Large Scale Proteomic Data and Network-Based Systems Biology Approaches to Explore the Plant World




Abstract
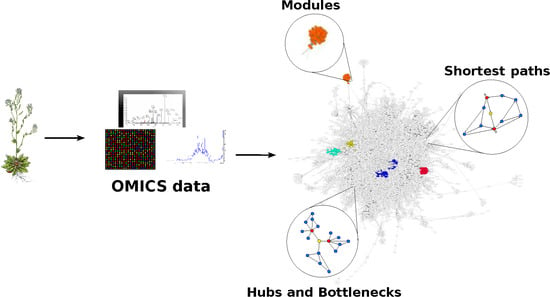
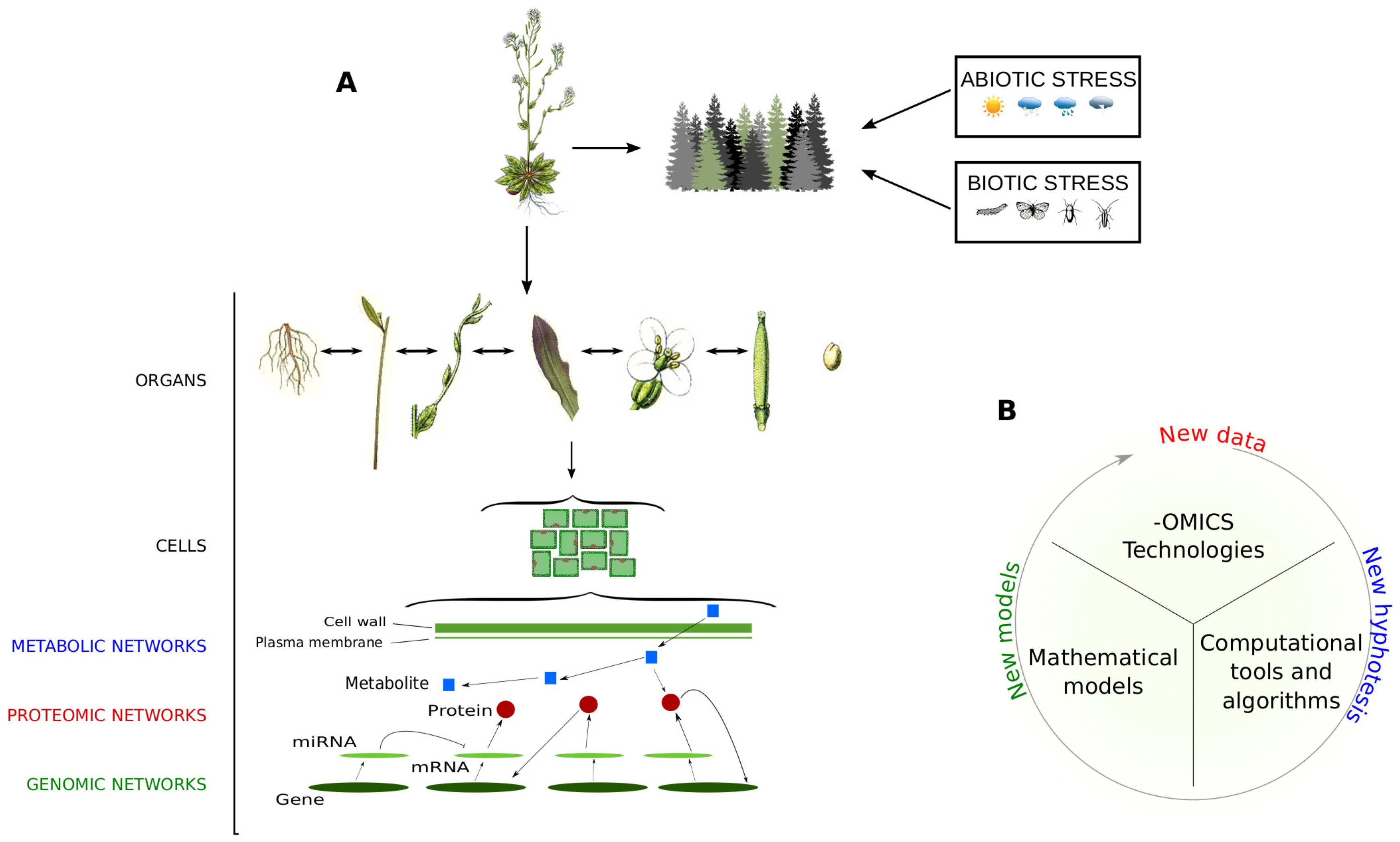
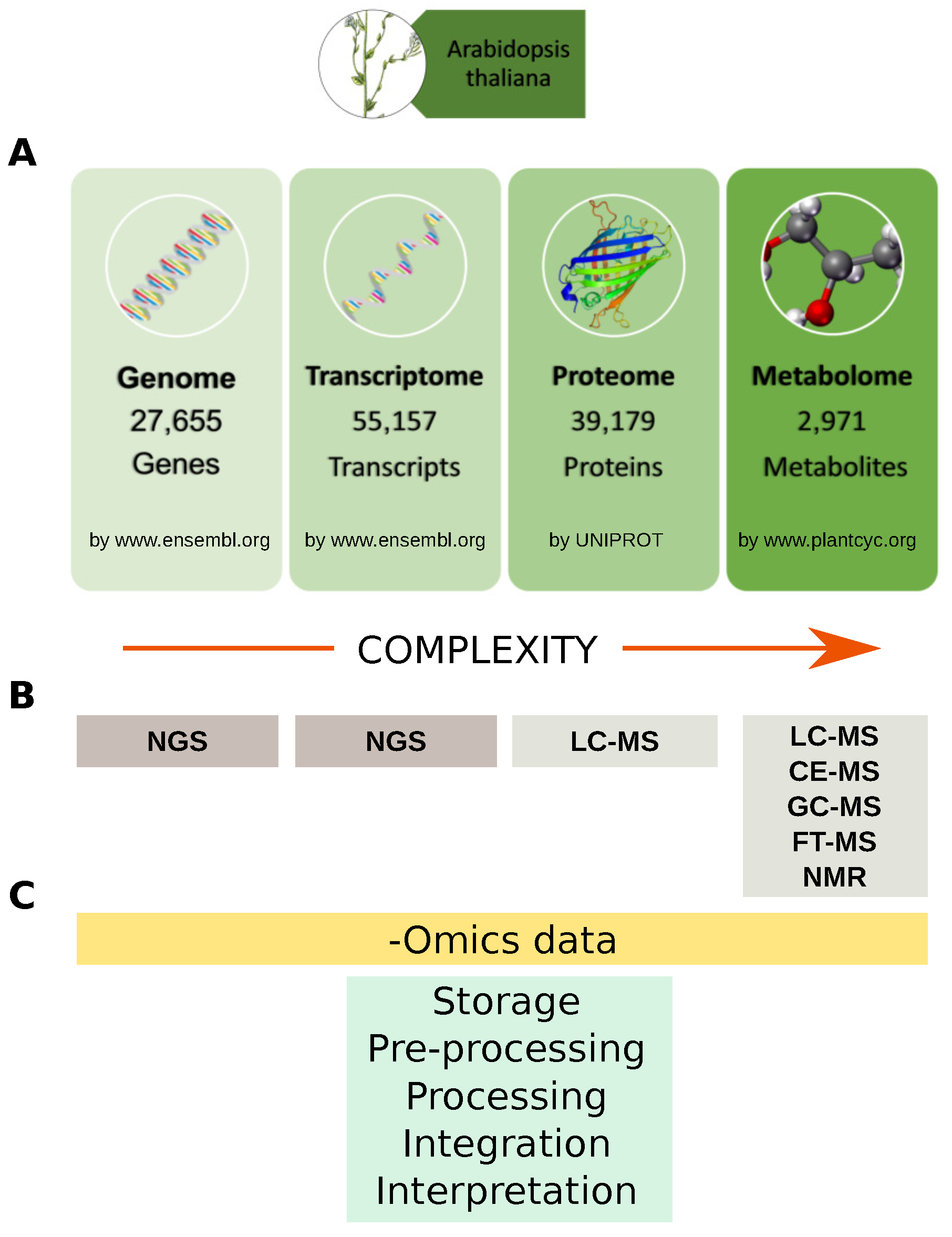
:
1. Introduction
2. Omic Technologies in the Plant World: From Genomics to Metabolomics by Way of Proteomics
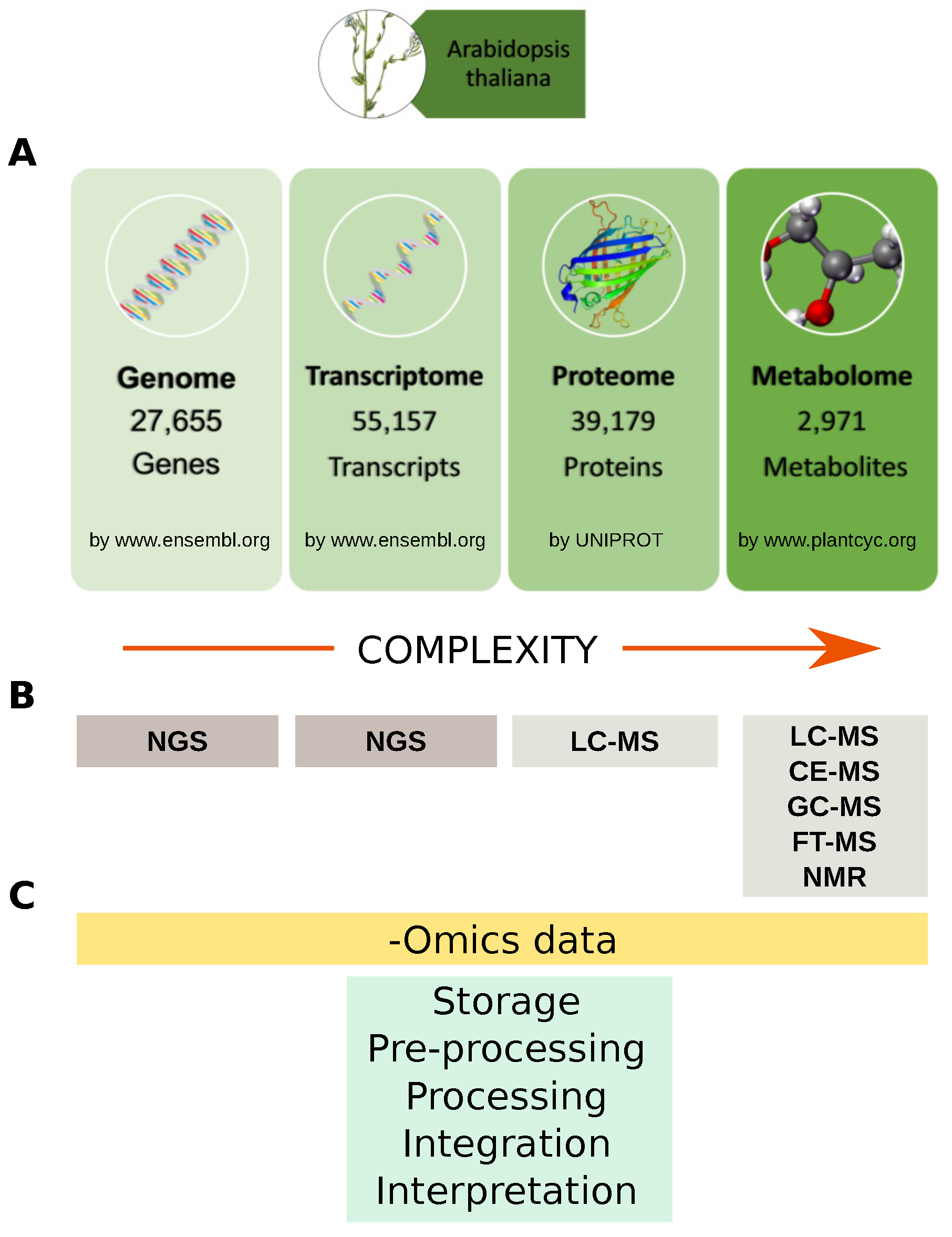
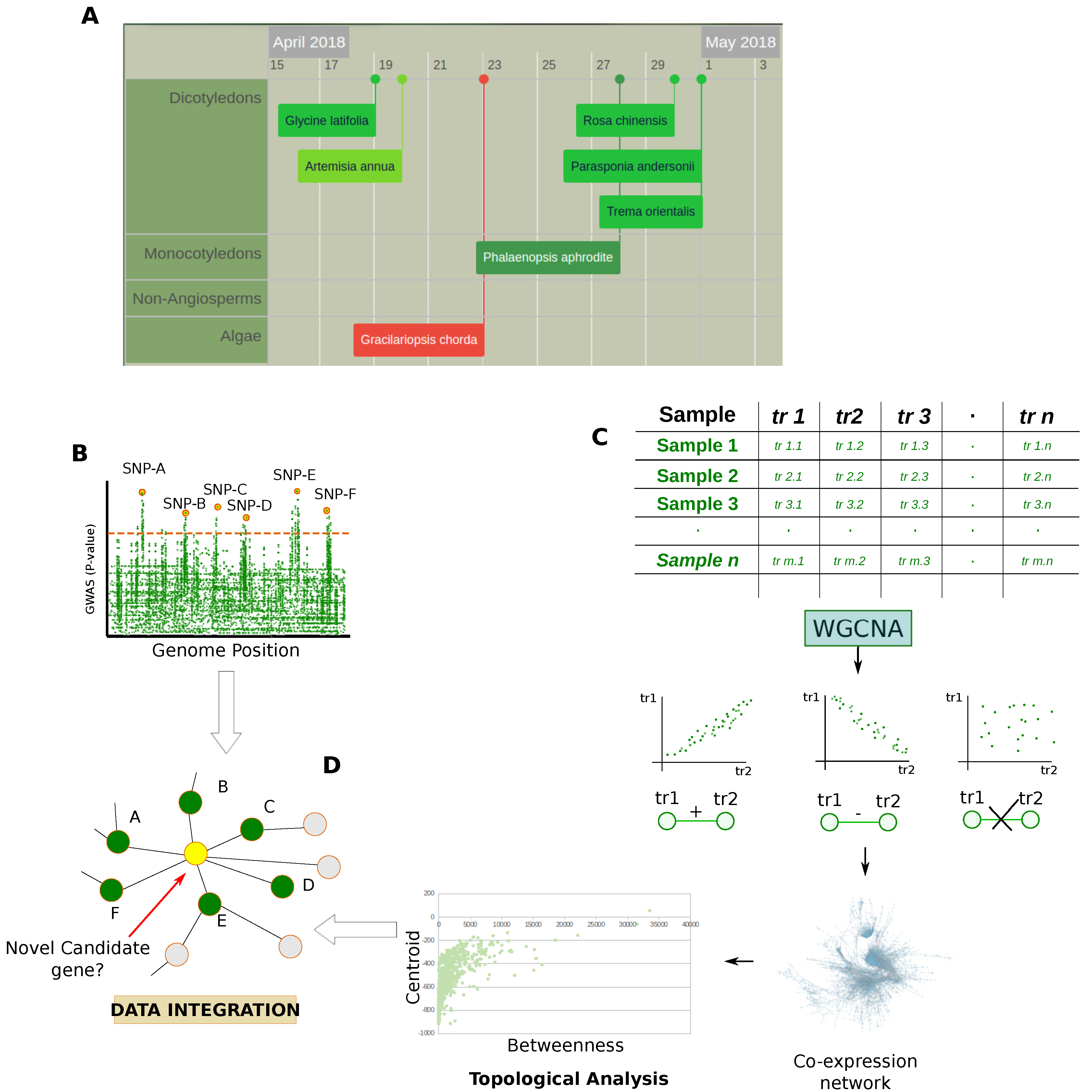
2.1. Genomics
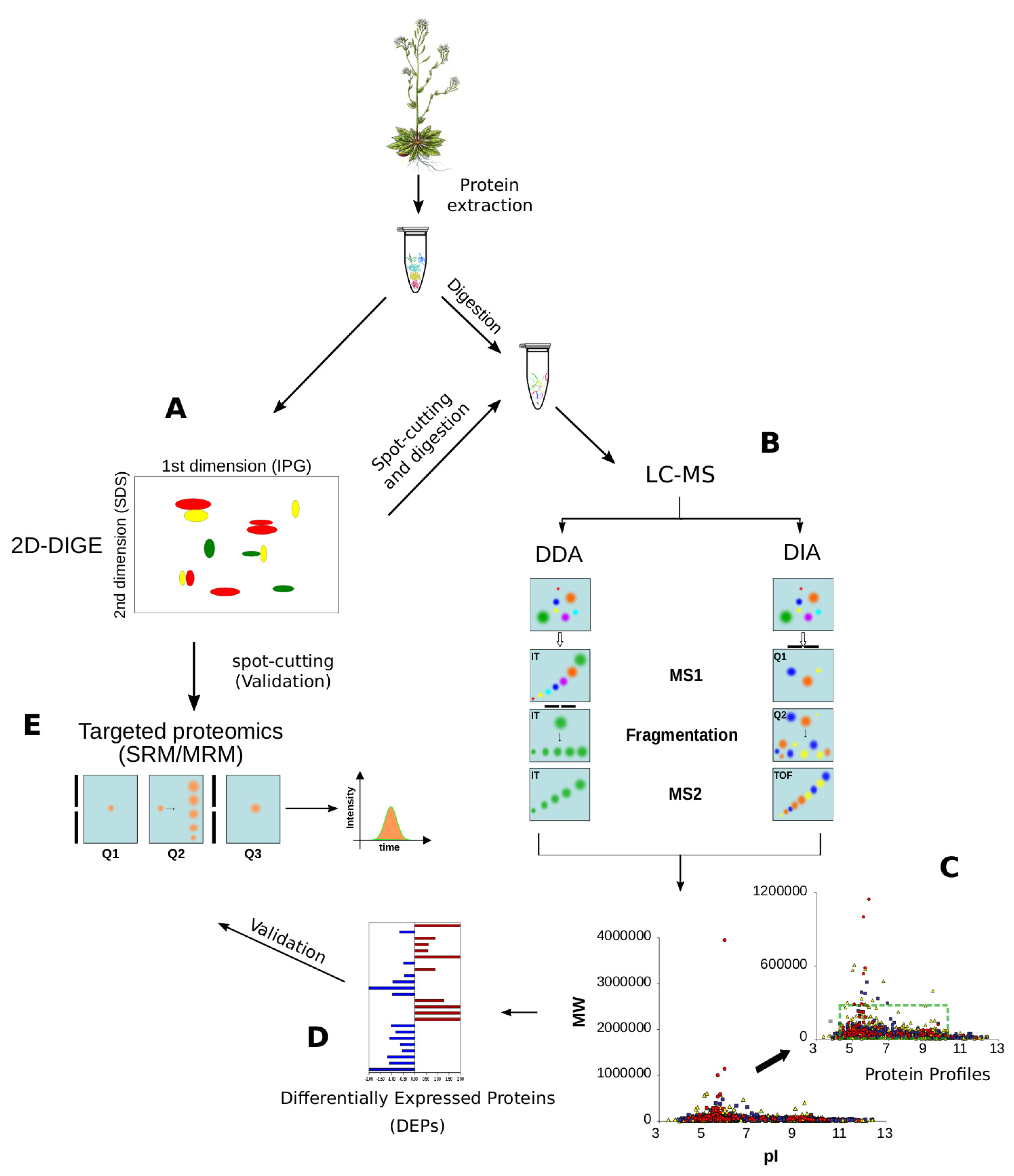
2.2. Proteomics
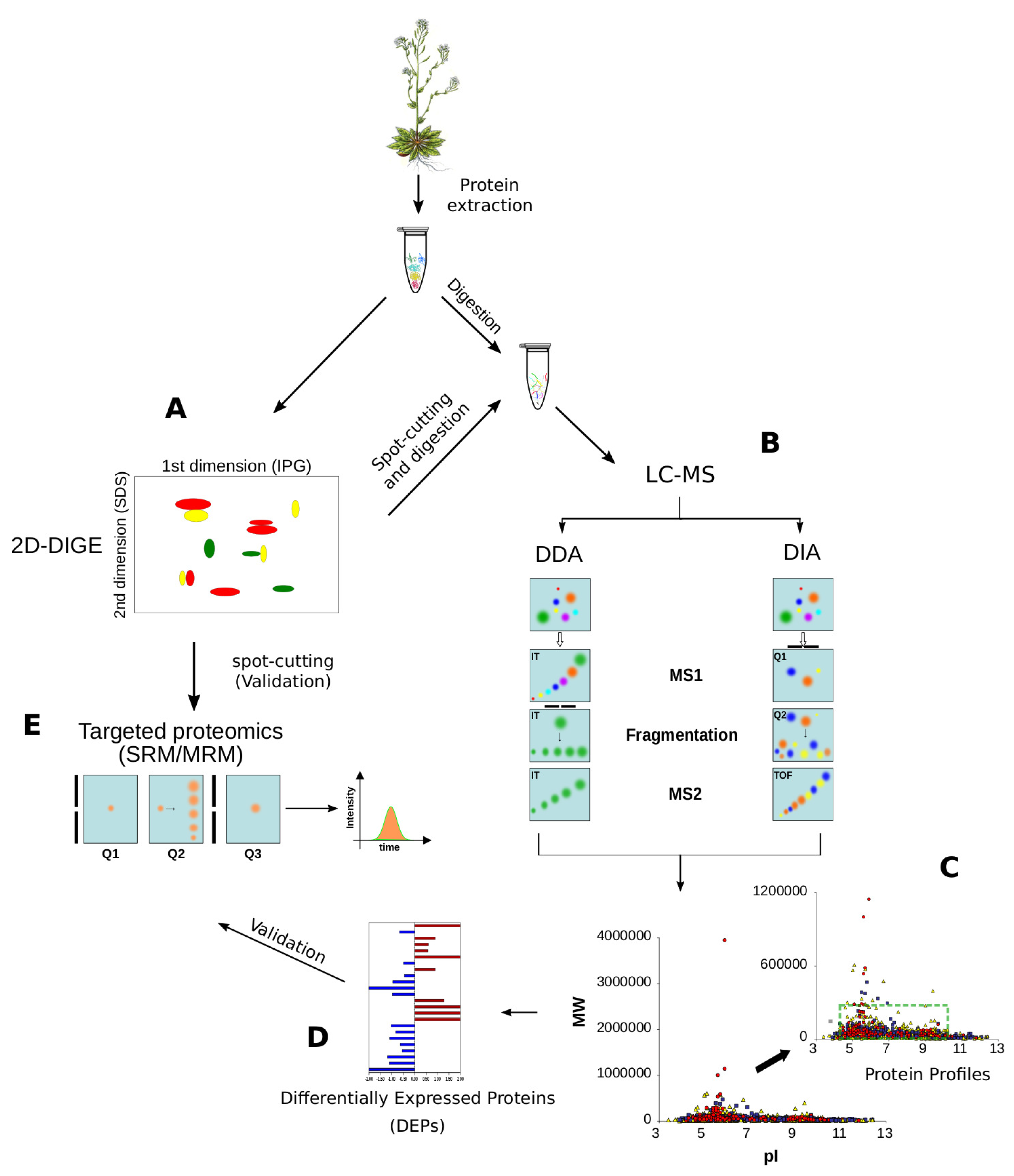
2.2.1. Shotgun Proteomics
2.2.2. Selected- and Multiple-Reaction Monitoring (SRM/MRM)
2.2.3. DIA/SWATH
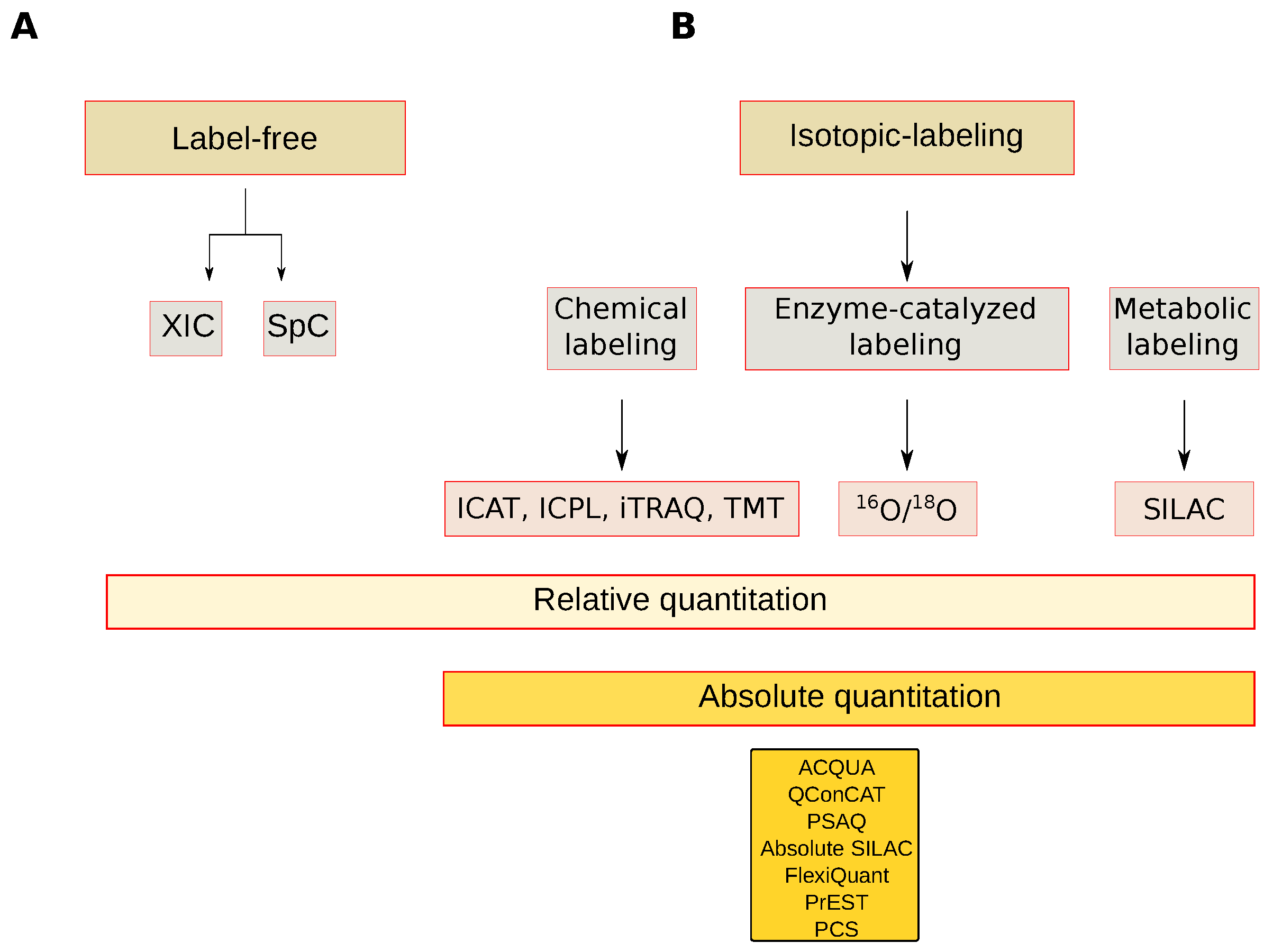
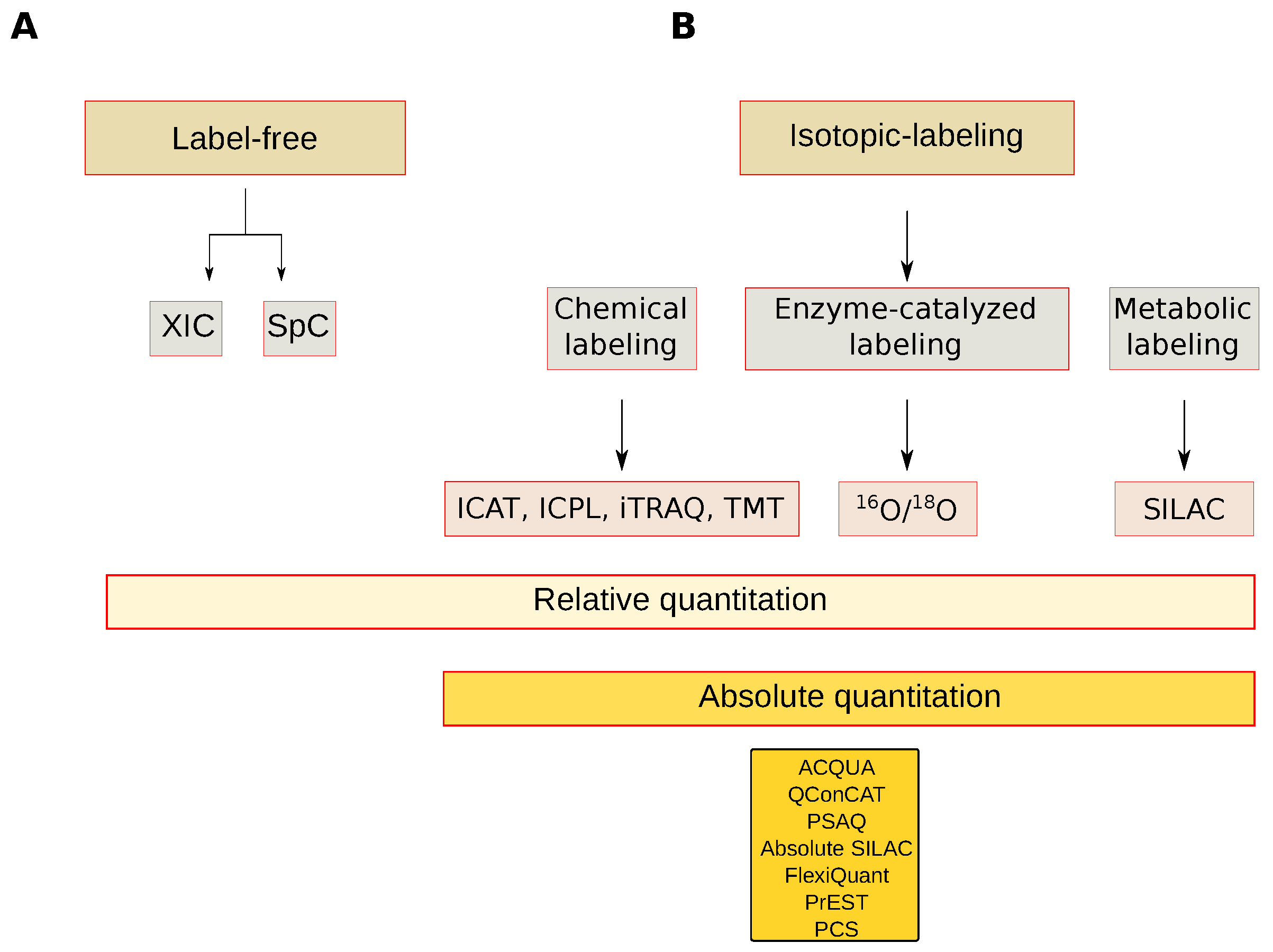
2.2.4. MS-Based Quantification Strategies
Label-Free Quantitation
Isotope-Labeling Quantitation
Absolute Quantitation
2.3. Metabolomics
3. Co-Expression and PPI Networks as Models to Investigate Plant Organisms
3.1. Gene Co-Expression Networks
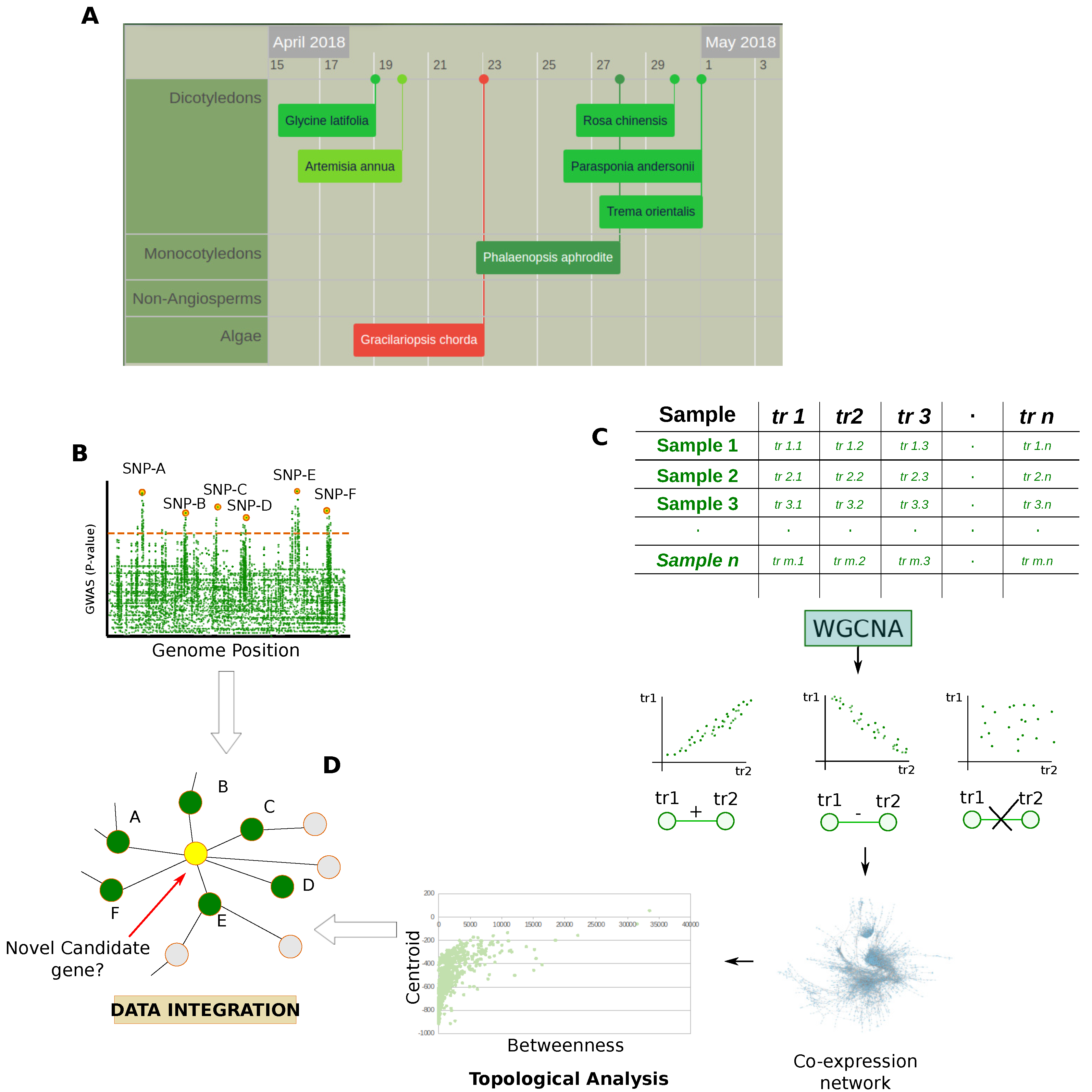
Gene Co-Expression Network Combined with GWAS and QTL Data
3.2. Protein Co-Expression Networks
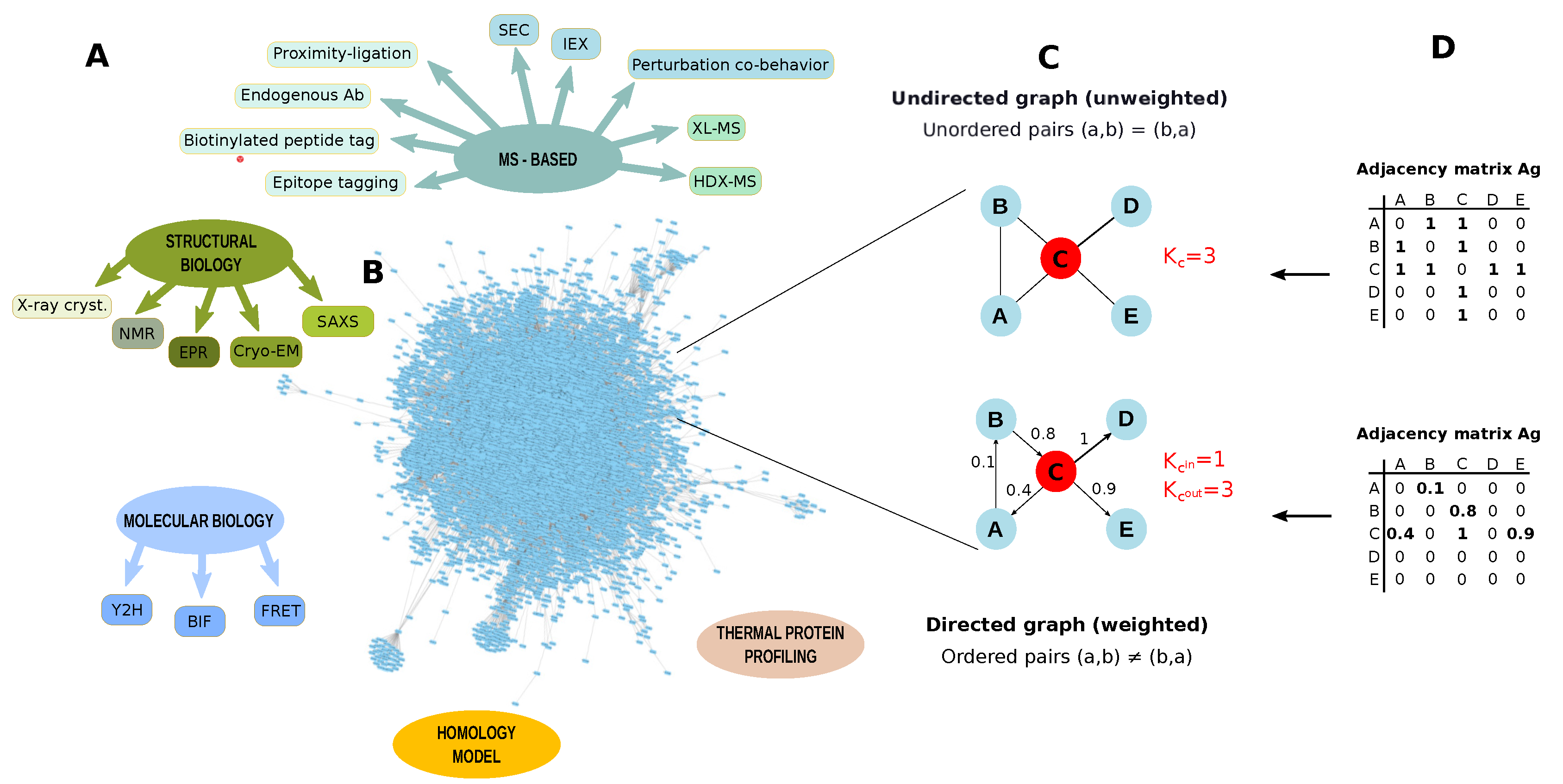
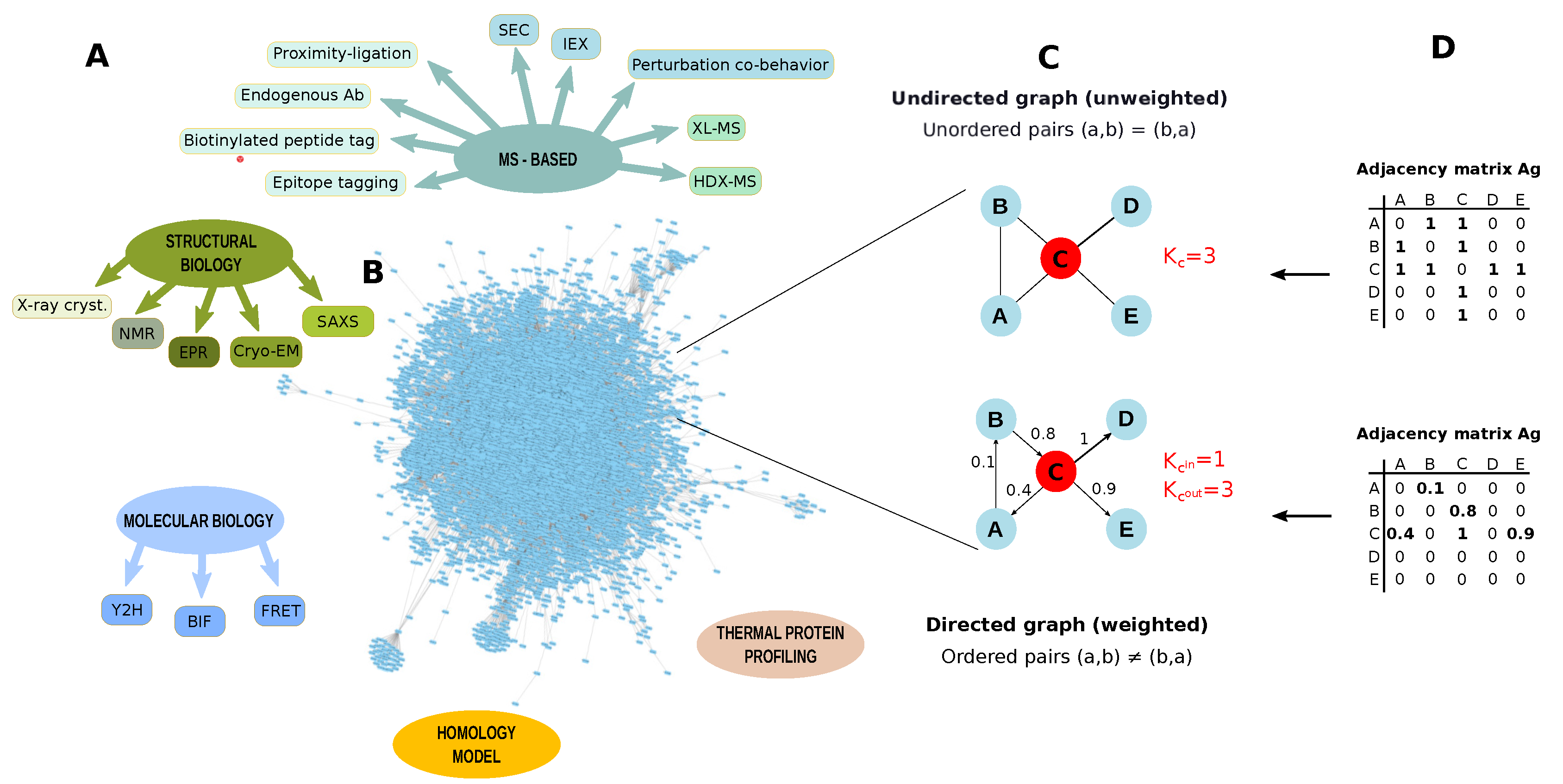
3.3. PPIs Identification
3.4. PPI Networks
Studies Combining PPIs and Network Topology
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2DE | 2 Dimensional Gel Electrophoresis |
| 2D-DIGE | 2 Dimensional Fluorescence Difference Gel Electrophoresis |
| AP-MS | Affinity Purification Mass Spectrometry |
| AQUA | Absolute Quantification |
| BiFC | Bimolecular Fluorescence Complementation |
| CAM | Crassulacean Acid Metabolism |
| CE | Capillary Electrophoresis |
| DDA | Data Dependent Acquisition |
| DEGs | Differentially Expressed Genes |
| DEPs | Differentially Expressed Proteins |
| DIA | Data Independent Acquisition |
| emPAI | Exponentially Modified Protein Abundance Index |
| FlexiQuant | Full-Length Expressed Stable Isotope-labeled Proteins for Quantification |
| FT-MS | Fourier Transform-Mass Spectrometry |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| FRET | Förster Resonance Energy Transfer |
| GC | Gas Chromatography |
| GO | Gene Ontology |
| GRN | Gene Regulatory Network |
| GS | Gene Significance |
| GWAS | Genome Wide Association Study |
| HILIC | Hydrophilic Interaction Liquid Chromatography |
| HMDB | Human Metabolome Database |
| HPLC | High Performance Liquid Chromatography |
| ICAT | Isotope-Coded Affinity Tag |
| ICPL | Isotope-Coded Protein Label |
| kME | Eigengene-Based Connectivity |
| LC | Liquid Chromatography |
| MeRy-B | Metabolomic Repository Bordeaux |
| miRNA | microRNA |
| MRM | Multiple-Reaction Monitoring |
| mRNA | messenger RNA |
| MS | Mass Spectrometry |
| MS/MS | tandem mass spectra |
| MudPIT | Multidimensional Protein Identification Technology |
| MW | Molecular Weight |
| NGS | Next Generation Sequencing |
| NMR | Nuclear Magnetic Resonance |
| NSAF | Normalized spectral abundance factor |
| ODEs | Ordinary Differential Equations |
| PC | Pearson’s Correlation |
| PCS | Peptide-Concatenated Standards |
| pI | Isoelectric point |
| PMN | Plant Metabolic Network Database |
| PPI | Protein-Protein Interaction |
| PPIN | Plant-Pathogen Interaction Network |
| PRM | Parallel Reaction Monitoring |
| PTMs | Post Translational Modifications |
| PrEST | Protein Epitope Signature Tag |
| QconCAT | Quantification Concatemer |
| QTL | Quantitative Trait Locus |
| RNA-Seq | RNA Sequencing |
| siRNA | small interference RNA |
| SpC | Spectral Count |
| SEC | Size-Exclusion Chromatography |
| SNP | Single Nucleotide Polymorphism |
| SRM | Selected Reaction Monitoring |
| SVM | Support Vector Machine |
| SWATH | Sequential Windowed Acquisition of All Theoretical Fragment Ion Mass Spectra |
| SWIM | SWItchMiner |
| TAP-MS | Tandem Affinity Purification coupled with Mass Spectrometry |
| TMT | Tandem Mass Tags |
| WGCNA | Weighted Gene Co-expression Network Analysis |
| XICs | Extracted Ion Chromatogram |
| XL | Cross Linking |
| Y2H | Yeast Two Hybrid |
References
- Brigandt, I.; Love, A. Reductionism in biology. In The Stanford Encyclopedia of Philosophy, Spring 2017 ed.; Zalta, E.N., Ed.; Metaphysics Research Lab, Stanford University: Stanford, CA, USA, 2017. [Google Scholar]
- Oltvai, Z.N.; Barabási, A.L. Life’s complexity pyramid. Science 2002, 298, 763–764. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, T.; Mann, M.; Aebersold, R.; Yates, J.R., III; Bairoch, A.; Bergeron, J.J. Mass spectrometry in high-throughput proteomics: ready for the big time. Nat. Methods 2010, 7, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Soon, W.W.; Hariharan, M.; Snyder, M.P. High-throughput sequencing for biology and medicine. Mol. Syst. Biol. 2013, 9, 640. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, A.; Chaturvedi, P.; Weckwerth, W. Metabolomics in plant stress physiology. Adv. Biochem. Eng. Biotechnol. 2018. [Google Scholar]
- Emmert-Streib, F.; Dehmer, M. Networks for systems biology: Conceptual connection of data and function. IET Syst. Biol. 2011, 5, 185–207. [Google Scholar] [CrossRef] [PubMed]
- Chasman, D.; Fotuhi Siahpirani, A.; Roy, S. Network-based approaches for analysis of complex biological systems. Curr. Opin. Biotechnol. 2016, 39, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vella, D.; Zoppis, I.; Mauri, G.; Mauri, P.; Di Silvestre, D. From protein-protein interactions to protein co-expression networks: A new perspective to evaluate large-scale proteomic data. EURASIP J. Bioinform. Syst. Biol. 2017, 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Menche, J.; Barabási, A.L.; Sharma, A. Human symptoms-disease network. Nat. Commun. 2014, 5, 4212. [Google Scholar] [CrossRef] [PubMed]
- Guney, E.; Menche, J.; Vidal, M.; Barábasi, A.L. Network-based in silico drug efficacy screening. Nat. Commun. 2016, 7, 10331. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, T.K.; Bashir, T.; Hashem, A.; Abd Allah, E.F. Systems biology approach in plant abiotic stresses. Plant Physiol. Biochem. 2017, 121, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, R.; Dubiella, U.; Barbacci, A.; Genin, S.; Raffaele, S.; Roby, D. Advances on plant-pathogen interactions from molecular toward systems biology perspectives. Plant J. Cell Mol. Biol. 2017, 90, 720–737. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, R.J.; Michno, J.M.; Myers, C.L. Unraveling gene function in agricultural species using gene co-expression networks. Biochim. Biophys. Acta 2017, 1860, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Alseekh, S.; Cuadros-Inostroza, Á.; Fusari, C.M.; Mutwil, M.; Kooke, R.; Keurentjes, J.B.; Fernie, A.R.; Willmitzer, L.; Brotman, Y. Combined use of genome-wide association data and correlation networks unravels key regulators of primary metabolism in Arabidopsis thaliana. PLos Genetics 2016, 12, e1006363. [Google Scholar] [CrossRef] [PubMed]
- Angelovici, R.; Batushansky, A.; Deason, N.; Gonzalez-Jorge, S.; Gore, M.A.; Fait, A.; DellaPenna, D. Network-guided GWAS improves identification of genes affecting free amino acids. Plant Physiol. 2016, 173, 872–886. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tohge, T.; Cuadros-Inostroza, Á.; Tong, H.; Tenenboim, H.; Kooke, R.; Méret, M.; Keurentjes, J.B.; Nikoloski, Z.; Fernie, A.R.; et al. Mapping the Arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Mol. Plant 2018, 11, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Perez-Fons, L.; Wells, T.; Corol, D.I.; Ward, J.L.; Gerrish, C.; Beale, M.H.; Seymour, G.B.; Bramley, P.M.; Fraser, P.D. A genome-wide metabolomic resource for tomato fruit from Solanum pennellii. Sci. Rep. 2014, 4, 3859. [Google Scholar] [CrossRef] [PubMed]
