alcon, M.; Garcia-Fuster, M.J.; La Harpe, R.; Garcia-Sevilla, J.A. Long-term regulation of signalling components of adenylyl cyclase and mitogen-activated protein kinase in the pre-frontal cortex of human opiate addicts. J. Neurochem. 2004, 90, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Smith, F.L.; Javed, R.R.; Elzey, M.J.; Dewey, W.L. The expression of a high level of morphine antinociceptive tolerance in mice involves both PKC and PKA. Brain Res. 2003, 985, 78–88. [Google Scholar] [CrossRef]
- Hull, L.C.; Llorente, J.; Gabra, B.H.; Smith, F.L.; Kelly, E.; Bailey, C.; Henderson, G.; Dewey, W.L. The effect of protein kinase C and G protein-coupled receptor kinase inhibition on tolerance induced by mu-opioid agonists of different efficacy. J. Pharmacol. Exp. Ther. 2010, 332, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.P.; Smith, F.L.; Kelly, E.; Dewey, W.L.; Henderson, G. How important is protein kinase C in mu-opioid receptor desensitization and morphine tolerance? Trends Pharmacol. Sci. 2006, 27, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Nishizuka, Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature 1988, 334, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Poised to signal. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E395–402. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.P.; Oldfield, S.; Llorente, J.; Caunt, C.J.; Teschemacher, A.G.; Roberts, L.; McArdle, C.A.; Smith, F.L.; Dewey, W.L.; Kelly, E.; et al. Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br. J. Pharmacol. 2009, 158, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Malmberg, A.B.; Chen, C.; Tonegawa, S.; Basbaum, A.I. Preserved acute pain and reduced neuropathic pain in mice lacking PKCgamma. Science 1997, 278, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Zou, W.; Liu, C.; Guo, Q. Gene knockdown with lentiviral vector-mediated intrathecal RNA interference of protein kinase C gamma reverses chronic morphine tolerance in rats. J. Gene Med. 2010, 12, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Sacktor, T.C.; Hell, J.W. The genetics of PKMzeta and memory maintenance. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.K.; Alkon, D.L. Protein kinase C activators as synaptogenic and memory therapeutics. Arch. Pharm. (Weinheim) 2009, 342, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, C.F.; Pei, G.; Xu, N.J. Reversal of morphine-induced memory impairment in mice by withdrawal in Morris water maze: Possible involvement of cholinergic system. Pharmacol. Biochem. Behav. 2001, 68, 507–513. [Google Scholar] [CrossRef]
- Zhu, X.; Birnbaumer, L. G protein subunits and the stimulation of phospholipase C by Gs-and Gi-coupled receptors: Lack of receptor selectivity of Galpha(16) and evidence for a synergic interaction between Gbeta gamma and the alpha subunit of a receptor activated G protein. Proc. Natl. Acad. Sci. USA 1996, 93, 2827–2831. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.P.; Kelly, E.; Henderson, G. Protein kinase C activation enhances morphine-induced rapid desensitization of mu-opioid receptors in mature rat locus ceruleus neurons. Mol. Pharmacol. 2004, 66, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Gresset, A.; Sondek, J.; Harden, T.K. The phospholipase C isozymes and their regulation. Subcell. Biochem. 2012, 58, 61–94. [Google Scholar] [PubMed]
- Bekhite, M.M.; Finkensieper, A.; Binas, S.; Muller, J.; Wetzker, R.; Figulla, H.R.; Sauer, H.; Wartenberg, M. VEGF-mediated PI3K class IA and PKC signaling in cardiomyogenesis and vasculogenesis of mouse embryonic stem cells. J. Cell Sci. 2011, 124, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.R.; Perez-Chanona, E.; Yadav, P.N.; Whistler, J.; Roth, B.; Jobin, C. Intestinal epithelial cell-derived mu-opioid signaling protects against ischemia reperfusion injury through PI3K signaling. Am. J. Pathol. 2013, 182, 776–785. [Google Scholar] [CrossRef] [PubMed]
