Regional Diversity in the Postsynaptic Proteome of the Mouse Brain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dissections of Mouse Brain Regions
2.2. PSD Isolation and Protein Preparations for Mass Spectrometry
2.3. Sample Preparation and LC-MS/MS Analysis
2.4. Mass Spectrometry and Data Analysis
2.5. Bioinformatic Analyses
2.6. Protein Interaction Identification and Mapping
3. Results
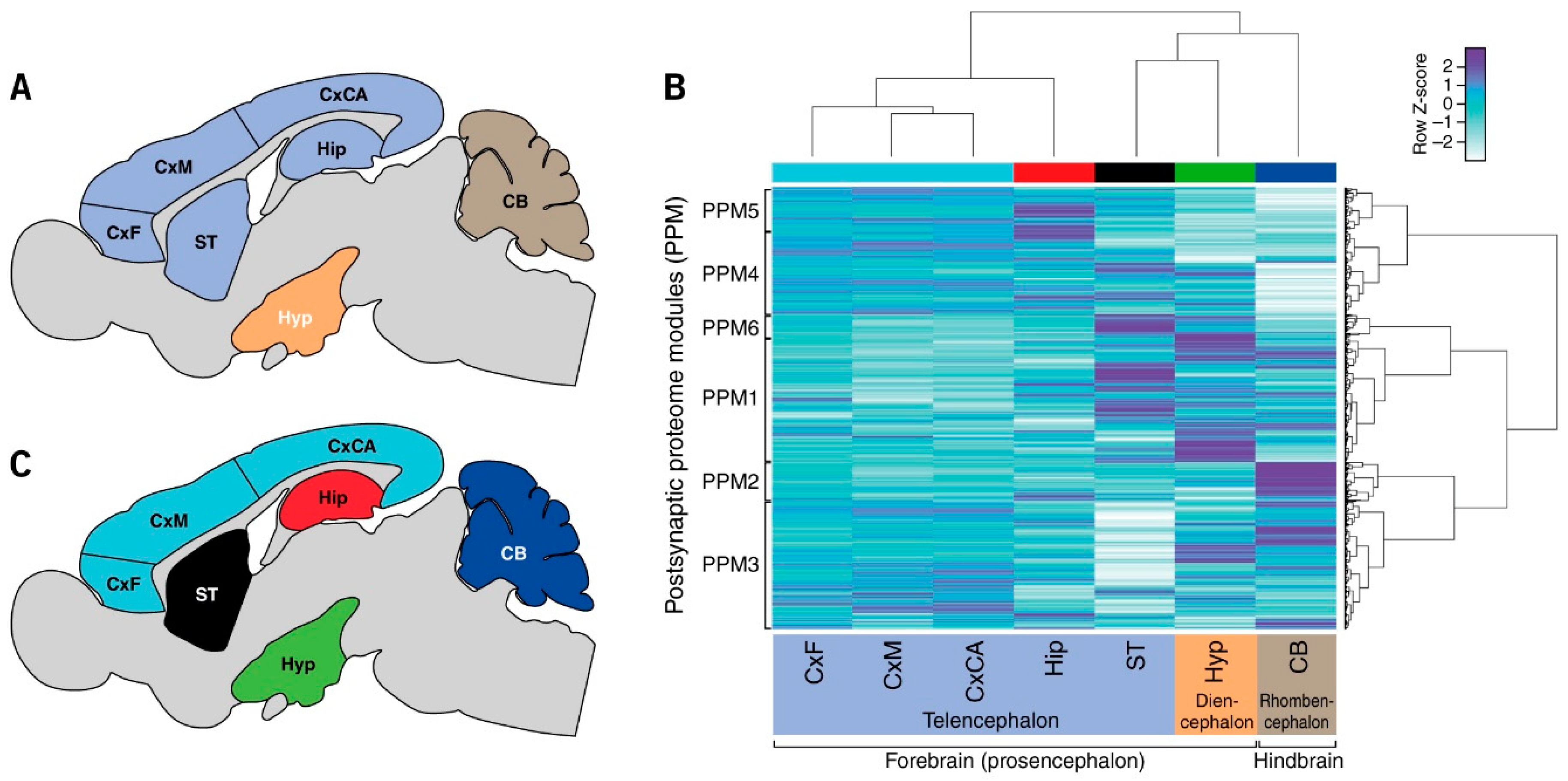
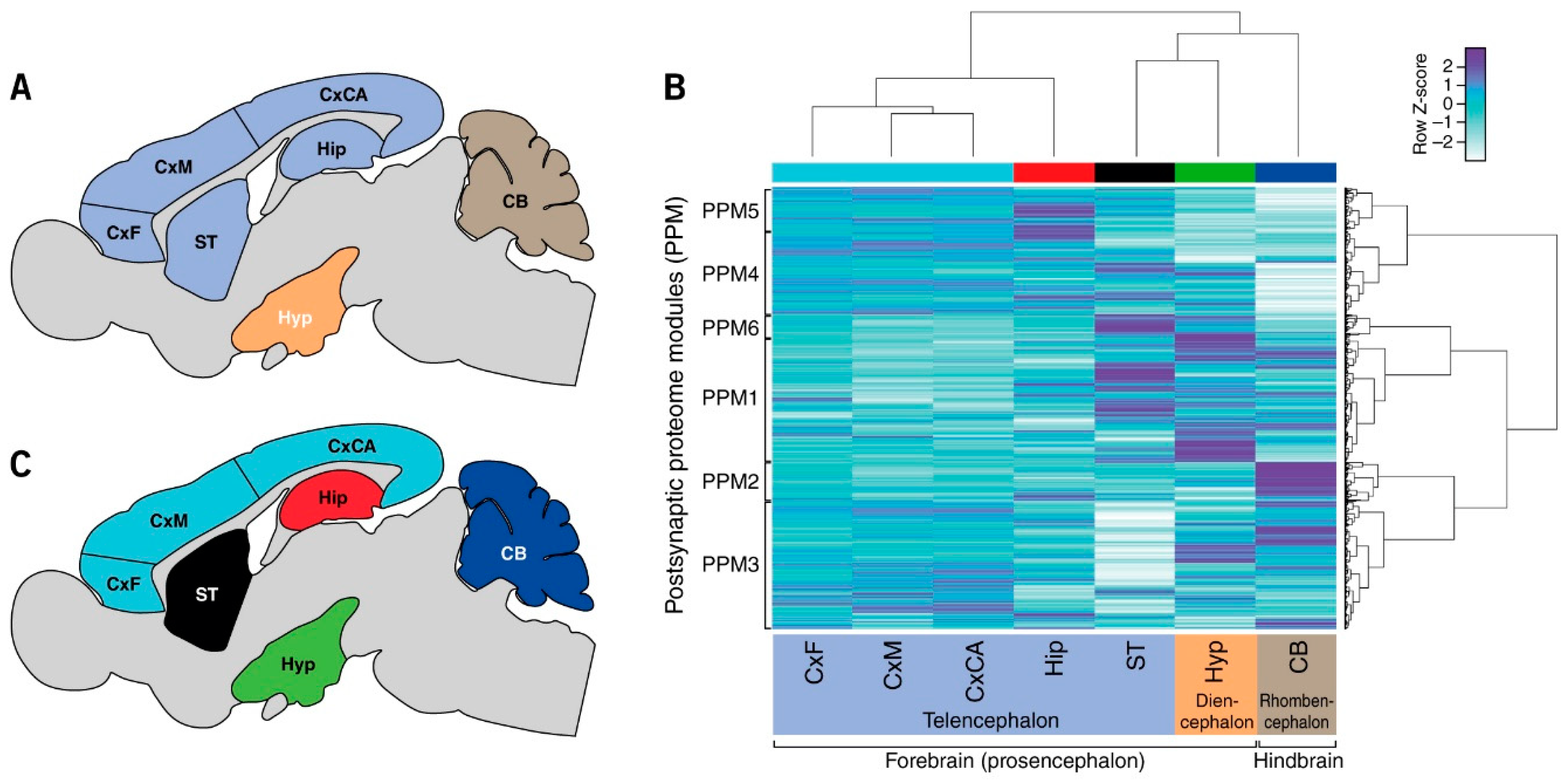
3.1. Quantification of Postsynaptic Proteins from Brain Regions
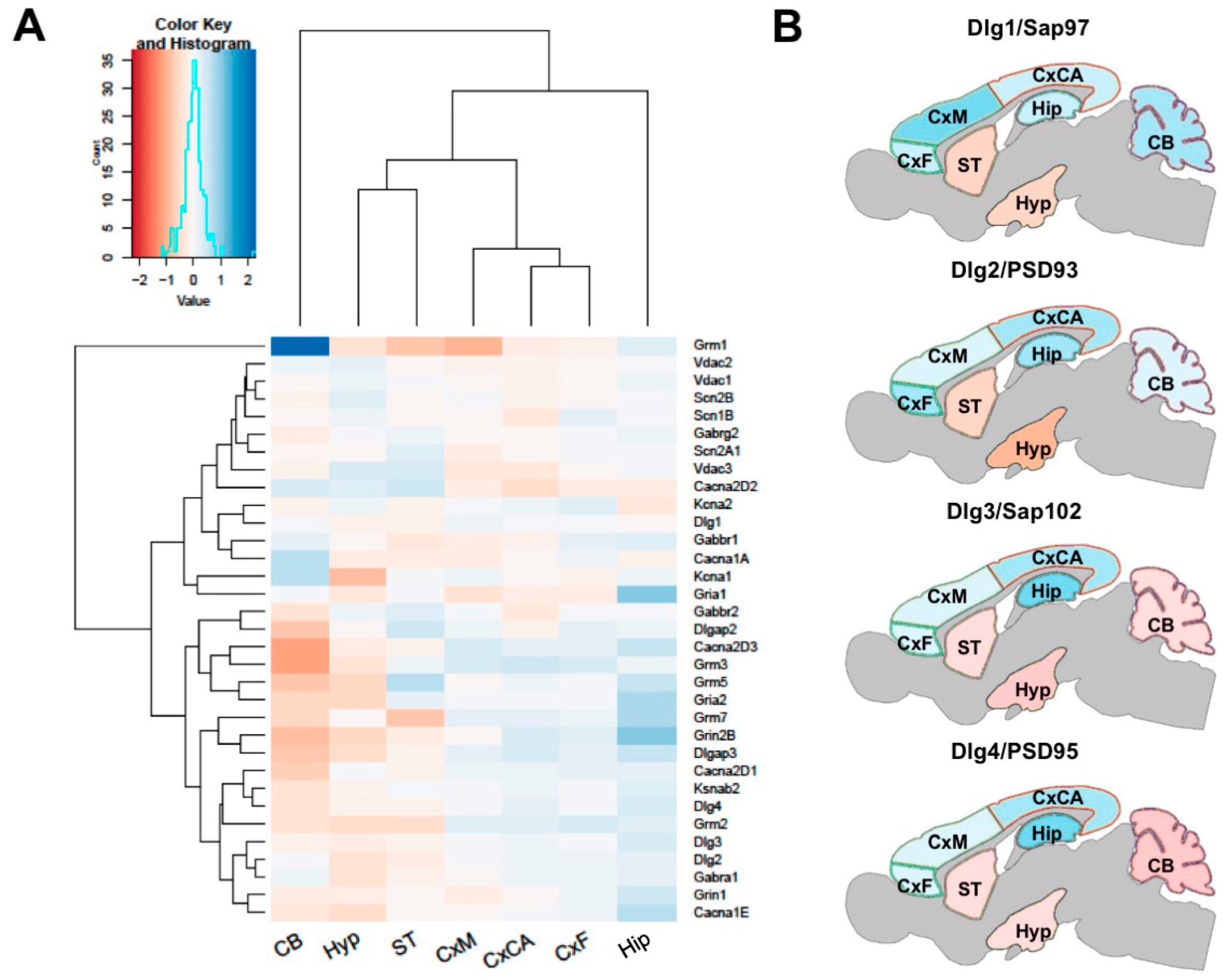
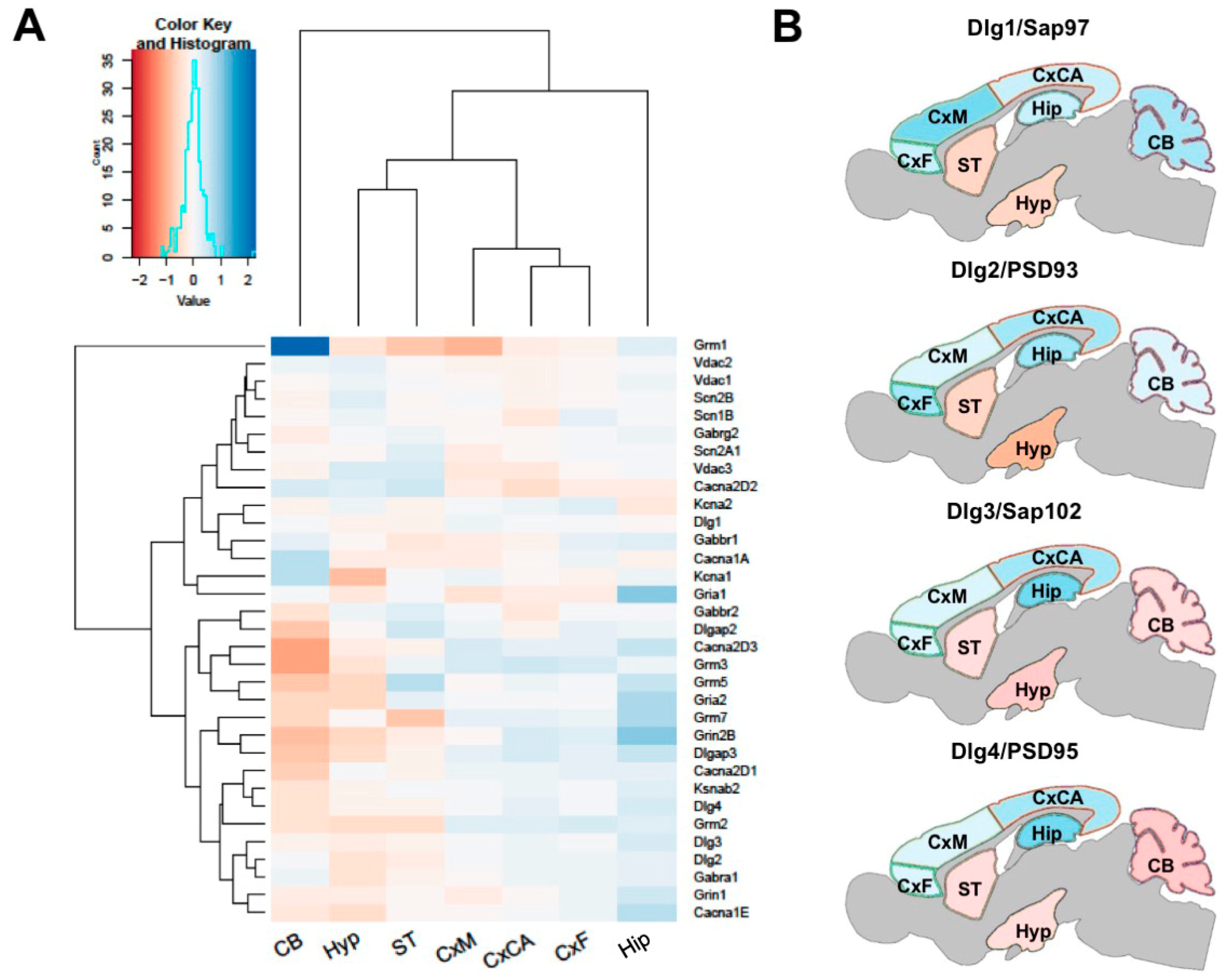
3.2. Regional Differences in Postsynaptic Proteome Composition
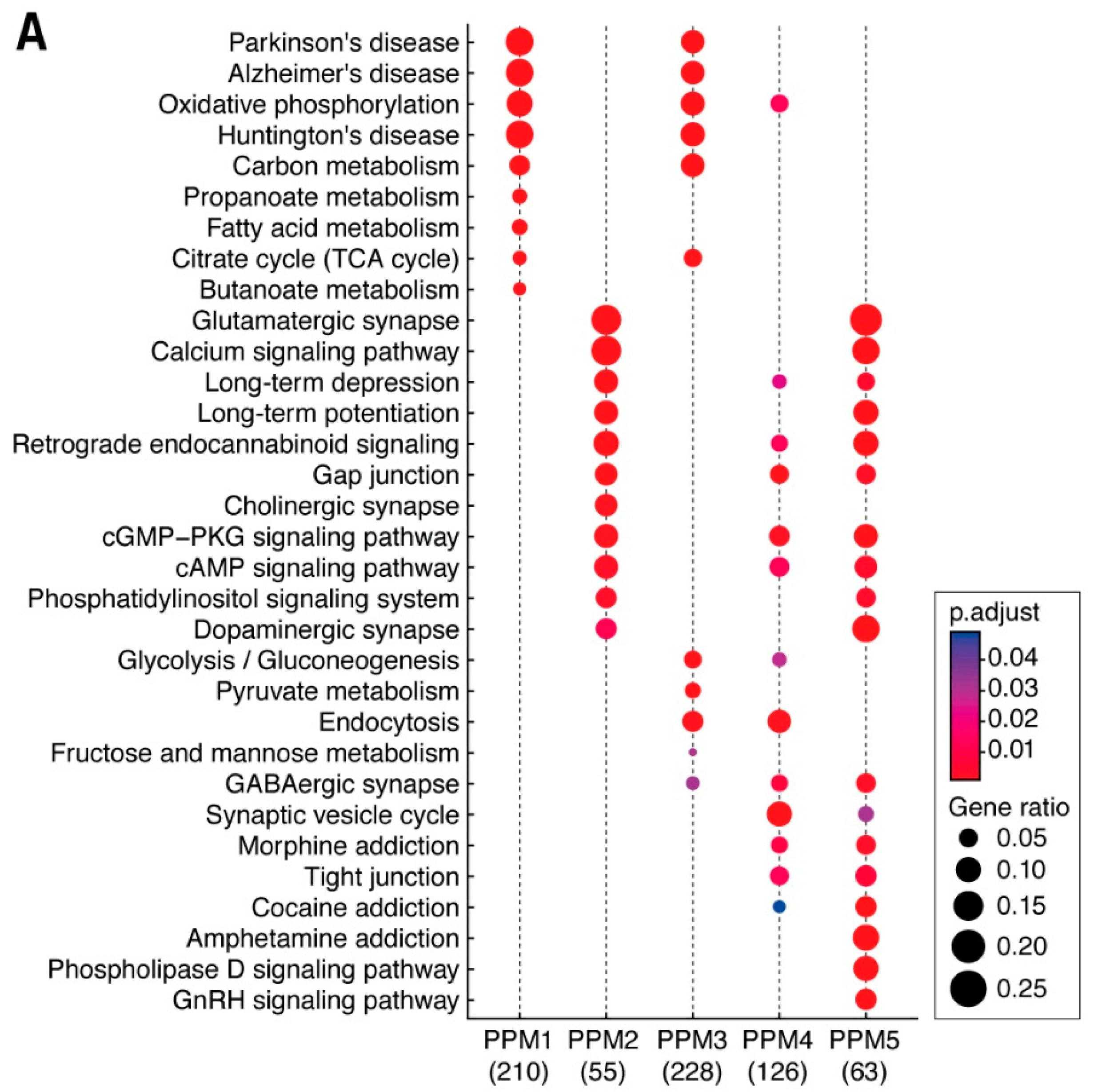
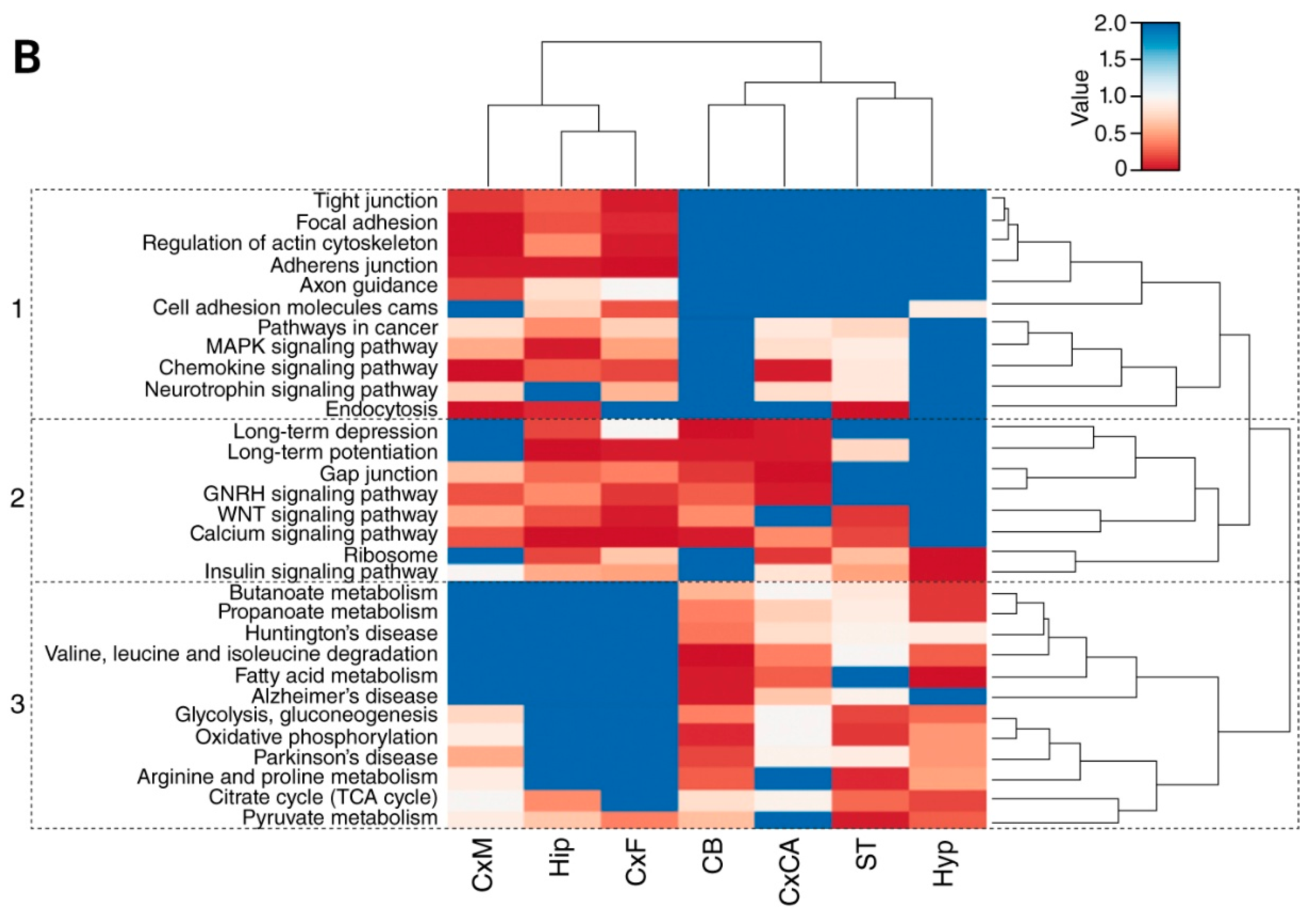
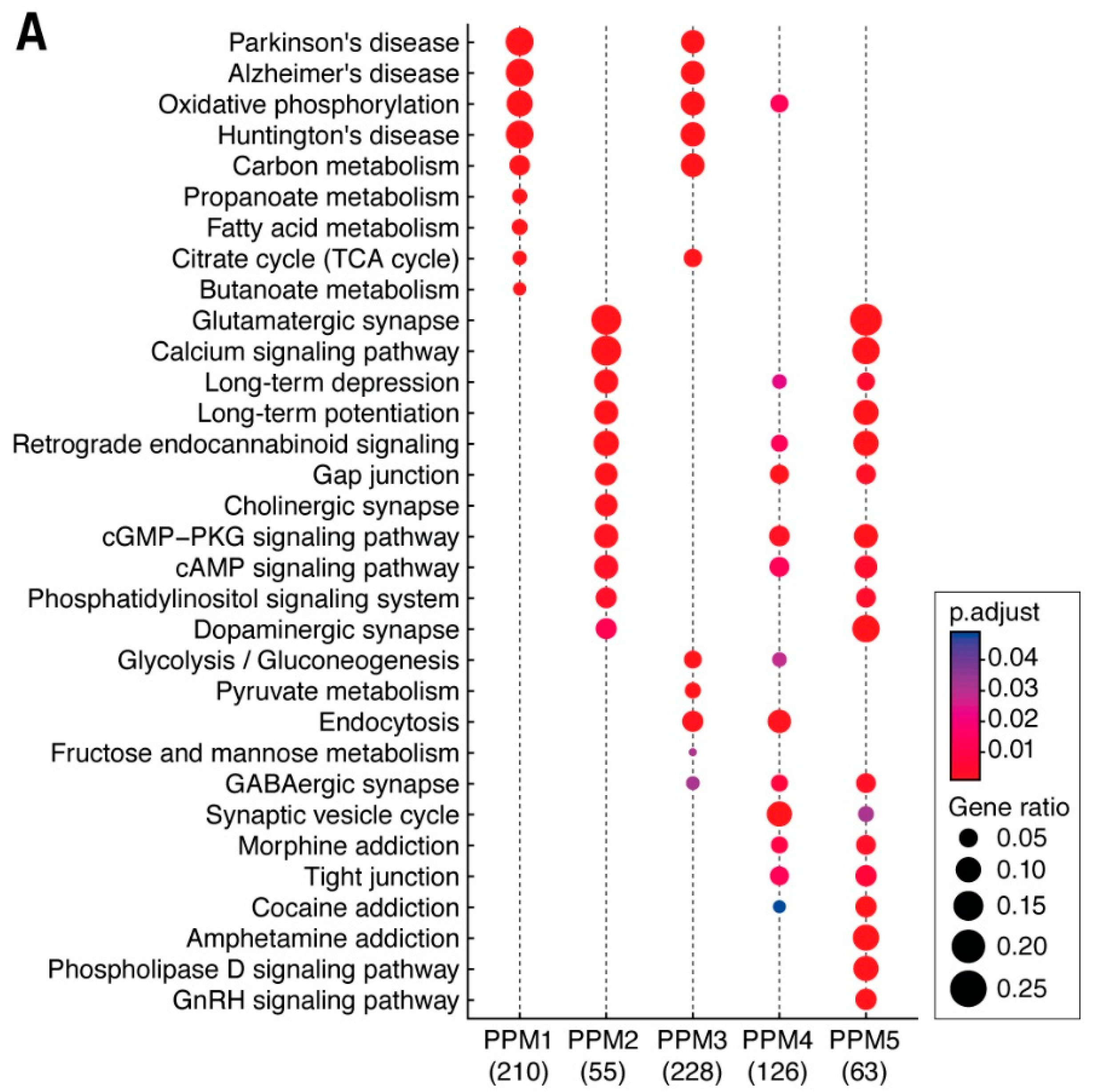
3.3. Distribution of Mechanism of Cognition and Protein Complexes
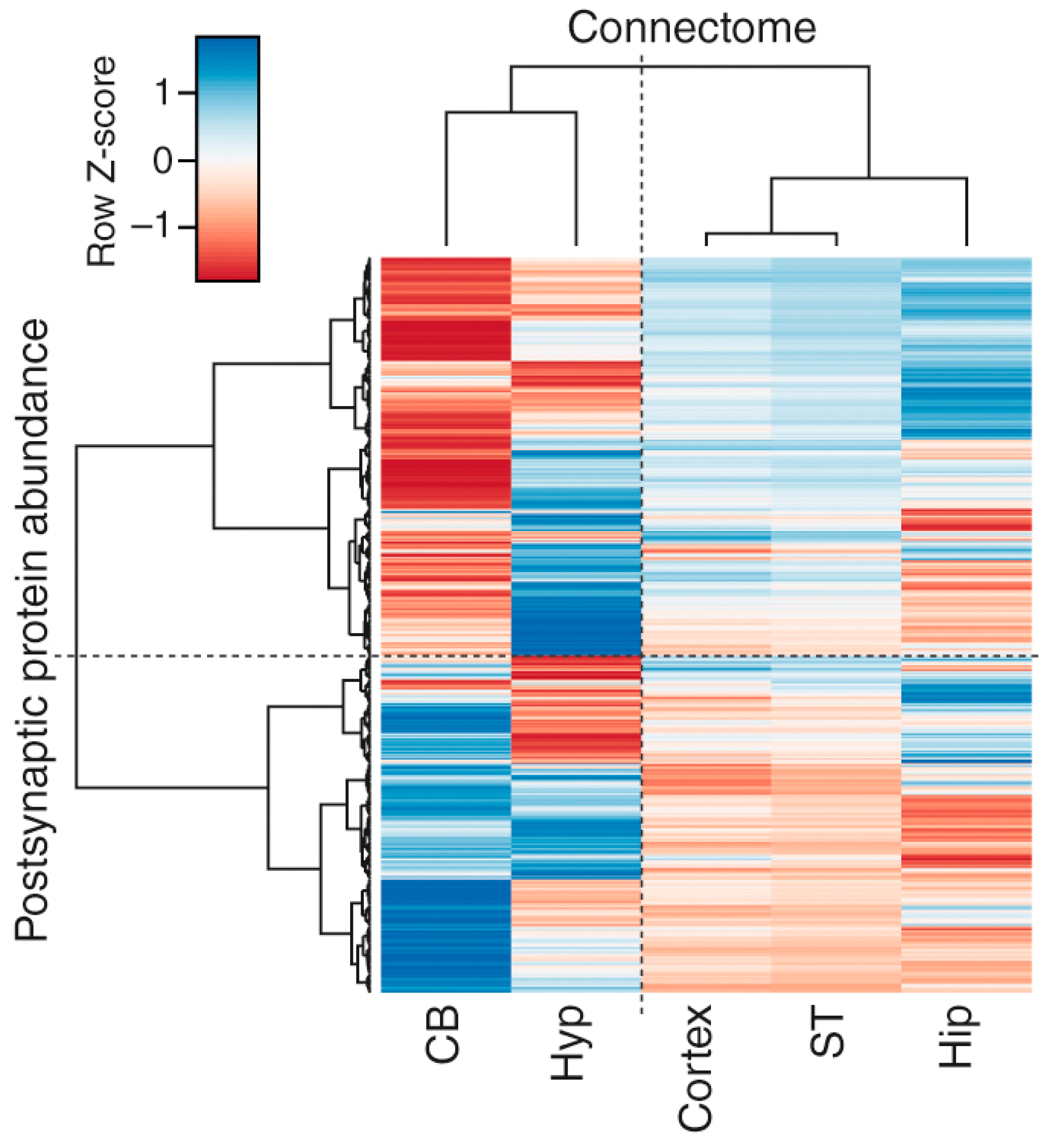
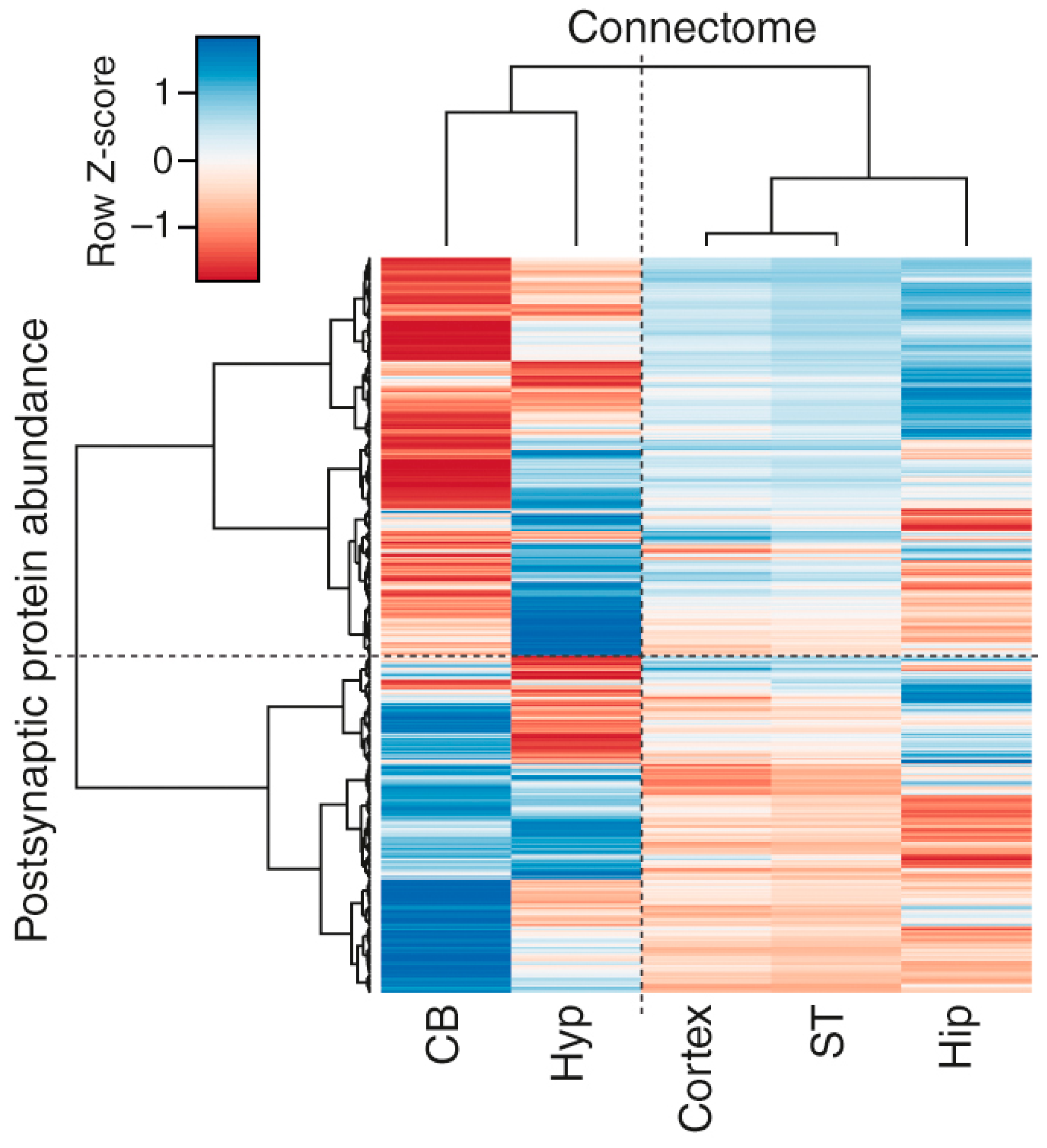
3.4. Correlations of Regional Synapse Proteomes with the Connectome
3.5. Regional Differences in Postsynaptic Protein Interaction Networks
3.6. Identifying a Stable Core Network
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Husi, H.; Ward, M.A.; Choudhary, J.S.; Blackstock, W.P.; Grant, S.G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 2000, 3, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Skene, N.G.; Roy, M.; Grant, S.G. A genomic lifespan program that reorganises the young adult brain is targeted in schizophrenia. eLife 2017, 6, e17915. [Google Scholar] [CrossRef] [PubMed]
- Bayes, A.; Collins, M.O.; Reig-Viader, R.; Gou, G.; Goulding, D.; Izquierdo, A.; Choudhary, J.S.; Emes, R.D.; Grant, S.G. Evolution of complexity in the zebrafish synapse proteome. Nat. Commun. 2017, 8, 14613. [Google Scholar] [CrossRef] [PubMed]