- Mähler, N.; Wang, J.; Terebieniec, B.K.; Ingvarsson, P.K.; Street, N.R.; Hvidsten, T.R. Gene co-expression network connectivity is an important determinant of selective constraint. Plos Genetics 2017, 13, e1006402. [Google Scholar] [CrossRef] [PubMed]
- Dhawi, F.; Datta, R.; Ramakrishna, W. Proteomics provides insights into biological pathways altered by plant growth promoting bacteria and arbuscular mycorrhiza in Sorghum grown in marginal soil. Biochim. Biophys. Acta 2017, 1865, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Aiello, G.; Fasoli, E.; Boschin, G.; Lammi, C.; Zanoni, C.; Citterio, A.; Arnoldi, A. Proteomic characterization of hempseed (Cannabis sativa L.). J. Proteomics 2016, 147, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Colzani, M.; Altomare, A.; Caliendo, M.; Aldini, G.; Righetti, P.G.; Fasoli, E. The secrets of oriental panacea: Panax ginseng. J. Proteom. 2016, 130, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Blasi, A.R.; Buffon, G.; Rativa, A.G.; Lopes, M.C.B.; Berger, M.; Santi, L.; Lavallée-Adam, M.; Yates, J.R.; Schwambach, J.; Beys-da Silva, W.O.; et al. High infestation levels of Schizotetranychus oryzae severely affects rice metabolism. J. Plant Physiol. 2017, 219, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Pi, E.; Qu, L.; Hu, J.; Huang, Y.; Qiu, L.; Lu, H.; Jiang, B.; Liu, C.; Peng, T.; Zhao, Y.; et al. Mechanisms of Soybean Roots’ Tolerances to Salinity Revealed by Proteomic and Phosphoproteomic Comparisons Between Two Cultivars. Mol. Cell. Proteomics 2016, 219, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Buffon, G.; Blasi, A.R.; Adamski, J.M.; Ferla, N.J.; Berger, M.; Santi, L.; Lavallée-Adam, M.; Yates, J.R.; Beys-da Silva, W.O.; Sperotto, R.A. Physiological and molecular alterations promoted by Schizotetranychus oryzae mite infestation in rice leaves. J. Proteome Res. 2016, 15, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Vigani, G.; Di Silvestre, D.; Agresta, A.M.; Donnini, S.; Mauri, P.; Gehl, C.; Bittner, F.; Murgia, I. Molybdenum and iron mutually impact their homeostasis in cucumber (Cucumis sativus) plants. New Phytol. 2017, 213, 1222–1241. [Google Scholar] [CrossRef] [PubMed]
- Islam, N.; Li, G.; Garrett, W.M.; Lin, R.; Sriram, G.; Cooper, B.; Coleman, G.D. Proteomics of nitrogen remobilization in poplar bark. J. Proteome Res. 2015, 14, 1112–1126. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tian, X.; Wang, C.; Zeng, X.; Xing, Y.; Ling, H.; Yin, W.; Tian, L.; Meng, Z.; Zhang, J.; et al. SWATH label-free proteomics analyses revealed the roles of oxidative stress and antioxidant defensing system in sclerotia formation of Polyporus umbellatus. Sci. Rep. 2017, 7, 41283. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.Y.; Chan, W.L.; Chen, M.X.; Kong, R.P.W.; Cai, C.; Wang, Q.; Zhang, J.H.; Lo, C. SWATH-MS quantitative proteomic investigation reveals a role of jasmonic acid during lead response in Arabidopsis. J. Proteome Res. 2016, 15, 3528–3539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, D.; Yu, J.; Li, M.; Damaris, R.N.; Gupta, R.; Kim, S.T.; Yang, P. Analysis of dynamic protein carbonylation in rice embryo during germination through AP-SWATH. Proteomics 2016, 16, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Osman, K.; Yang, J.; Roitinger, E.; Lambing, C.; Heckmann, S.; Howell, E.; Cuacos, M.; Imre, R.; Dürnberger, G.; Mechtler, K.; et al. Affinity proteomics reveals extensive phosphorylation of the Brassica chromosome axis protein ASY1 and a network of associated proteins at prophase I of meiosis. Plant J. 2018, 93, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Senkler, J.; Senkler, M.; Eubel, H.; Hildebrandt, T.; Lengwenus, C.; Schertl, P.; Schwarzländer, M.; Wagner, S.; Wittig, I.; Braun, H.P. The mitochondrial complexome of Arabidopsis thaliana. Plant J. 2017, 89, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Ishikawa, S.; Matsunami, E.; Yamauchi, J.; Homma, K.; Faulkner, C.; Oparka, K.; Jisaka, M.; Nagaya, T.; Yokota, K.; et al. New gateway-compatible vectors for a high-throughput protein-protein interaction analysis by a bimolecular fluorescence complementation (BiFC) assay in plants and their application to a plant clathrin structure analysis. Biosci. Biotechnol. Biochem. 2015, 79, 1995–2006. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Liu, S.; Li, L.; Zuo, K.; Zhao, L.; Zhang, L. Genome-wide inference of protein-protein interaction networks identifies crosstalk in abscisic acid signaling. Plant Physiol. 2016, 171, 1511–1522. [Google Scholar] [PubMed]
- Sahu, S.S.; Weirick, T.; Kaundal, R. Predicting genome-scale Arabidopsis-Pseudomonas syringae interactome using domain and interolog-based approaches. BMC Bioinformatics 2014, 15, S13. [Google Scholar] [CrossRef] [PubMed]
- Rodgers-Melnick, E.; Culp, M.; DiFazio, S.P. Predicting whole genome protein interaction networks from primary sequence data in model and non-model organisms using ENTS. BMC Genomics 2013, 14, 608. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Gu, H.; Jiao, Y.; Huang, D.; Chen, M. Computational identification of protein-protein interactions in rice based on the predicted rice interactome network. Genomics Proteomics & Bioinformatics 2011, 9, 128–137. [Google Scholar]
- Scardoni, G.; Tosadori, G.; Faizan, M.; Spoto, F.; Fabbri, F.; Laudanna, C. Biological network analysis with CentiScaPe: centralities and experimental dataset integration. F1000Res. 2014, 3, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, A.; Blank, L.M. Systems biology: Hypothesis-driven omics integration. Nat. Chem. Biol. 2010, 6, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Serin, E.A.R.; Nijveen, H.; Hilhorst, H.W.M.; Ligterink, W. Learning from co-expression networks: Possibilities and challenges. Front. Plant Sci. 2016, 7, 444. [Google Scholar] [CrossRef] [PubMed]
- Pajoro, A.; Biewers, S.; Dougali, E.; Leal Valentim, F.; Mendes, M.A.; Porri, A.; Coupland, G.; Van de Peer, Y.; Van Dijk, A.D.; Colombo, L.; et al. The (r) evolution of gene regulatory networks controlling Arabidopsis plant reproduction: A two-decade history. J. Exp. Bot. 2014, 65, 4731–4745. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bhadauria, V.; Ma, B. ChIP-Seq: A powerful tool for studying protein-DNA interactions in plants. Curr. Issues Mol. Biol. 2017, 27, 171–180. [Google Scholar] [PubMed]
- Hartmann, A.; Jozefowicz, A.M. VANTED: A tool for integrative visualization and analysis of -omics data. Methods Mol. Biol. 2018, 1696, 261–278. [Google Scholar] [PubMed]
- Zhu, L.; Zhang, Y.H.; Su, F.; Chen, L.; Huang, T.; Cai, Y.D. A shortest-path-based method for the analysis and prediction of fruit-related genes in Arabidopsis thaliana. PLoS ONE 2016, 11, e0159519. [Google Scholar] [CrossRef] [PubMed]
- Unamba, C.I.N.; Nag, A.; Sharma, R.K. Next generation sequencing technologies: The toorway to the unexplored genomics of non-model plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef] [PubMed]