- Halls, M.L.; Yeatman, H.R.; Nowell, C.J.; Thompson, G.L.; Gondin, A.B.; Civciristov, S.; Bunnett, N.W.; Lambert, N.A.; Poole, D.P.; Canals, M. Plasma membrane localization of the mu-opioid receptor controls spatiotemporal signaling. Sci. Signal. 2016, 9, ra16. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.J.; Wung, B.S.; Chao, Y.J.; Wang, D.L. Sequential activation of protein kinase C (PKC)-alpha and PKC-epsilon contributes to sustained Raf/ERK1/2 activation in endothelial cells under mechanical strain. J. Biol. Chem. 2001, 276, 31368–31375. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Debing, Y.; Zhou, X.; Yin, Y.; Xu, L.; Herrera Carrillo, E.; Brandsma, J.H.; Poot, R.A.; Berkhout, B.; et al. Biological or pharmacological activation of protein kinase C alpha constrains hepatitis E virus replication. Antivir. Res. 2017, 140, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.A.; Oldfield, S.; Braksator, E.; Gonzalez-Cuello, A.; Couch, D.; Hall, K.J.; Mundell, S.J.; Bailey, C.P.; Kelly, E.; Henderson, G. Agonist-selective mechanisms of mu-opioid receptor desensitization in human embryonic kidney 293 cells. Mol. Pharmacol. 2006, 70, 676–685. [Google Scholar] [CrossRef] [PubMed]
- El Kouhen, R.; Burd, A.L.; Erickson-Herbrandson, L.J.; Chang, C.Y.; Law, P.Y.; Loh, H.H. Phosphorylation of Ser363, Thr370, and Ser375 residues within the carboxyl tail differentially regulates mu-opioid receptor internalization. J. Biol. Chem. 2001, 276, 12774–12780. [Google Scholar] [CrossRef] [PubMed]
- Arttamangkul, S.; Heinz, D.A.; Bunzow, J.R.; Song, X.; Williams, J.T. Cellular tolerance at the micro-opioid receptor is phosphorylation dependent. eLife 2018, 7, e34989. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Li, Z.; Wang, J.B. Protein kinase C-mediated phosphorylation of the mu-opioid receptor and its effects on receptor signaling. Mol. Pharmacol. 2011, 79, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.K.; Trester-Zedlitz, M.; Trinidad, J.C.; Kotowski, S.J.; Krutchinsky, A.N.; Burlingame, A.L.; von Zastrow, M. Quantitative encoding of the effect of a partial agonist on individual opioid receptors by multisite phosphorylation and threshold detection. Sci. Signal. 2011, 4, ra52. [Google Scholar] [CrossRef] [PubMed]
- Pena, D.A.; Andrade, V.P.; Silva, G.A.; Neves, J.I.; Oliveira, P.S.; Alves, M.J.; Devi, L.A.; Schechtman, D. Rational design and validation of an anti-protein kinase C active-state specific antibody based on conformational changes. Sci. Rep. 2016, 6, 22114. [Google Scholar] [CrossRef] [PubMed]
- Hefti, F.F.; Rosenthal, A.; Walicke, P.A.; Wyatt, S.; Vergara, G.; Shelton, D.L.; Davies, A.M. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol. Sci. 2006, 27, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, A.; Lammers, R.; Wiesmuller, K.H.; Jung, G.; Schlessinger, J.; Ullrich, A. Identification of Trk binding sites for SHC and phosphatidylinositol 3’-kinase and formation of a multimeric signaling complex. J. Biol. Chem. 1993, 268, 22963–22966. [Google Scholar] [PubMed]