- Bayes, A.; Collins, M.O.; Croning, M.D.; van de Lagemaat, L.N.; Choudhary, J.S.; Grant, S.G. Comparative study of human and mouse postsynaptic proteomes finds high compositional conservation and abundance differences for key synaptic proteins. PLoS ONE 2012, 7, e46683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.O.; Husi, H.; Yu, L.; Brandon, J.M.; Anderson, C.N.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 2006, 97 (Suppl. 1), 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Distler, U.; Schmeisser, M.J.; Pelosi, A.; Reim, D.; Kuharev, J.; Weiczner, R.; Baumgart, J.; Boeckers, T.M.; Nitsch, R.; Vogt, J.; et al. In-depth protein profiling of the postsynaptic density from mouse hippocampus using data-independent acquisition proteomics. Proteomics 2014, 14, 2607–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosemeci, A.; Tao-Cheng, J.H.; Vinade, L.; Jaffe, H. Preparation of postsynaptic density fraction from hippocampal slices and proteomic analysis. Biochem. Biophys. Res. Commun. 2006, 339, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.A.; Fernholz, B.D.; Boussac, M.; Xu, C.; Grigorean, G.; Ziff, E.B.; Neubert, T.A. Identification and verification of novel rodent postsynaptic density proteins. Mol. Cell. Proteom. 2004, 3, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Kim, M.J.; Cheng, D.; Duong, D.M.; Gygi, S.P.; Sheng, M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 2004, 279, 21003–21011. [Google Scholar] [CrossRef] [PubMed]
- Trinidad, J.C.; Thalhammer, A.; Specht, C.G.; Lynn, A.J.; Baker, P.R.; Schoepfer, R.; Burlingame, A.L. Quantitative analysis of synaptic phosphorylation and protein expression. Mol. Cell. Proteom. 2008, 7, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Trinidad, J.C.; Thalhammer, A.; Specht, C.G.; Schoepfer, R.; Burlingame, A.L. Phosphorylation state of postsynaptic density proteins. J. Neurochem. 2005, 92, 1306–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uezu, A.; Kanak, D.J.; Bradshaw, T.W.; Soderblom, E.J.; Catavero, C.M.; Burette, A.C.; Weinberg, R.J.; Soderling, S.H. Identification of an elaborate complex mediating postsynaptic inhibition. Science 2016, 353, 1123–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, Y.; Yamauchi, Y.; Shinkawa, T.; Taoka, M.; Donai, H.; Takahashi, N.; Isobe, T.; Yamauchi, T. Molecular constituents of the postsynaptic density fraction revealed by proteomic analysis using multidimensional liquid chromatography-tandem mass spectrometry. J. Neurochem. 