- Deyholos, M.K. Making the most of drought and salinity transcriptomics. Plant Cell Environ. 2010, 33, 648–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.; Kim, H.; Lee, I. Network-assisted crop systems genetics: network inference and integrative analysis. Curr. Opin. Plant Biol. 2015, 24, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Feltus, F.A. Systems genetics: a paradigm to improve discovery of candidate genes and mechanisms underlying complex traits. Plant Sci. 2014, 223, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Ma, Y.; Wu, S.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.; Zhou, Z.; Yu, H.; Zhang, M.; et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Langfelder, P.; Horvath, S. Comparison of co-expression measures: Mutual information, correlation, and model based indices. BMC Bioinformatics 2012, 13, 328. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Sadhukhan, A.; Tazib, T.; Nakano, Y.; Kusunoki, K.; Kamara, M.; Chaffai, R.; Iuchi, S.; Sahoo, L.; Kobayashi, M.; et al. Joint genetic and network analyses identify loci associated with root growth under NaCl stress in Arabidopsis thaliana. Plant Cell Environ. 2016, 39, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Lee, I. araGWAB: Network-based boosting of genome-wide association studies in Arabidopsis thaliana. Sci. Rep. 2018, 8, 2925. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gong, F.; Wang, W. Protein extraction from plant tissues for 2DE and its application in proteomic analysis. Proteomics 2014, 14, 645–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorrín-Novo, J.V.; Pascual, J.; Sánchez-Lucas, R.; Romero-Rodríguez, M.C.; Rodríguez-Ortega, M.J.; Lenz, C.; Valledor, L. Fourteen years of plant proteomics reflected in Proteomics: Moving from model species and 2DE-based approaches to orphan species and gel-free platforms. Proteomics 2015, 15, 1089–1112. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Hyun, T.K.; Kumar, D.; Rim, Y.; Chen, X.Y.; Jo, Y.; Kim, S.; Lee, K.W.; Park, Z.Y.; Lucas, W.J.; et al. Proteomic analysis to identify tightly-bound cell wall protein in rice calli. Mol. Cells 2015, 38, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, P.; Doerfler, H.; Jegadeesan, S.; Ghatak, A.; Pressman, E.; Castillejo, M.A.; Wienkoop, S.; Egelhofer, V.; Firon, N.; Weckwerth, W. Heat-Treatment-Responsive Proteins in Different Developmental Stages of Tomato Pollen Detected by Targeted Mass Accuracy Precursor Alignment (tMAPA). J. Proteome Res. 2015, 14, 4463–4471. [Google Scholar] [CrossRef] [PubMed]
- Distler, U.; Kuharev, J.; Navarro, P.; Levin, Y.; Schild, H.; Tenzer, S. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 2014, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Lu, X.; Yu, Z.; Li, X.; Ma, W.; Yan, Y. Rapid separation of seed gliadins by reversed-phase ultra performance liquid chromatography (RP-UPLC) and its application in wheat cultivar and germplasm identification. Biosci. Biotechnol. Biochem. 2015, 79, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, A.; Noble, W.S.; Wolf-Yadlin, A. Technical advances in proteomics: New developments in data-independent acquisition. F1000Res. 2016, 5, 419. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; McReynolds, M.R.; Walley, J.W. Sample preparation protocols for protein abundance, acetylome, and phosphoproteome profiling of plant tissues. Methods Mol. Biol. 2017, 1610, 123–133. [Google Scholar] [PubMed]
- Bilbao, A.; Varesio, E.; Luban, J.; Strambio-De-Castillia, C.; Hopfgartner, G.; Müller, M.; Lisacek, F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomics 2015, 15, 964–980. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, K.; Mbeunkui, F.; Mitra, S.K.; Mentzel, T.; Goshe, M.B. Improving protein and proteome coverage through data-independent multiplexed peptide fragmentation. J. Proteome Res. 2010, 9, 3621–3637. [Google Scholar] [CrossRef] [PubMed]
- Melo-Braga, M.N.; Verano-Braga, T.; León, I.R.; Antonacci, D.; Nogueira, F.C.; Thelen, J.J.; Larsen, M.R.; Palmisano, G. Modulation of protein phosphorylation, N-glycosylation and Lys-acetylation in grape (Vitis vinifera) mesocarp and exocarp owing to Lobesia botrana infection. Mol. Cell. Proteomics 2012, 11, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Bolger, M.E.; Weisshaar, B.; Scholz, U.; Stein, N.; Usadel, B.; Mayer, K.F.X. Plant genome sequencing-applications for crop improvement. Curr. Opin. Biotechnol. 2014, 26, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J.; Trapp, J.; Pible, O.; Geffard, O.; Chaumot, A.; Hartmann, E.M. Non-model organisms, a species endangered by proteogenomics. J. Proteomics 2014, 105, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I. Protein identification by tandem mass spectrometry and sequence database searching. Methods Mol. Biol. 2007, 367, 87–119. [Google Scholar] [PubMed]
- Muth, T.; Rapp, E.; Berven, F.S.; Barsnes, H.; Vaudel, M. Tandem mass spectrum sequencing: An alternative to database search engines in shotgun proteomics. Adv. Exp. Med. Biol. 2016, 919, 217–226. [Google Scholar] [PubMed]
- Ye, X.; Zhao, N.; Yu, X.; Han, X.; Gao, H.; Zhang, X. Extensive characterization of peptides from Panax ginseng CA Meyer using mass spectrometric approach. Proteomics 2016, 16, 2788–2791. [Google Scholar] [CrossRef] [PubMed]
- Grobei, M.A.; Qeli, E.; Brunner, E.; Rehrauer, H.; Zhang, R.; Roschitzki, B.; Basler, K.; Ahrens, C.H.; Grossniklaus, U. Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res. 2009, 19, 1786–1800. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fonslow, B.R.; Shan, B.; Baek, M.C.; Yates, J.R., III. Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 2013, 113, 2343–2394. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, C.; Di Silvestre, D.; Fischer-Schliebs, E.; Homann, U.; De Palma, A.; Comunian, C.; Mauri, P.L.; Thiel, G. Proteomic analysis of Mesembryanthemum crystallinum leaf microsomal fractions finds an imbalance in V-ATPase stoichiometry during the salt-induced transition from C3 to CAM. Biochem. J. 2013, 450, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.K.; Chen, X.Y.; Chu, H.; Rim, Y.; Kim, S.; Kim, S.T.; Kim, S.W.; Park, Z.Y.; Kim, J.Y. Proteomic analysis of the secretome of rice calli. Physiol. Plant 2009, 135, 331–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.; Li, L.; Zhou, X.; Stanley, B.; Ma, H. Analysis of the Arabidopsis floral proteome: Detection of over 2000 proteins and evidence for posttranslational modifications. J. Integr. Plant Biol. 2009, 51, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.K.; Huang, E.L.; Low-Décarie, E.; Lefsrud, M.G. Comparative shotgun proteomic analysis of wastewater-cultured microalgae: Nitrogen sensing and carbon fixation for growth and nutrient removal in Chlamydomonas reinhardtii. J. Proteome Res. 2015, 14, 3051–3067. [Google Scholar] [CrossRef] [PubMed]
- Barkla, BJ.; Vera-Estrella, R.; Hernández-Coronado, M.; Pantoja, O. Quantitative proteomics of the tonoplast reveals a role for glycolytic enzymes in salt tolerance. Plant Cell. 2009, 21, 4044–4058. [Google Scholar] [CrossRef] [PubMed]
- Donnini, S.; Prinsi, B.; Negri, A.S.; Vigani, G.; Espen, L.; Zocchi, G. Comparative shotgun proteomic analysis of wastewater-cultured microalgae: Nitrogen sensing and carbon fixation for growth and nutrient removal in Chlamydomonas reinhardtii. BMC Plant Biol. 2010, 10, 268. [Google Scholar]
- Di Silvestre, D.; Brambilla, F.; Agnetti, G.; Mauri, P. Bottom-up proteomics. In Manual of Cardiovascular Proteomics; Springer: Berlin, Germany, 2016; pp. 155–185. [Google Scholar]
- Parsons, H.T.; Heazlewood, J.L. Beyond the western front: targeted proteomics and organelle abundance profiling. Front. Plant Sci. 2015, 6, 301. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Mohareb, F.; Jones, A.M.E.; Bessant, C. MRMaid: The SRM assay design tool for Arabidopsis and other species. Front. Plant Sci. 2012, 3, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majovsky, P.; Naumann, C.; Lee, C.W.; Lassowskat, I.; Trujillo, M.; Dissmeyer, N.; Hoehenwarter, W. Targeted proteomics analysis of protein degradation in plant signaling on an LTQ-Orbitrap mass spectrometer. J. Proteome Res. 2014, 13, 4246–4258. [Google Scholar] [CrossRef] [PubMed]