- Chuang, H.H.; Prescott, E.D.I; Kong, H.; Shields, S.; Jordt, S.E.; Basbaum, A.I.; Chao, M.V.; Julius, D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 2001, 411, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; McNaughton, P.A. Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron 2008, 59, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Gao, Y.; Yang, L.; Kong, X.; Yu, J.; Hou, W.; Hua, B. The mechanism of mu-opioid receptor (MOR)-TRPV1 crosstalk in TRPV1 activation involves morphine anti-nociception, tolerance and dependence. Channels (Austin) 2015, 9, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.; Cheng, W.; Peiris, M.; Wyse, B.D.; Roberts-Thomson, S.J.; Zheng, J.; Monteith, G.R.; Cabot, P.J. Rapid, opioid-sensitive mechanisms involved in transient receptor potential vanilloid 1 sensitization. J. Biol. Chem. 2008, 283, 19540–19550. [Google Scholar] [CrossRef] [PubMed]
- Endres-Becker, J.; Heppenstall, P.A.; Mousa, S.A.; Labuz, D.; Oksche, A.; Schafer, M.; Stein, C.; Zollner, C. Mu-opioid receptor activation modulates transient receptor potential vanilloid 1 (TRPV1) currents in sensory neurons in a model of inflammatory pain. Mol. Pharmacol. 2007, 71, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Geis, C.; Sommer, C. Activation of TRPV1 contributes to morphine tolerance: involvement of the mitogen-activated protein kinase signaling pathway. J. Neurosci. 2008, 28, 5836–5845. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Pan, H.L. Blocking mu opioid receptors in the spinal cord prevents the analgesic action by subsequent systemic opioids. Brain Res. 2006, 1081, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, L.; Bansal, Y.; Singh, R.; Saroj, P.; Bhandari, R.; Kuhad, A. TRP channels: Potential drug target for neuropathic pain. Inflammopharmacology 2016, 24, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Kuroda, Y.; Murata, E. NGF/TrkA Signaling as a Therapeutic Target for Pain. Pain Pract. 2016, 16, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Mahal, B.A. NGF–The TrkA to successful pain treatment. J. Pain Res. 2012, 5, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Stockton, S.D., Jr.; Gomes, I.; Liu, T.; Moraje, C.; Hipolito, L.; Jones, M.R.; Ma'ayan, A.; Moron, J.A.; Li, H.; Devi, L.A. Morphine Regulated Synaptic Networks Revealed by Integrated Proteomics and Network Analysis. Mol. Cell Proteom. 2015, 14, 2564–2576. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.K.; Tang, L.; Wang, Z.J. Phosphorylation of neurogranin, protein kinase C, and Ca2+/calmodulin dependent protein kinase II in opioid tolerance and dependence. Neurosci. Lett. 2006, 404, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Deiss, K.; Kisker, C.; Lohse, M.J.; Lorenz, K. Raf kinase inhibitor protein (RKIP) dimer formation controls its target switch from Raf1 to G protein-coupled receptor kinase (GRK) 2. J. Biol. Chem. 2012, 287, 23407–23417. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Bailey, C.P.; Henderson, G. Agonist-selective mechanisms of GPCR desensitization. Br. J. Pharmacol. 2008, 153, S379–S388. [Google Scholar] [CrossRef] [PubMed]
- Brackley, A.D.; Gomez, R.; Akopian, A.N.; Henry, M.A.; Jeske, N.A. GRK2 Constitutively Governs Peripheral Delta Opioid Receptor Activity. Cell Rep. 2016, 16, 2686–2698. [Google Scholar] [CrossRef] [PubMed]
- Koese, M.; Koese, M.; Rentero, C.; Kota, B.P.; Hoque, M.; Cairns, R.; Wood, P.; Vila de Muga, S.; Reverter, M.; Alvarez-Guaita, A.; Monastyrskaya, K.; et al. Annexin A6 is a scaffold for PKCalpha to promote EGFR inactivation. Oncogene 2013, 32, 2858–2872. [Google Scholar] [CrossRef] [PubMed]