2004, 88, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Coba, M.P.; Pocklington, A.J.; Collins, M.O.; Kopanitsa, M.V.; Uren, R.T.; Swamy, S.; Croning, M.D.; Choudhary, J.S.; Grant, S.G. Neurotransmitters drive combinatorial multistate postsynaptic density networks. Sci. Signal. 2009, 2, ra19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, W.; Yang, H.; Howrigan, D.P.; Wilkinson, B.; Souaiaia, T.; Evgrafov, O.V.; Genovese, G.; Clementel, V.A.; Tudor, J.C.; et al. Spatiotemporal profile of postsynaptic interactomes integrates components of complex brain disorders. Nat. Neurosci. 2017, 20, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Bayes, A.; van de Lagemaat, L.N.; Collins, M.O.; Croning, M.D.; Whittle, I.R.; Choudhary, J.S.; Grant, S.G. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011, 14, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Kaizuka, T.; Takumi, T. Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J. Biochem. 2018, 163, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.; Collins, M.O.; Frank, R.A.W.; Zhu, F.; Kopanitsa, M.V.; Nithianantharajah, J.; Lempriere, S.A.; Fricker, D.; Elsegood, K.A.; McLaughlin, C.L.; et al. Arc Requires PSD95 for Assembly into Postsynaptic Complexes Involved with Neural Dysfunction and Intelligence. Cell Rep. 2017, 21, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.A.; Grant, S.G. Supramolecular organization of NMDA receptors and the postsynaptic density. Curr. Opin. Neurobiol. 2017, 45, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.A.W.; Zhu, F.; Komiyama, N.H.; Grant, S.G.N. Hierarchical organization and genetically separable subfamilies of PSD95 postsynaptic supercomplexes. J. Neurochem. 