- Bourmaud, A.; Gallien, S.; Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 2016, 15, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Kuharev, J.; Gillet, L.C.; Bernhardt, O.M.; MacLean, B.; Röst, H.L.; Tate, S.A.; Tsou, C.C.; Reiter, L.; Distler, U.; et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 2016, 34, 1130. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Du, L.; Li, L.; Palmer, L.C.; Forney, C.F.; Fillmore, S.; Zhang, Z.; Li, X. Targeted quantitative proteomic investigation employing multiple reaction monitoring on quantitative changes in proteins that regulate volatile biosynthesis of strawberry fruit at different ripening stages. J. Proteomics 2015, 126, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.B.B.; Sherwood, R.W.; Nicklay, J.J.; Yang, Y.; Muratore-Schroeder, T.L.; Anderson, E.T.; Thannhauser, T.W.; Rose, J.K.C.; Zhang, S. Application of wide selected-ion monitoring data-independent acquisition to identify tomato fruit proteins regulated by the CUTIN DEFICIENT2 transcription factor. Proteomics 2016, 16, 2081–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Ness, L.K.; Jayaraman, D.; Maeda, J.; Barrett-Wilt, G.A.; Sussman, M.R.; Ané, J.M. Mass spectrometric-based selected reaction monitoring of protein phosphorylation during symbiotic signaling in the model legume, Medicago truncatula. PLoS ONE 2016, 11, e0155460. [Google Scholar] [CrossRef] [PubMed]
- Rogniaux, H.; Pavlovic, M.; Lupi, R.; Lollier, V.; Joint, M.; Mameri, H.; Denery, S.; Larré, C. Allergen relative abundance in several wheat varieties as revealed via a targeted quantitative approach using MS. Proteomics 2015, 15, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Colgrave, M.L.; Goswami, H.; Byrne, K.; Blundell, M.; Howitt, C.A.; Tanner, G.J. Proteomic profiling of 16 cereal grains and the application of targeted proteomics to detect wheat contamination. J. Proteome Res. 2015, 14, 2659–2668. [Google Scholar] [CrossRef] [PubMed]
- Tsou, C.C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.C.; Nesvizhskii, A.I. DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258. [Google Scholar] [CrossRef] [PubMed]
- Fabre, B.; Korona, D.; Mata, C.I.; Parsons, H.T.; Deery, M.J.; Hertog, M.L.A.T.M.; Nicolaï, B.M.; Russell, S.; Lilley, K.S. Spectral libraries for SWATH-MS assays for Drosophila melanogaster and Solanum lycopersicum. Proteomics 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.Y.; Chen, M.X.; Su, Y.W.; Xu, X.; Ye, N.H.; Cao, Y.Y.; Lin, S.; Liu, T.Y.; Li, H.X.; Wang, G.Q.; et al. SWATH-MS quantitative analysis of proteins in the rice inferior and superior spikelets during grain filling. Front. Plant Sci. 2016, 7, 1926. [Google Scholar] [CrossRef] [PubMed]
- Kersten, B.; Feilner, T. Generation of plant protein microarrays and investigation of antigen-antibody interactions. In Plant Proteomics In Methods in Molecular Biology; Springer: Berlin, Germany, 2007. [Google Scholar]
- Nelson, C.J.; Li, L.; Millar, A.H. Quantitative analysis of protein turnover in plants. Proteomics 2014, 14, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Blein-Nicolas, M.; Zivy, M. Thousand and one ways to quantify and compare protein abundances in label-free bottom-up proteomics. Biochim. Biophys. Acta 2016, 1864, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, C.; Thomanek, N.; Hundt, F.; Lerari, T.; Meyer, H.E.; Wolters, D.; Marcus, K. Strategies in relative and absolute quantitative mass spectrometry based proteomics. Biol. Chem. 2017, 398, 687–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Lam, H. Graph-based peak alignment algorithms for multiple liquid chromatography-mass spectrometry datasets. Bioinformatics 2013, 29, 2469–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Silvestre, D.; Brambilla, F.; Motta, S.; Mauri, P. Evaluation of Proteomic Data: From Profiling to Network Analysis by Way of Biomarker Discovery. In Biomarker Validation, Technological, Clinical and Commercial Aspects; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015. [Google Scholar]
- Gokce, E.; Shuford, C.M.; Franck, W.L.; Dean, R.A.; Muddiman, D.C. Evaluation of normalization methods on GeLC-MS/MS label-free spectral counting data to correct for variation during proteomic workflows. J. Am. Soc. Mass Spectrom. 2011, 22, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Nestler, H.; Groh, K.J.; Schönenberger, R.; Eggen, R.I.; Suter, M.J.F. Linking proteome responses with physiological and biochemical effects in herbicide-exposed Chlamydomonas reinhardtii. J. Proteomics 2012, 75, 5370–5385. [Google Scholar] [CrossRef] [PubMed]
- Ponnala, L.; Wang, Y.; Sun, Q.; Wijk, K.J. Correlation of mRNA and protein abundance in the developing maize leaf. Plant J. 2014, 78, 424–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.W.; Hsu, Y.K.; Cheng, Y.H.; Yen, H.C.; Wu, Y.P.; Wang, C.S.; Lai, C.C. Proteomic analysis of salt-responsive ubiquitin-related proteins in rice roots. Rapid Commun. Mass Spectrom. 2012, 26, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Ferro, M.; Brugière, S.; Salvi, D.; Seigneurin-Berny, D.; Court, M.; Moyet, L.; Ramus, C.; Miras, S.; Mellal, M.; Le Gall, S.; et al. AT CHLORO, a comprehensive chloroplast proteome database with subplastidial localization and curated information on envelope proteins. Mol. Cell. Proteomics 2010, 9, 1063–1084. [Google Scholar] [CrossRef] [PubMed]
- Gammulla, C.G.; Pascovici, D.; Atwell, B.J.; Haynes, P.A. Differential proteomic response of rice (Oryza sativa) leaves exposed to high- and low-temperature stress. Proteomics 2011, 11, 2839–2850. [Google Scholar] [CrossRef] [PubMed]
- Salvato, F.; Havelund, J.F.; Chen, M.; Rao, R.S.P.; Rogowska-Wrzesinska, A.; Jensen, O.N.; Gang, D.R.; Thelen, J.J.; Møller, I.M. The potato tuber mitochondrial proteome. Plant Physiol. 2014, 164, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Luo, Z.; Huang, X.; Zhang, L.; Zhao, P.; Ma, H.; Li, X.; Ban, Z.; Liu, X. Label-free quantitative proteomics to investigate strawberry fruit proteome changes under controlled atmosphere and low temperature storage. J. Proteomics 2015, 120, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Fesenko, I.; Seredina, A.; Arapidi, G.; Ptushenko, V.; Urban, A.; Butenko, I.; Kovalchuk, S.; Babalyan, K.; Knyazev, A.; Khazigaleeva, R.; et al. The Physcomitrella patens chloroplast proteome changes in response to protoplastation. Front. Plant Sci. 2016, 7, 1661. [Google Scholar] [CrossRef] [PubMed]
- Fares, A.; Nespoulous, C.; Rossignol, M.; Peltier, J.B. Simultaneous identification and quantification of nitrosylation sites by combination of biotin switch and ICAT labeling. Plant Proteom. 2014, 1072, 609–620. [Google Scholar]