- Schott, M.B.; Grove, B. Receptor-mediated Ca2+ and PKC signaling triggers the loss of cortical PKA compartmentalization through the redistribution of gravin. Cell Signal. 2013, 25, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Grewal, T.; Evans, R.; Rentero, C.; Tebar, F.; Cubells, L.; de Diego, I.; Kirchhoff, M.F.; Hughes, W.E.; Heeren, J.; Rye, K.A.; et al. Annexin A6 stimulates the membrane recruitment of p120GAP to modulate Ras and Raf-1 activity. Oncogene 2005, 24, 5809–5820. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Wang, H.Y.; Malbon, C.C. Protein kinase A regulates AKAP250 (gravin) scaffold binding to the beta2-adrenergic receptor. EMBO J. 2003, 22, 6419–6429. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, E.; Luongo, L.; de Novellis, V.; Rossi, F.; Marabese, I.; Maione, S. Transient receptor potential vanilloid type 1 and pain development. Curr. Opin. Pharmacol. 2012, 12, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Shukla, P.K.; Wang, Z.J. Disruption of acute opioid dependence by antisense oligodeoxynucleotides targeting neurogranin. Brain Res. 2007, 1143, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Cherry, T.; Bies, C.E.; Florence, M.A.; Gerges, N.Z. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009, 28, 3027–3039. [Google Scholar] [CrossRef] [PubMed]
- Cheppudira, B.P.; Trevino, A.V.; Petz, L.N.; Christy, R.J.; Clifford, J.L. Anti-nerve growth factor antibody attenuates chronic morphine treatment-induced tolerance in the rat. BMC Anesthesiol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ma, X.; Luo, L.; Huang, M.; Dong, J.; Zhang, X.; Jiang, W.; Xu, T. Exchange factor directly activated by cAMP-PKCepsilon signalling mediates chronic morphine-induced expression of purine P2X3 receptor in rat dorsal root ganglia. Br. J. Pharmacol. 2018, 175, 1760–1769. [Google Scholar] [CrossRef] [PubMed]
- Storch, U.; Straub, J.; Erdogmus, S.; Gudermann, T.; Mederos, Y.; Schnitzler, M. Dynamic monitoring of Gi/o-protein-mediated decreases of intracellular cAMP by FRET-based Epac sensors. Pflugers Arch. 2017, 469, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Mardy, S.; Miura, Y.; Endo, F.; Matsuda, I.; Indo, Y. Congenital insensitivity to pain with anhidrosis (CIPA), effect of TRKA (NTRK1) missense mutations on autophosphorylation of the receptor tyrosine kinase for nerve growth factor. Hum. Mol. Genet. 2001, 10, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Mardy, S.; Awaya, Y.; Nihei, K.; Endo, F.; Matsuda, I.; Indo, Y. Mutation and polymorphism analysis of the TRKA (NTRK1) gene encoding a high-affinity receptor for nerve growth factor in congenital insensitivity to pain with anhidrosis (CIPA) families. Hum. Genet. 2000, 106, 116–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Omerbasic, D.; Smith, E.S.; Moroni, M.; Homfeld, J.; Eigenbrod, O.; Bennett, N.C.; Reznick, J.; Faulkes, C.G.; Selbach, M.; Lewin, G.R. Hypofunctional TrkA Accounts for the Absence of Pain Sensitization in the African Naked Mole-Rat. Cell Rep. 2016, 17, 748–758. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pena, D.A.; Duarte, M.L.; Pramio, D.T.; Devi, L.A.; Schechtman, D. Exploring Morphine-Triggered PKC-Targets and Their Interaction with Signaling Pathways Leading to Pain via TrkA. Proteomes 2018, 6, 39. https://doi.org/10.3390/proteomes6040039
Pena DA, Duarte ML, Pramio DT, Devi LA, Schechtman D. Exploring Morphine-Triggered PKC-Targets and Their Interaction with Signaling Pathways Leading to Pain via TrkA. Proteomes. 2018; 6(4):39. https://doi.org/10.3390/proteomes6040039
Chicago/Turabian StylePena, Darlene A., Mariana Lemos Duarte, Dimitrius T. Pramio, Lakshmi A. Devi, and Deborah Schechtman. 2018. "Exploring Morphine-Triggered PKC-Targets and Their Interaction with Signaling Pathways Leading to Pain via TrkA" Proteomes 6, no. 4: 39. https://doi.org/10.3390/proteomes6040039
APA StylePena, D. A., Duarte, M. L., Pramio, D. T., Devi, L. A., & Schechtman, D. (2018). Exploring Morphine-Triggered PKC-Targets and Their Interaction with Signaling Pathways Leading to Pain via TrkA. Proteomes, 6(4), 39. https://doi.org/10.3390/proteomes6040039