2017, 142, 504–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbas, A.I.; Yadav, P.N.; Yao, W.D.; Arbuckle, M.I.; Grant, S.G.; Caron, M.G.; Roth, B.L. PSD-95 is essential for hallucinogen and atypical antipsychotic drug actions at serotonin receptors. J. Neurosci. 2009, 29, 7124–7136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, E.; Collins, M.O.; Uren, R.T.; Kopanitsa, M.V.; Komiyama, N.H.; Croning, M.D.; Zografos, L.; Armstrong, J.D.; Choudhary, J.S.; Grant, S.G. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol. Syst. Biol. 2009, 5, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, R.A.; Komiyama, N.H.; Ryan, T.J.; Zhu, F.; O’Dell, T.J.; Grant, S.G. NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat. Commun. 2016, 7, 11264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, L.W. Brain Maps III: Structure of the Rat Brain: An Atlas with Printed and Electronic Templates for Data, Models, and Schematics, 3rd rev. ed.; Elsevier, Academic Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Roy, M.; Sorokina, O.; Skene, N.; Simonnet, C.; Mazzo, F.; Zwart, R.; Sher, E.; Smith, C.; Armstrong, J.D.; Grant, S.G.N. Proteomic analysis of postsynaptic proteins in regions of the human neocortex. Nat. Neurosci. 2018, 21, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Cizeron, M.; Qiu, Z.; Benavides-Piccione, R.; Kopanitsa, M.V.; Skene, N.; Koniaris, B.; DeFelipe, J.; Fransén, E.; Komiyama, N.H.; et al. Architecture of the mouse brain synaptome. Neuron 2018, 172, 143–150. [Google Scholar] [CrossRef]
- Palkovits, M. General Neurochemical Techniques; Boulton, A.A., Baker, G.B., Eds.; Springer: Berlin, Germany, 1985; Volume 1, pp. 1–17. [Google Scholar]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar]
- Hawrylycz, M.; Miller, J.A.; Menon, V.; Feng, D.; Dolbeare, T.; Guillozet-Bongaarts, A.L.; Jegga, A.G.; Aronow, B.J.; Lee, C.K.; Bernard, A.; et al. Canonical genetic signatures of the adult human brain. Nat. Neurosci. 