- Puyaubert, J.; Fares, A.; Rézé, N.; Peltier, J.B.; Baudouin, E. Identification of endogenously S-nitrosylated proteins in Arabidopsis plantlets: Effect of cold stress on cysteine nitrosylation level. Plant Sci. 2014, 215, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Sun, F.; Gao, X.; Xie, K.; Zhang, C.; Liu, S.; Xi, Y. Proteomic analysis of melatonin-mediated osmotic tolerance by improving energy metabolism and autophagy in wheat (Triticum aestivum L.). Planta 2018, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.A.; Zhang, K.; Islam, F.; Wang, J.; Athar, H.U.; Nawaz, A.; Ullah Zafar, Z.; Xu, J.; Zhou, W. Physiological and iTRAQ based quantitative proteomics analysis of methyl jasmonate induced tolerance in Brassica napus under arsenic stress. Proteomics 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.X.; Luo, Y.M.; Ye, Z.Q.; Cao, X.; Liang, J.N.; Wang, Q.; Wu, Y.; Wu, J.H.; Wang, H.Y.; Zhang, M.; et al. iTRAQ-based proteomics analysis of autophagy-mediated immune responses against the vascular fungal pathogen Verticillium dahliae in Arabidopsis. Autophagy 2018, 14, 598–618. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhang, S.; Wang, J.; Hu, Y. Quantitative proteomic analysis of wheat seeds during artificial ageing and priming using the isobaric tandem mass tag labeling. PLoS ONE 2016, 11, e0162851. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, F.C.; Palmisano, G.; Schwämmle, V.; Soares, E.L.; Soares, A.A.; Roepstorff, P.; Domont, G.B.; Campos, F.A. Isotope labeling-based quantitative proteomics of developing seeds of castor oil seed (Ricinus communis L.). J. Proteome Res. 2013, 12, 5012–5024. [Google Scholar] [CrossRef] [PubMed]
- Vélez-Bermúdez, I.C.; Wen, T.N.; Lan, P.; Schmidt, W. Isobaric tag for relative and absolute quantitation (iTRAQ)-based protein profiling in plants. Methods Mol. Biol. 2016, 1450, 213–221. [Google Scholar] [PubMed]
- Nelson, C.J.; Hegeman, A.D.; Harms, A.C.; Sussman, M.R. A quantitative analysis of Arabidopsis plasma membrane using trypsin-catalyzed 18O labeling. Mol. Cell. Proteomics 2006, 5, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, D.; ten Have, S.; Hodge, K.; Tillemans, V.; Lamond, A.I.; Brown, J.W. Plant SILAC: Stable-isotope labelling with amino acids of Arabidopsis seedlings for quantitative proteomics. PLoS ONE 2013, 8, e72207. [Google Scholar] [CrossRef] [PubMed]
- Matthes, A.; Köhl, K.; Schulze, W.X. SILAC and alternatives in studying cellular proteomes of plants. In Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC); Springer: Berlin, Germany, 2014; pp. 65–83. [Google Scholar]
- Calderón-Celis, F.; Encinar, J.R.; Sanz-Medel, A. Standardization approaches in absolute quantitative proteomics with mass spectrometry. Mass Spectrom. Rev. 2017, 9999, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, N.; Rao, R.S.P.; Gruppuso, P.A.; Ramratnam, B.; Salomon, A.R. Targeted proteomics: current status and future perspectives for quantification of food allergens. J. Proteomics 2016, 143, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hanke, S.; Besir, H.; Oesterhelt, D.; Mann, M. Absolute SILAC for accurate quantitation of proteins in complex mixtures down to the attomole level. J. Proteome Res. 2008, 7, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kirchner, M.; Steen, J.A.; Steen, H. A.; Steen, H. A practical guide to the FLEXIQuant method. In Quantitative Methods in Proteomics; Springer: Berlin, Germany, 2012; pp. 295–319. [Google Scholar]
- Zeiler, M.; Straube, W.L.; Lundberg, E.; Uhlen, M.; Mann, M. A protein epitope signature tag (PrEST) library allows SILAC-based absolute quantification and multiplexed determination of protein copy numbers in cell lines. Mol. Cell. Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Kito, K.; Okada, M.; Ishibashi, Y.; Okada, S.; Ito, T. A strategy for absolute proteome quantification with mass spectrometry by hierarchical use of peptide-concatenated standards. Proteomics 2016, 16, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. Bioessays 2013, 35, 1050–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiśniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Heo, M.; Maslov, S.; Shakhnovich, E. Topology of protein interaction network shapes protein abundances and strengths of their functional and nonspecific interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 4258–4263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weckwerth, W.; Kahl, G. The Handbook of Plant Metabolomics; John Wiley Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- Yonekura-Sakakibara, K.; Saito, K. Functional genomics for plant natural product biosynthesis. Nat. Prod. Rep. 2009, 26, 1466–1487. [Google Scholar] [CrossRef] [PubMed]
- Schläpfer, P.; Zhang, P.; Wang, C.; Kim, T.; Banf, M.; Chae, L.; Dreher, K.; Chavali, A.K.; Nilo-Poyanco, R.; Bernard, T.; et al. Genome-wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiol. 2017, 173, 2041–2059. [Google Scholar] [CrossRef] [PubMed]
- Moco, S.; Bino, R.J.; Vorst, O.; Verhoeven, H.A.; de Groot, J.; van Beek, T.A.; Vervoort, J.; De Vos, C.R. A liquid chromatography-mass spectrometry-based metabolome database for tomato. Plant Physiol. 2006, 141, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Joshi, T.; Yao, Q.; Levi, D.F.; Brechenmacher, L.; Valliyodan, B.; Stacey, G.; Nguyen, H.; Xu, D. SoyMetDB: the soybean metabolome database. In Proceedings of the 2010 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Hong Kong, China, 18–21 December 2010; pp. 203–208. [Google Scholar]
- Fukushima, A.; Kusano, M.; Mejia, R.F.; Iwasa, M.; Kobayashi, M.; Hayashi, N.; Watanabe-Takahashi, A.; Narisawa, T.; Tohge, T.; Hur, M.; et al. Metabolomic characterization of knockout mutants in Arabidopsis: Development of a metabolite profiling database for knockout mutants in Arabidopsis. Plant Physiol. 2014, 165, 948–961. [Google Scholar] [CrossRef] [PubMed]
- Ferry-Dumazet, H.; Gil, L.; Deborde, C.; Moing, A.; Bernillon, S.; Rolin, D.; Nikolski, M.; De Daruvar, A.; Jacob, D. MeRy-B: A web knowledgebase for the storage, visualization, analysis and annotation of plant NMR metabolomic profiles. BMC Plant Biol. 2011, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Commisso, M.; Bianconi, M.; Di Carlo, F.; Poletti, S.; Bulgarini, A.; Munari, F.; Negri, S.; Stocchero, M.; Ceoldo, S.; Avesani, L.; et al. Multi-approach metabolomics analysis and artificial simplified phytocomplexes reveal cultivar-dependent synergy between polyphenols and ascorbic acid in fruits of the sweet cherry (Prunus avium L.). PLoS ONE 2017, 12, e0180889. [Google Scholar] [CrossRef] [PubMed]
- Boiteau, R.M.; Hoyt, D.W.; Nicora, C.D.; Kinmonth-Schultz, H.A.; Ward, J.K.; Bingol, K. Structure elucidation of unknown metabolites in metabolomics by combined NMR and MS/MS prediction. Metabolites 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; de Souza, L.P.; Fernie, A.R. Genome-enabled plant metabolomics. J. Chromatogr. B 2014, 966, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Mounet, F.; Moing, A.; Garcia, V.; Petit, J.; Maucourt, M.; Deborde, C.; Bernillon, S.; Le Gall, G.; Colquhoun, I.; Defernez, M.; et al. Gene and metabolite regulatory network analysis of early developing fruit tissues highlights new candidate genes for the control of tomato fruit composition and development. Plant Physiol. 2009, 149, 1505–1528. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant metabolomics: An indispensable system biology tool for plant science. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Trimigno, A.; Marincola, F.C.; Dellarosa, N.; Picone, G.; Laghi, L. Definition of food quality by NMR-based foodomics. Curr. Opin. Food Sci. 2015, 4, 99–104. [Google Scholar] [CrossRef]
- Le Gall, H.; Fontaine, J.X.; Molinié, R.; Pelloux, J.; Mesnard, F.; Gillet, F.; Fliniaux, O. NMR-based metabolomics to study the cold-acclimation strategy of two Miscanthus genotypes. Phytochem. Anal. 2017, 28, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, O.; Annarao, S.; Pathre, U.; Snehi, S.; Raj, S.; Roy, R.; Tuli, R.; Khetrapal, C. Metabolic and histopathological alterations of Jatropha mosaic begomovirus-infected Jatropha curcas L. by HR-MAS NMR spectroscopy and magnetic resonance imaging. Planta 2010, 232, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, D.R.; Wang, Q.; Friedt, W.; Spengler, B.; Gottwald, S.; Römpp, A. High resolution mass spectrometry imaging of plant tissues: Towards a plant metabolite atlas. Analyst 2015, 140, 7696–7709. [Google Scholar] [CrossRef] [PubMed]