2015, 18, 1832–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galili, T. Dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.W.; Harris, J.A.; Ng, L.; Winslow, B.; Cain, N.; Mihalas, S.; Wang, Q.; Lau, C.; Kuan, L.; Henry, A.M.; et al. A mesoscale connectome of the mouse brain. Nature 2014, 508, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatr-Aryamontri, A.; Oughtred, R.; Boucher, L.; Rust, J.; Chang, C.; Kolas, N.K.; O’Donnell, L.; Oster, S.; Theesfeld, C.; Sellam, A.; et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017, 45, D369–D379. [Google Scholar] [CrossRef] [PubMed]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Xenarios, I.; Salwinski, L.; Duan, X.J.; Higney, P.; Kim, S.M.; Eisenberg, D. DIP, the Database of Interacting Proteins: A research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002, 30, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.E. Finding community structure in networks using the eigenvectors of matrices. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 2006, 74, 036104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.E.; Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 2004, 69, 026113. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.I.; Armstrong, J.D.; Jarman, A.P. Merged consensus clustering to assess and improve class discovery with microarray data. BMC Bioinform. 2010, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.S.; Cornelisse, L.N.; Toonen, R.F.; Min, J.L.; Hultman, C.M.; International Schizophrenia; Holmans, P.A.; O’Donovan, M.C.; Purcell, S.M.; Smit, A.B.; et al. Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol. Psychiatry 2012, 17, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronson, A.R.; Lang, F.M. An overview of MetaMap: Historical perspective and recent advances. J. Am. Med. Inform. Assoc. 2010, 17, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Musen, M.A.; Noy, N.F.; Shah, N.H.; Whetzel, P.L.; Chute, C.G.; Story, M.A.; Smith, B.; Team, N. The National Center for Biomedical Ontology. J. Am. Med. Inform. Assoc. 2012, 19, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whetzel, P.L.; Noy, N.F.; Shah, N.H.; Alexander, P.R.; Nyulas, C.; Tudorache, T.; Musen, M.A. BioPortal: Enhanced functionality via new Web services from the National Center for Biomedical Ontology to access and use ontologies in software applications. Nucleic Acids Res. 2011, 39, W541–W545. [Google Scholar] [CrossRef] [PubMed]