- Allwood, J.W.; Parker, D.; Beckmann, M.; Draper, J.; Goodacre, R. Fourier transform ion cyclotron resonance mass spectrometry for plant metabolite profiling and metabolite identification. In Plant Metabolomics; Springer: Berlin, Germany, 2011; pp. 157–176. [Google Scholar]
- Jorge, T.F.; Mata, A.T.; António, C. Mass spectrometry as a quantitative tool in plant metabolomics. Philos. Trans. A Math. Phys. Eng. Sci. 2016, 374, 20150370. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Nägele, T.; Doerfler, H.; Fragner, L.; Chaturvedi, P.; Nukarinen, E.; Bellaire, A.; Huber, W.; Weiszmann, J.; Engelmeier, D.; et al. System level analysis of cacao seed ripening reveals a sequential interplay of primary and secondary metabolism leading to polyphenol accumulation and preparation of stress resistance. Plant J. 2016, 87, 318–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Sun, X.; Weiszmann, J.; Weckwerth, W. System-level and granger network analysis of integrated proteomic and metabolomic dynamics identifies key points of grape berry development at the interface of primary and secondary metabolism. Front. Plant Sci. 2017, 8, 1066. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.Q.; Zou, L.; Yin, X.X.; Ong, C.N. HILIC-MS for metabolomics: An attractive and complementary approach to RPLC-MS. Mass Spectrom. Rev. 2016, 35, 574–600. [Google Scholar] [CrossRef] [PubMed]
- Lisec, J.; Schauer, N.; Kopka, J.; Willmitzer, L.; Fernie, A.R. Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387. [Google Scholar] [CrossRef] [PubMed]
- Meijón, M.; Feito, I.; Oravec, M.; Delatorre, C.; Weckwerth, W.; Majada, J.; Valledor, L. Exploring natural variation of Pinus pinaster Aiton using metabolomics: Is it possible to identify the region of origin of a pine from its metabolites? Mol. Ecol. 2016, 25, 959–976. [Google Scholar] [CrossRef] [PubMed]
- Hoermiller, I.I.; Naegele, T.; Augustin, H.; Stutz, S.; Weckwerth, W.; Heyer, A.G. Subcellular reprogramming of metabolism during cold acclimation in Arabidopsis thaliana. Plant Cell Environ. 2017, 40, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Lam, H.; Pi, E.; Zhan, Q.; Tsai, S.; Wang, C.; Kwan, Y.; Ngai, S. Comparative metabolomics in Glycine max and Glycine soja under salt stress to reveal the phenotypes of their offspring. J. Agric. Food Chem. 2013, 61, 8711–8721. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, Y.; Hu, C.; Zhao, C.; Zhang, J.; Li, L.; Zeng, J.; Peng, X.; Lu, X.; Xu, G. Metabolic profiling with gas chromatography—mass spectrometry and capillary electrophoresis–mass spectrometry reveals the carbon—nitrogen status of tobacco leaves across different planting areas. J. Proteome Res. 2016, 15, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Begou, O.; Gika, H.; Wilson, I.; Theodoridis, G. Hyphenated MS-based targeted approaches in metabolomics. Analyst 2017, 142, 3079–3100. [Google Scholar] [CrossRef] [PubMed]
- Dinh, J.L.; Farcot, E.; Hodgman, C. The logic of the floral transition: Reverse-engineering the switch controlling the identity of lateral organs. PLoS Comput. Biol. 2017, 13, e1005744. [Google Scholar] [CrossRef] [PubMed]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Lavallée-Adam, M.; Rauniyar, N.; McClatchy, D.B.; Yates, J.R. PSEA-Quant: A protein set enrichment analysis on label-free and label-based protein quantification data. J. Proteome Res. 2014, 13, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P. Reverse engineering of genome-wide gene regulatory networks from gene expression data. Curr. Genomics 2015, 16, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Weirauch, M.T. Gene coexpression networks for the analysis of DNA microarray data. In Applied Statistics for Network Biology: Methods in Systems Biology; Wiley-Blackwell: Hoboken, NJ, USA, 2011; pp. 215–250. [Google Scholar]
- Shen, P.C.; Hour, A.L.; Liu, L.Y.D. Microarray meta-analysis to explore abiotic stress-specific gene expression patterns in Arabidopsis. Bot. Stud. 2017, 58, 22. [Google Scholar] [CrossRef] [PubMed]
- Arhondakis, S.; Bita, C.E.; Perrakis, A.; Manioudaki, M.E.; Krokida, A.; Kaloudas, D.; Kalaitzis, P. In silico transcriptional regulatory networks involved in tomato fruit ripening. Front. Plant Sci. 2016, 7, 1234. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ball, G.; Hodgman, C.; Coules, A.; Zhao, H.; Lu, C. Analysis of gene regulatory networks of maize in response to nitrogen. Genes 2018, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.Y.; Mutwil, M. Towards revealing the functions of all genes in plants. Trends Plant Sci. 2014, 19, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Cheng, D.; Yang, Y.; Zhang, G.; Qin, M.; Chen, J.; Chen, Y.; Jiang, M. Co-expression network analysis of the transcriptomes of rice roots exposed to various cadmium stresses reveals universal cadmium-responsive genes. BMC Plant Biol. 2017, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Cao, M.; Zheng, J.; Zhao, Y.; Zheng, Z.L. Gene co-expression network analysis of fruit transcriptomes uncovers a possible mechanistically distinct class of sugar/acid ratio-associated genes in sweet orange. BMC Plant Biol. 2017, 17, 186. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Zhang, Z.; Wang, Z.; Yu, J.; Qin, H.; Mao, X.; Jiang, H.; Xin, D.; Yin, Z.; Zhu, R.; et al. Meta-analysis and transcriptome profiling reveal hub genes for soybean seed storage composition during seed development. Plant Cell Environ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Paci, P.; Colombo, T.; Fiscon, G.; Gurtner, A.; Pavesi, G.; Farina, L. SWIM: A computational tool to unveiling crucial nodes in complex biological networks. Sci. Rep. 2017, 7, 44797. [Google Scholar] [CrossRef] [PubMed]
- Massonnet, M.; Fasoli, M.; Tornielli, G.B.; Altieri, M.; Sandri, M.; Zuccolotto, P.; Paci, P.; Gardiman, M.; Zenoni, S.; Pezzotti, M. Ripening transcriptomic program in red and white grapevine varieties correlates with berry skin anthocyanin accumulation. Plant Physiol. 2017, 174, 2376–2396. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.N.; DeClerck, G.; Greenberg, A.; Clark, R.; McCouch, S. Next-generation phenotyping: requirements and strategies for enhancing our understanding of genotype–phenotype relationships and its relevance to crop improvement. Theor. Appl. Genet. 2013, 126, 867–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.L.; Baratt, A.; Baric, R.S.; Kawaoka, Y.; Smith, R.D.; Orwoll, E.S.; Katze, M.G.; McWeeney, S.K. Protein co-expression network analysis (ProCoNA). J. Clin. Bioinform. 2013, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, C.; Gatto, L.; Ferro, M.; Bruley, C.; Burger, T. Accounting for the multiple natures of missing values in label-free quantitative proteomics data sets to compare imputation strategies. J. Proteome Res. 2016, 15, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.L.; Gralinski, L.; Baric, R.S.; McWeeney, S.K. Multi-omic network signatures of disease. Front. Genet. 2014, 4, 309. [Google Scholar] [CrossRef] [PubMed]
- Braun, P.; Aubourg, S.; Van Leene, J.; De Jaeger, G.; Lurin, C. Plant protein interactomes. Annu. Rev. Plant Biol. 2013, 64, 161–187. [Google Scholar] [CrossRef] [PubMed]