- Schriml, L.M.; Arze, C.; Nadendla, S.; Chang, Y.W.; Mazaitis, M.; Felix, V.; Feng, G.; Kibbe, W.A. Disease Ontology: A backbone for disease semantic integration. Nucleic Acids Res. 2012, 40, D940–D946. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenfuhrer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamanut, R.; Kennedy, H.; Toroczkai, Z.; Ercsey-Ravasz, M.; Van Essen, D.C.; Knoblauch, K.; Burkhalter, A. The Mouse Cortical Connectome, Characterized by an Ultra-Dense Cortical Graph, Maintains Specificity by Distinct Connectivity Profiles. Neuron 2018, 97, 698–715. [Google Scholar] [CrossRef] [PubMed]
- Pocklington, A.J.; Cumiskey, M.; Armstrong, J.D.; Grant, S.G. The proteomes of neurotransmitter receptor complexes form modular networks with distributed functionality underlying plasticity and behaviour. Mol. Syst. Biol. 2006, 2, 2006.0023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, C.; He, X.; Simpson, T.I.; Armstrong, J.D. Improved Functional Enrichment Analysis of Biological Networks using Scalable Modularity Based Clustering. J. Proteom. Bioinform. 2016, 9, 9–18. [Google Scholar] [CrossRef]
- Valor, L.M.; Charlesworth, P.; Humphreys, L.; Anderson, C.N.; Grant, S.G. Network activity-independent coordinated gene expression program for synapse assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 4658–4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, L.W. Brain Architecture: Understanding the Basic Plan, 2nd ed.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Dietrichs, E.; Haines, D.E. Demonstration of hypothalamo-cerebellar and cerebello-hypothalamic fibres in a prosimian primate (Galago crassicaudatus). Anat. Embryol. (Berl.) 1984, 170, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Dietrichs, E.; Haines, D.E.; Roste, G.K.; Roste, L.S. Hypothalamocerebellar and cerebellohypothalamic projections--circuits for regulating nonsomatic cerebellar activity? Histol. Histopathol. 1994, 9, 603–614. [Google Scholar] [PubMed]