- Legrand, J.; Léger, J.B.; Robin, S.; Vernoux, T.; Guédon, Y. Modelling the influence of dimerisation sequence dissimilarities on the auxin signalling network. BMC Syst. Biol. 2016, 10, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, S.; Wallmeroth, N.; Berendzen, K.W.; Grefen, C. Techniques for the Analysis of Protein-Protein Interactions in Vivo. Plant Physiol. 2015, 171, 727–758. [Google Scholar] [CrossRef] [PubMed]
- Schäferling, M.; Nagl, S. Förster resonance energy transfer methods for quantification of protein-protein interactions on microarrays. Methods Mol. Biol. 2011, 723, 303–320. [Google Scholar] [PubMed]
- Smits, A.H.; Vermeulen, M. Characterizing protein-protein interactions using mass spectrometry: Challenges and opportunities. Trends Biotechnol. 2016, 34, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Larsen, B.; Lin, Z.Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.C.; Nesvizhskii, A.I. SAINT: Probabilistic scoring of affinity purification–mass spectrometry data. Nat. Methods 2011, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.H.; Jansen, P.W.; Poser, I.; Hyman, A.A.; Vermeulen, M. Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res. 2012, 41, e28–e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holding, A.N. XL-MS: Protein cross-linking coupled with mass spectrometry. Methods 2015, 89, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Yu, F.; Yang, Z.; Liu, S.; Dai, C.; Lu, X.; Liu, C.; Yu, W.; Li, N. In planta chemical cross-linking and mass spectrometry analysis of protein structure and interaction in Arabidopsis. Proteomics 2016, 16, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Aryal, U.K.; McBride, Z.; Chen, D.; Xie, J.; Szymanski, D.B. Analysis of protein complexes in Arabidopsis leaves using size exclusion chromatography and label-free protein correlation profiling. J. Proteomics 2017, 166, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.H.; Go, K.D.; Bisteau, X.; Dai, L.; Yong, C.H.; Prabhu, N.; Ozturk, M.B.; Lim, Y.T.; Sreekumar, L.; Lengqvist, J.; et al. Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 2018, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Lei, D.; Lin, R.; Yin, C.; Li, P.; Zheng, A. Global protein-protein interaction network of rice sheath blight pathogen. J. Proteome Res. 2014, 13, 3277–3293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, L.; Liu, Y.; Zhang, Y.; Wang, S. Identifying novel fruit-related genes in Arabidopsis thaliana based on the random walk with restart algorithm. PLoS ONE 2017, 12, e0177017. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Zhao, J.; Cai, S.; Qian, H.; Zuo, K.; Zhao, L.; Zhang, L. A computational interactome for prioritizing genes associated with complex agronomic traits in rice (Oryza sativa). Plant J. 2017, 90, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; He, X.; Chan, E.; Xu, H.; Zhang, J. Measuring the evolutionary rate of protein-protein interaction. Proc. Natl. Acad. Sci. USA 2011, 108, 8725–8730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kshirsagar, M.; Schleker, S.; Carbonell, J.; Klein-Seetharaman, J. Techniques for transferring host-pathogen protein interactions knowledge to new tasks. Front. Microbiol. 2015, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Bosque, G.; Folch-Fortuny, A.; Picó, J.; Ferrer, A.; Elena, S.F. Topology analysis and visualization of Potyvirus protein-protein interaction network. BMC Syst. Biol. 2014, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Song, Q.; Tao, H.; Harrison, A.; Wang, S.; Liu, W.; Lin, S.; Zhang, Z.; Ai, Y.; He, H. Prediction of protein-protein interactions between fungus (Magnaporthe grisea) and rice (Oryza sativa L.). Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhou, Y.; Zhang, Z. Network analysis reveals a common host-pathogen interaction pattern in Arabidopsis immune responses. Front. Plant Sci. 2017, 8, 893. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Huang, J.; Liu, Y.; Rao, J.; Luo, D.; He, M. Exploring potential new floral organ morphogenesis genes of Arabidopsis thaliana using systems biology approach. Front. Plant Sci. 2015, 6, 829. [Google Scholar] [CrossRef] [PubMed]
- Santoni, D.; Swiercz, A.; Zmieńko, A.; Kasprzak, M.; Blazewicz, M.; Bertolazzi, P.; Felici, G. An integrated approach (CLuster Analysis Integration Method) to combine expression data and protein-protein interaction networks in agrigenomics: Application on Arabidopsis thaliana. OMICS 2014, 18, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Duan, G.; Walther, D.; Schulze, W.X. Reconstruction and analysis of nutrient-induced phosphorylation networks in Arabidopsis thaliana. Front. Plant Sci. 2013, 4, 540. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Dong, X.; Zhang, Z. Network-based comparative analysis of Arabidopsis immune responses to Golovinomyces orontii and Botrytis cinerea infections. Sci. Rep. 2016, 6, 19149. [Google Scholar] [CrossRef] [PubMed]
- Naseem, M.; Kunz, M.; Dandekar, T. Probing the unknowns in cytokinin-mediated immune defense in Arabidopsis with systems biology approaches. Bioinform. Biol. Insights 2014, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Lysenko, A.; Defoin-Platel, M.; Hassani-Pak, K.; Taubert, J.; Hodgman, C.; Rawlings, C.J.; Saqi, M. Assessing the functional coherence of modules found in multiple-evidence networks from Arabidopsis. BMC Bioinformatics 2011, 12, 203. [Google Scholar] [CrossRef] [PubMed]
- Van Leene, J.; Boruc, J.; De Jaeger, G.; Russinova, E.; De Veylder, L. A kaleidoscopic view of the Arabidopsis core cell cycle interactome. Trends Plant Sci. 2011, 16, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Lu, L.J. Investigating the validity of current network analysis on static conglomerate networks by protein network stratification. BMC Bioinformatics 2010, 11, 466. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yin, Y.; Dam, P.; Xu, Y. Identification of novel proteins Involved in plant cell-wall synthesis based on protein-protein interaction data. J. Proteome Res. 2010, 9, 5025–5037. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.G.; He, F.; Zhang, Z.; Peng, Y.L. Prediction of protein–protein interactions between Ralstonia solanacearum and Arabidopsis thaliana. Amino Acids 2012, 42, 2363–2371. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Zhu, P.; Jiao, Y.; Meng, Y.; Chen, M. PRIN: A predicted rice interactome network. BMC Bioinformatics 2011, 12, 161. [Google Scholar] [CrossRef] [PubMed]