- Haines, D.E.; Dietrichs, E.; Mihailoff, G.A.; McDonald, E.F. The cerebellar-hypothalamic axis: Basic circuits and clinical observations. Int. Rev. Neurobiol. 1997, 41, 83–107. [Google Scholar] [PubMed]
- Zhu, J.N.; Wang, J.J. The cerebellum in feeding control: Possible function and mechanism. Cell. Mol. Neurobiol. 2008, 28, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Zaccai, J.; McCracken, C.; Brayne, C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing 2005, 34, 561–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitwell, J.L.; Weigand, S.D.; Shiung, M.M.; Boeve, B.F.; Ferman, T.J.; Smith, G.E.; Knopman, D.S.; Petersen, R.C.; Benarroch, E.E.; Josephs, K.A.; et al. Focal atrophy in dementia with Lewy bodies on MRI: A distinct pattern from Alzheimer’s disease. Brain 2007, 130, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Fontes, M.; Zhou, Y.; Denisov, V.; Paulsen, J.S.; Kirik, D.; Petersen, A.; Huntington Study Group. Early changes in the hypothalamic region in prodromal Huntington disease revealed by MRI analysis. Neurobiol. Dis. 2010, 40, 531–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabery, S.; Murphy, K.; Schultz, K.; Loy, C.T.; McCusker, E.; Kirik, D.; Halliday, G.; Petersen, A. Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropathological analyses. Acta Neuropathol. 2010, 120, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, S.E.; Reeves, J.L.; Lucas, E.K.; Gamble, K.L.; Lesort, M.; Cowell, R.M. Disruption of Purkinje cell function prior to huntingtin accumulation and cell loss in an animal model of Huntington disease. Exp. Neurol. 2012, 236, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Fennema-Notestine, C.; Archibald, S.L.; Jacobson, M.W.; Corey-Bloom, J.; Paulsen, J.S.; Peavy, G.M.; Gamst, A.C.; Hamilton, J.M.; Salmon, D.P.; Jernigan, T.L. In vivo evidence of cerebellar atrophy and cerebral white matter loss in Huntington disease. Neurology 2004, 63, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Devys, D.; Goldenberg, A.; Maltete, D.; Herve, C.; Hannequin, D.; Guyant-Marechal, L. Juvenile Huntington disease in an 18-month-old boy revealed by global developmental delay and reduced cerebellar volume. Am. J. Med. Genet. A 2011, 155A, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Sakazume, S.; Yoshinari, S.; Oguma, E.; Utsuno, E.; Ishii, T.; Narumi, Y.; Shiihara, T.; Ohashi, H. A patient with early onset Huntington disease and severe cerebellar atrophy. Am. J. Med. Genet. A 2009, 149A, 598–601. [Google Scholar] [CrossRef] [PubMed]
- Wilson, H.; Giordano, B.; Turkheimer, F.E.; Chaudhuri, K.R.; Politis, M. Serotonergic dysregulation is linked to sleep problems in Parkinson’s disease. Neuroimage Clin. 2018, 18, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Joutsa, J.; Horn, A.; Hsu, J.; Fox, M.D. Localizing parkinsonism based on focal brain lesions. Brain 2018, 141, 2445–2456. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, M.; Sorokina, O.; McLean, C.; Tapia-González, S.; DeFelipe, J.; Armstrong, J.D.; Grant, S.G.N. Regional Diversity in the Postsynaptic Proteome of the Mouse Brain. Proteomes 2018, 6, 31. https://doi.org/10.3390/proteomes6030031
Roy M, Sorokina O, McLean C, Tapia-González S, DeFelipe J, Armstrong JD, Grant SGN. Regional Diversity in the Postsynaptic Proteome of the Mouse Brain. Proteomes. 2018; 6(3):31. https://doi.org/10.3390/proteomes6030031
Chicago/Turabian StyleRoy, Marcia, Oksana Sorokina, Colin McLean, Silvia Tapia-González, Javier DeFelipe, J. Douglas Armstrong, and Seth G. N. Grant. 2018. "Regional Diversity in the Postsynaptic Proteome of the Mouse Brain" Proteomes 6, no. 3: 31. https://doi.org/10.3390/proteomes6030031
APA StyleRoy, M., Sorokina, O., McLean, C., Tapia-González, S., DeFelipe, J., Armstrong, J. D., & Grant, S. G. N. (2018). Regional Diversity in the Postsynaptic Proteome of the Mouse Brain. Proteomes, 6(3), 31. https://doi.org/10.3390/proteomes6030031