- Sperotto, R.A.; de Araújo Junior, A.T.; Adamski, J.M.; Cargnelutti, D.; Ricachenevsky, F.K.; de Oliveira, B.H.N.; da Cruz, R.P.; dos Santos, R.P.; da Silva, L.P.; Fett, J.P. Deep RNAseq indicates protective mechanisms of cold-tolerant indica rice plants during early vegetative stage. Plant Cell Rep. 2017, 37, 347–375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Li, Y.; Li, T.; Li, Z.G.; Hsiang, T.; Zhang, Z.; Sun, W. Pathogenicity genes in Ustilaginoidea virens revealed by a predicted protein-protein interaction network. J. Proteome Res. 2017, 16, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, J.; Gangwar, I.; Panzade, G.; Shankar, R.; Yadav, S.K. Global De Novo protein-protein interactome elucidates interactions of drought-responsive proteins in horse gram (Macrotyloma uniflorum). J. Proteome Res. 2016, 15, 1794–1809. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Wu, A.; Xu, X.J.; Xiao, P.; Lu, L.; Liu, J.; Cao, Y.; Chen, L.; Wu, J.; Zhao, X.M. PPIM: A protein-protein interaction database for maize. Plant Physiol. 2015, 170, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.D.; Chang, J.W.; Guo, J.; Chen, D.; Li, S.; Xu, Q.; Deng, X.X.; Cheng, Y.J.; Chen, L.L. Prediction and functional analysis of the sweet orange protein-protein interaction network. BMC Plant Biol. 2014, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhang, Y.; Guo, W.; Wang, Q. Gleditsia sinensis: Transcriptome sequencing, construction, and application of its protein-protein interaction network. BioMed Res. Int. 2014, 2014, 404578. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Cao, X.; Yao, C.; Xue, S.; Xiu, Z. Protein-protein interaction network of the marine microalga Tetraselmis subcordiformis: Prediction and application for starch metabolism analysis. J. Ind. Microbiol. Biotechnol. 2014, 41, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Thanasomboon, R.; Kalapanulak, S.; Netrphan, S.; Saithong, T. Prediction of cassava protein interactome based on interolog method. Sci. Rep. 2017, 7, 17206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Zhao, M.; Rotgans, B.A.; Ni, G.; Dean, J.F.; Nahrung, H.F.; Cummins, S.F. Proteomic analysis of the venom and venom sac of the woodwasp, Sirex noctilio-Towards understanding its biological impact. J. Proteomics 2016, 146, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Zhang, D.; Ban, R.; Ma, X.; Chen, D.; Li, G.; Liu, J.; Wisniewski, M.; Droby, S.; Liu, Y. PCPPI: A comprehensive database for the prediction of Penicillium–crop protein–protein interactions. Database 2017, 2017. [Google Scholar]
- Verheggen, K.; Ræder, H.; Berven, F.S.; Martens, L.; Barsnes, H.; Vaudel, M. Anatomy and evolution of database search engines—A central component of mass spectrometry based proteomic workflows. Mass Spectrom. Rev. 2017, 1–15. [Google Scholar] [CrossRef] [PubMed]
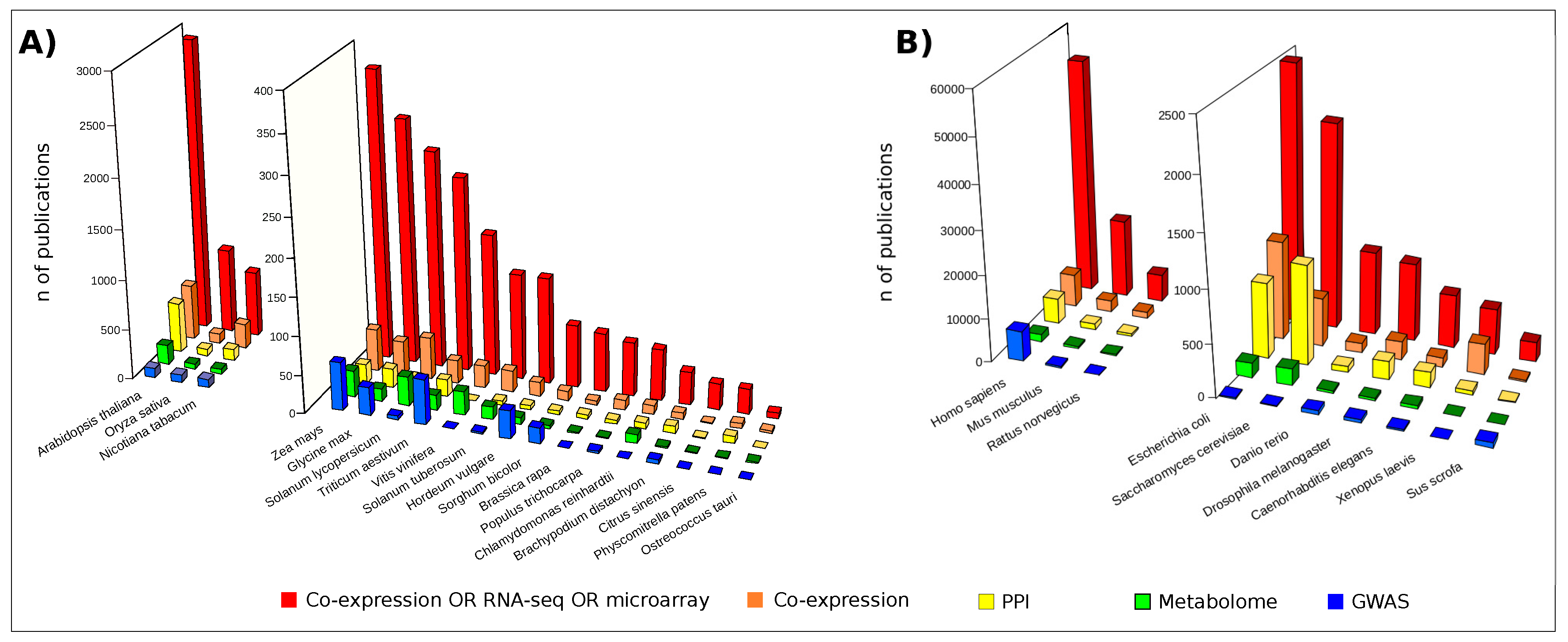
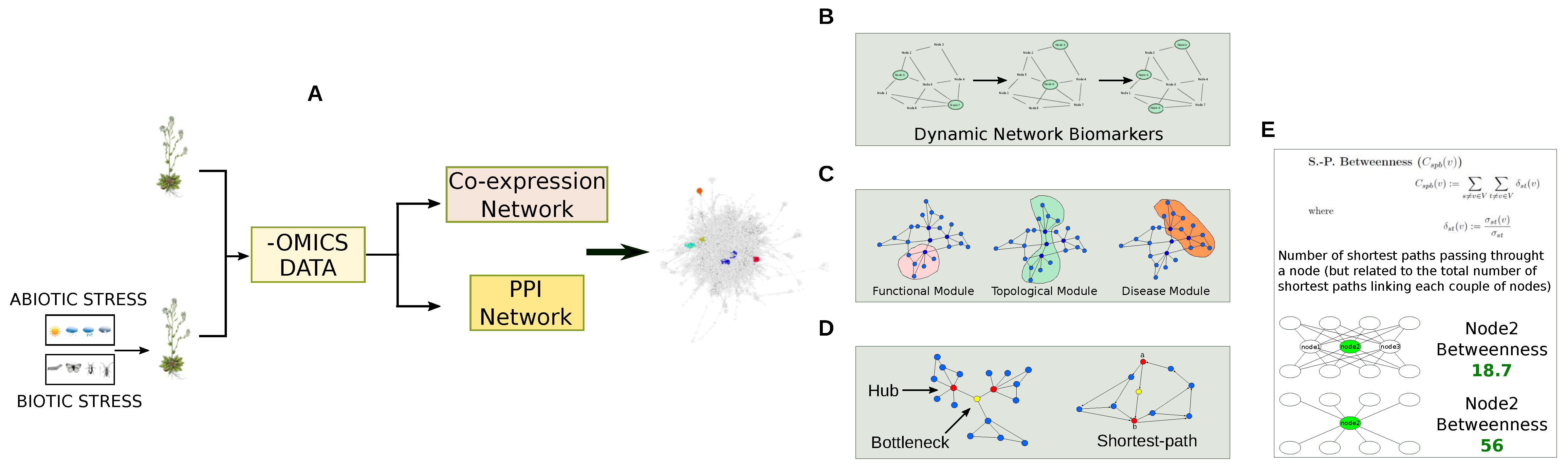

{kind=link}
{kind=link}
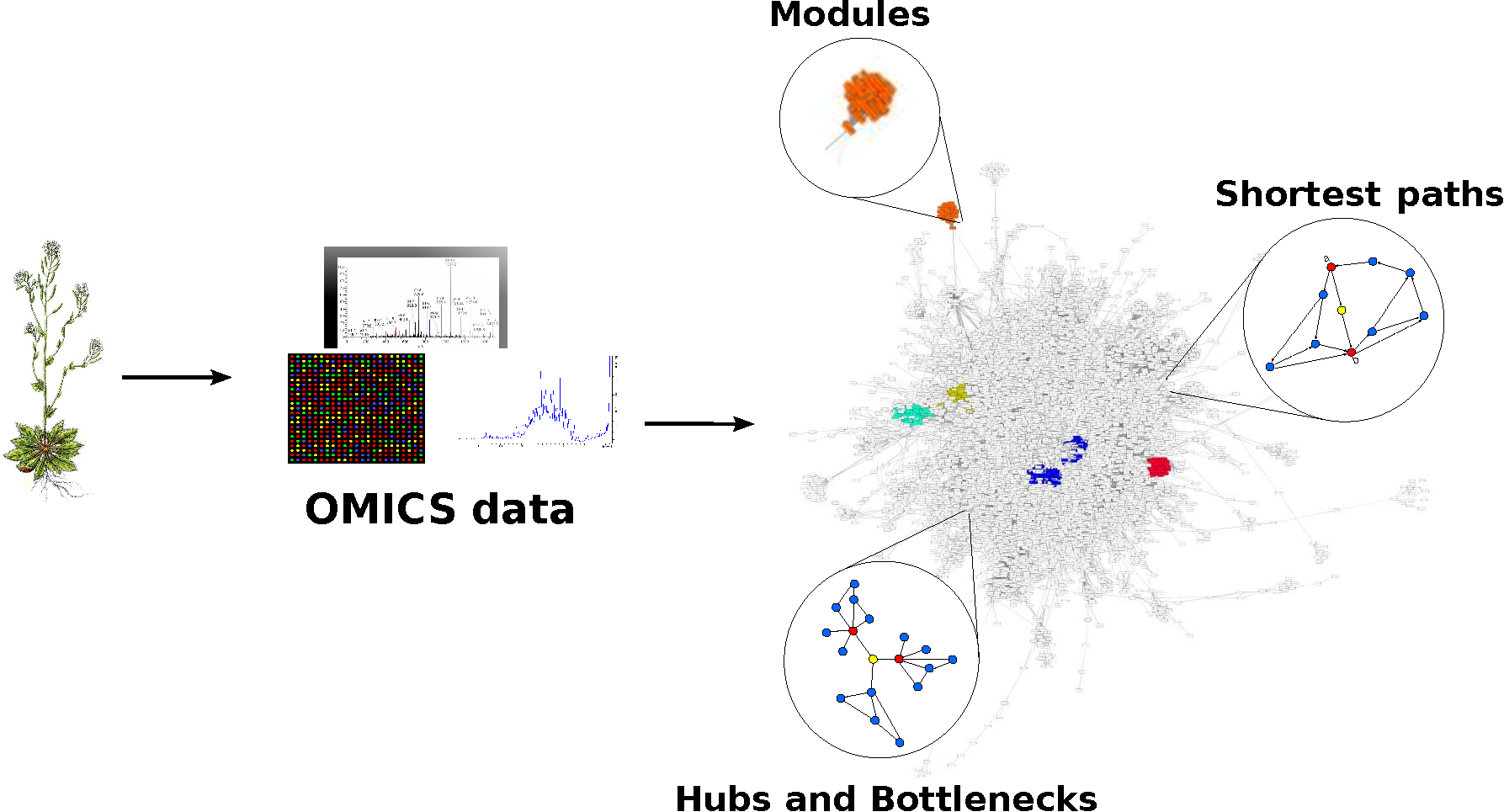
{kind=link}
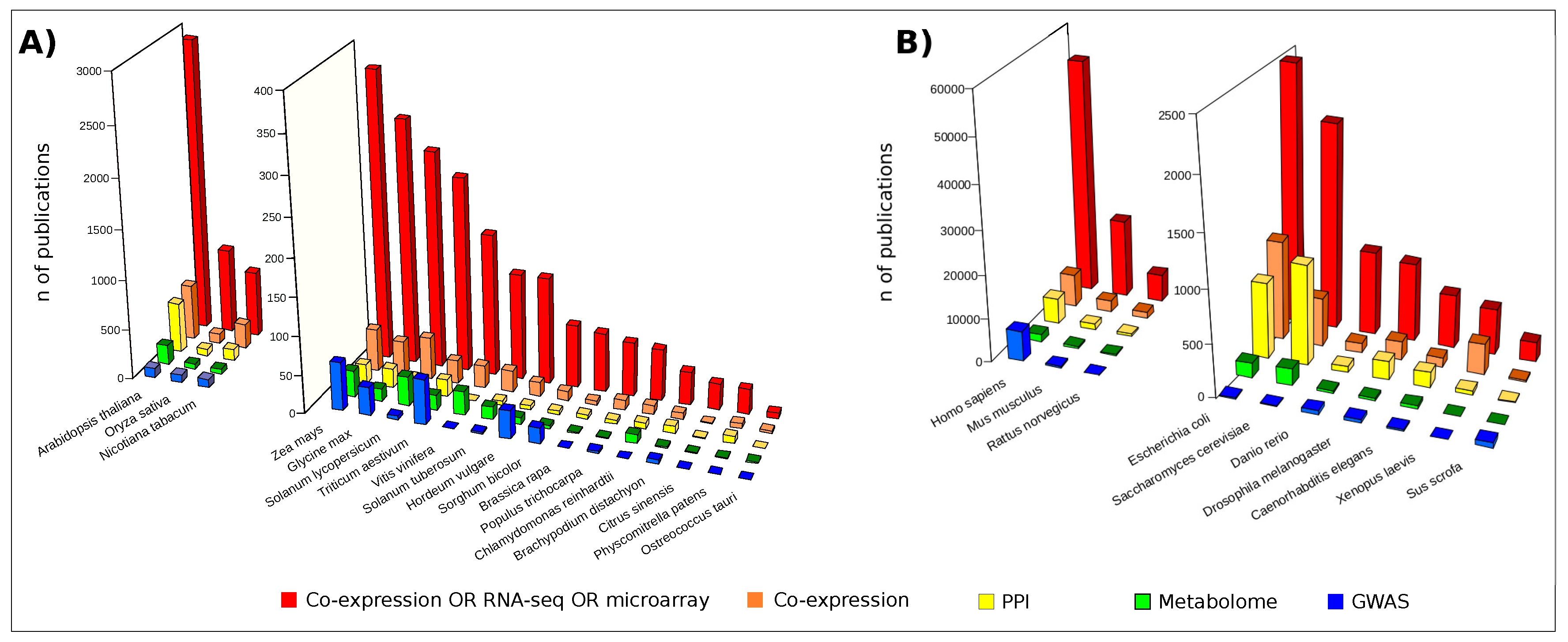
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | db Link | Proteins | PPIs | Organism |
|---|---|---|---|---|
| MIND | http://biodb.lumc.edu/mind/index.php | - | 12,102 (E) | A. thaliana |
| PAIR | http://www.cls.zju.edu.cn/pair/home.pair | - | 329,044 (P) and 6257 (E) | A. thaliana |
| AI1 | http://signal.salk.edu/interactome/AI1.html | 2774 | 6205 (P) | A. thaliana |
| SUBA | http://suba.live/ | - | 19,933 (P) | A. thaliana |
| AraPPINet | http://netbio.sjtu.edu.cn/arappinet/ | 12,574 | 316,747 (P) | A. thaliana |
| AtPID | http://www.megabionet.org/ atpid/webfile/index.php | 5562 | 28,062 (P) | A. thaliana |
| AtPIN | http://bioinfo.esalq.usp.br/atpin | - | 96,000 (E/P) | A. thaliana |
| PRIN | http://bis.zju.edu.cn/prin/ | 5049 | 76,585 (P) | O. sativa |
| DIPOS | http://comp-sysbio.org/dipos/ | 27,746 | 14,614,067 (P) | O. sativa |
| RicePPINet | http://netbio.sjtu.edu.cn/riceppinet/ | 16,895 | 708,819 (P) | O. sativa |
| PPIM | http://comp-sysbio.org/ppim/ | 14,000 | 2,762,560 (P) | Z. mays |
| PiZeaM | 6004 | 49,026 (P) | Z. mays | |
| PTIR | http://bdg.hfut.edu.cn/ptir/ | 10,626 | 357,946 (P) | S. lycopersicum |
| MauPIR | http://14.139.59.222:8080/MauPIR/ | 1812 | 6804 (P) | Macrotyloma uniflorum |
| PlaNet | http://bml.sbi.kmutt.ac.th/ppi/index.php | 7209 | 90,173 (P) | Manihot esculenta |
| Citrus sinensis Annotation project | http://citrus.hzau.edu.cn/orange/index.php | 8195 | 124,491 (P) | Fruit crop |
| FPPI | http://comp-sysbio.org/fppi/python/Default_PredictedPPIs.html | 7406 | 223,166 (P) | Gibberella zeae |
| Organisms | Proteins | PPIs | Topic | Exp. Val. | Comp. Pred. | Ref. |
|---|---|---|---|---|---|---|
| A. thaliana | 25,123 | - | Fruit development | Yes | - | [185] |
| A. thaliana | 5598 | 13,328 | Immune response | Yes | - | [195] |
| A. thaliana | 3056 | 18,233 | Immune response | Yes | - | [196] |
| A. thaliana | 2355 | 25,172 | Network topology | Yes | - | [197] |
| A. thaliana | 393 | 857 | Cell cycle | Yes | - | [198] |
| A. thaliana | 13,136 | 42,131 | Network analysis/Methods | - | Yes | [199] |
| A. thaliana | 13,347 | 45,058 | Cell-wall synthesis | - | Yes | [200] |
| A. thaliana/Pseudomonas syringae | - | 14,043 (A. thaliana) | Host-pathogen interaction | - | Yes | [35] |
| 1337 (Pseudomonas syringae) | ||||||
| A. thaliana/Ralstonia solanacearum | 1442 (A. thaliana) | 3074 | Host-pathogen interaction | - | Yes | [201] |
| 119 (Ralstonia solanacearum) | ||||||
| A. thaliana/Potyvirus | 5127 (A. thaliana) | 12,624 | Host-pathogen interaction | - | - | [189] |
| 11 (Potyvirus) | ||||||
| O. sativa | 16,895 | 708,819 | Network inference/Methods | - | Yes | [186] |
| O. sativa | 5049 | 76,585 | Network inference/Methods | - | Yes | [202] |
| O. sativa subsp. indica | 454 | 4114 | Abiotic stress response | - | Yes | [203] |
| O. sativa/Ustilaginoidea virens | 3305 | 20,217 | Host-pathogen interaction | - | Yes | [204] |
| O. sativa/Rhizoctonia solani | 1773 | 6705 | Host-pathogen interaction | Yes | - | [184] |
| Macrotyloma uniflorum | 1812 | 6804 | Abiotic stress response | - | Yes | [205] |
| Z. mays | 14,000 | 2,762,560 | Network inference/Methods | - | Yes | [206] |
| Citrus sinensis | 8195 | 124,491 | Network inference/Methods | - | Yes | [207] |
| Gleditsia sinensis | 1897 | 7078 | Network inference/Abiotic stress response | Yes | - | [208] |
| Tetraselmis subcordiformis | 938 | 12,887 | Network inference/Metabolism | Yes | - | [209] |
| Manihot esculenta | 7209 | 90,173 | Network inference/Methods | - | Yes | [210] |
| Pinus taeda/Sirex noctilio | 528 | 4363 | Host-pathogen interaction | Yes | - | [211] |
| apple, maize, pear, rice, strawberry and tomato/Penicillium expansum | 9911 | 439,904 | Host-pathogen interaction | - | Yes | [212] |
| Database | db Link | Proteins | PPIs | Organisms |
|---|---|---|---|---|
| PPIRA | http://protein.cau.edu.cn/ppira/ | - | 3074 | R. solanacearum and A. thaliana |
| PPIN-1 | http://signal.salk.edu/interactome/PPIN1.html | 926 | 3148 | Pseudomonas syringae and A. thaliana |
| PathoPlant | http://www.pathoplant.de/ | - | 350 | Plants and plant-pathogen |
| PHI-base | http://www.phi-base.org/ | - | 8046 | Multispecies host-pathogen |
| HPIDB | http://hpidb.igbb.msstate.edu/ | - | 62,653 | Multispecies host-pathogen |
| VirHostnet | http://virhostnet.prabi.fr/ | - | 28,000 | Multispecies host-virus |
| VirusMentha | http://virusmentha.uniroma2.it/ | 4426 | 9876 | Multispecies host-virus |
| EBI Intact | https://www.ebi.ac.uk/intact/ | 105,180 | 805,177 | Multispecies host-pathogen |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Silvestre, D.; Bergamaschi, A.; Bellini, E.; Mauri, P. Large Scale Proteomic Data and Network-Based Systems Biology Approaches to Explore the Plant World. Proteomes 2018, 6, 27. https://doi.org/10.3390/proteomes6020027
Di Silvestre D, Bergamaschi A, Bellini E, Mauri P. Large Scale Proteomic Data and Network-Based Systems Biology Approaches to Explore the Plant World. Proteomes. 2018; 6(2):27. https://doi.org/10.3390/proteomes6020027
Chicago/Turabian StyleDi Silvestre, Dario, Andrea Bergamaschi, Edoardo Bellini, and PierLuigi Mauri. 2018. "Large Scale Proteomic Data and Network-Based Systems Biology Approaches to Explore the Plant World" Proteomes 6, no. 2: 27. https://doi.org/10.3390/proteomes6020027
APA StyleDi Silvestre, D., Bergamaschi, A., Bellini, E., & Mauri, P. (2018). Large Scale Proteomic Data and Network-Based Systems Biology Approaches to Explore the Plant World. Proteomes, 6(2), 27. https://doi.org/10.3390/proteomes6020027