MetaGOmics: A Web-Based Tool for Peptide-Centric Functional and Taxonomic Analysis of Metaproteomics Data


Abstract
1. Introduction
2. Methods
2.1. Web Application Implementation
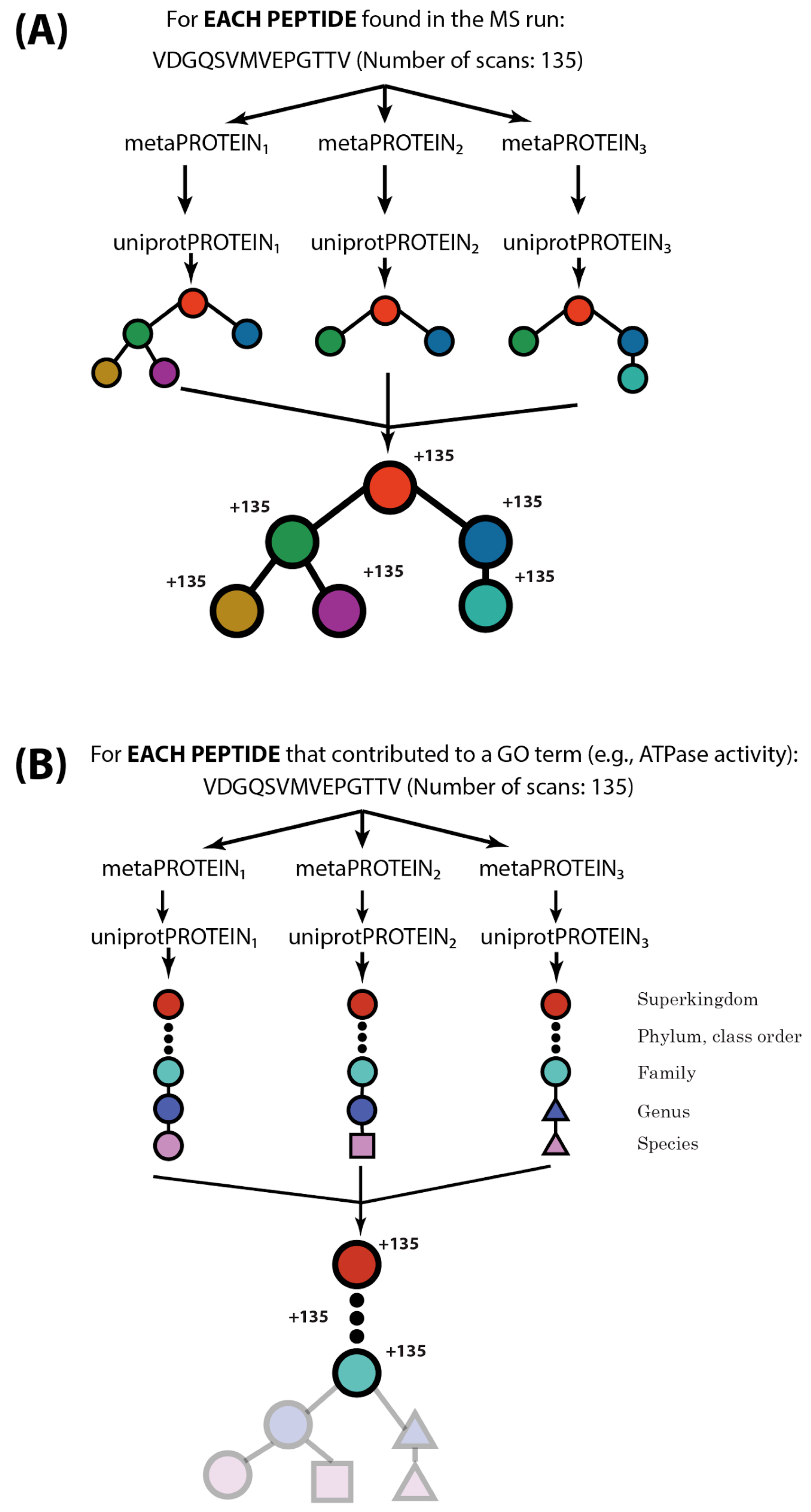
2.2. MetaGOmics Algorithm
2.3. Specifying Relative Abundance
2.4. Running BLAST
2.5. Expected Wait Times
2.6. Unknown Gene Ontology Annotations
2.7. Analysis of Ocean Metaproteomics Dataset
3. Results and Discussion
3.1. Web Application
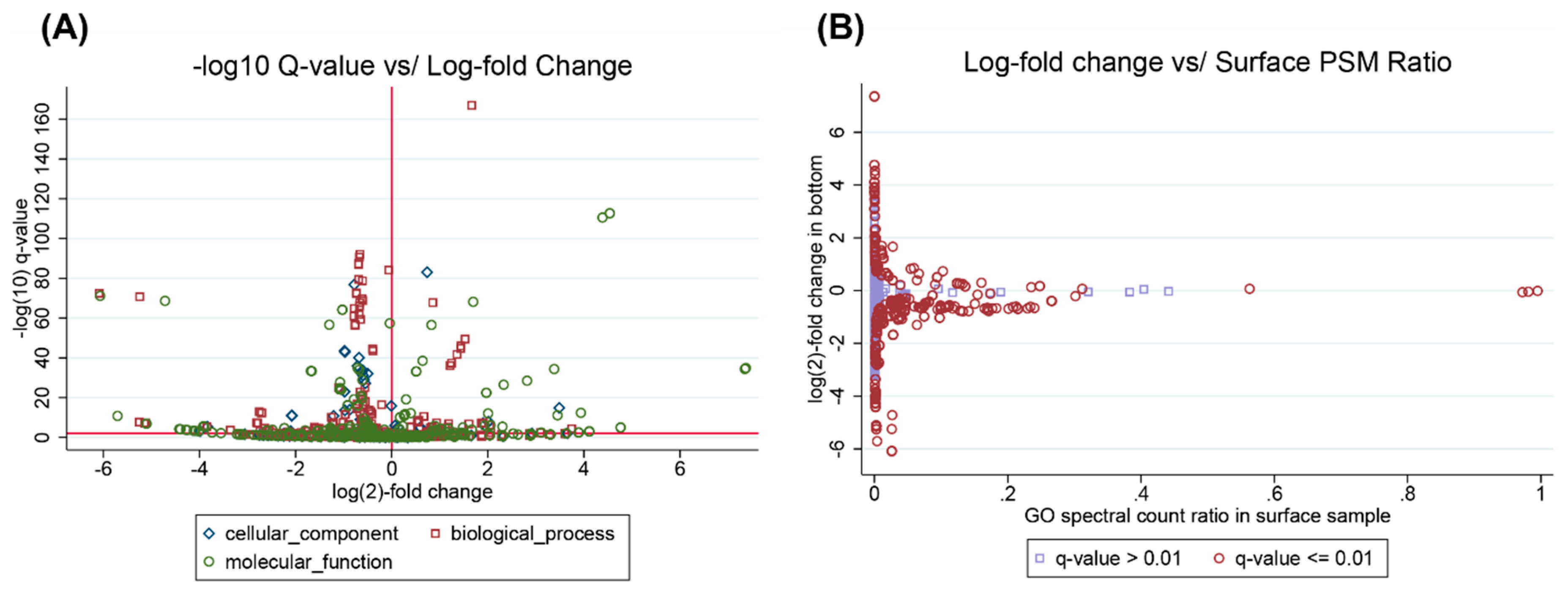
3.2. Example Analysis: Ocean Metaproteomics
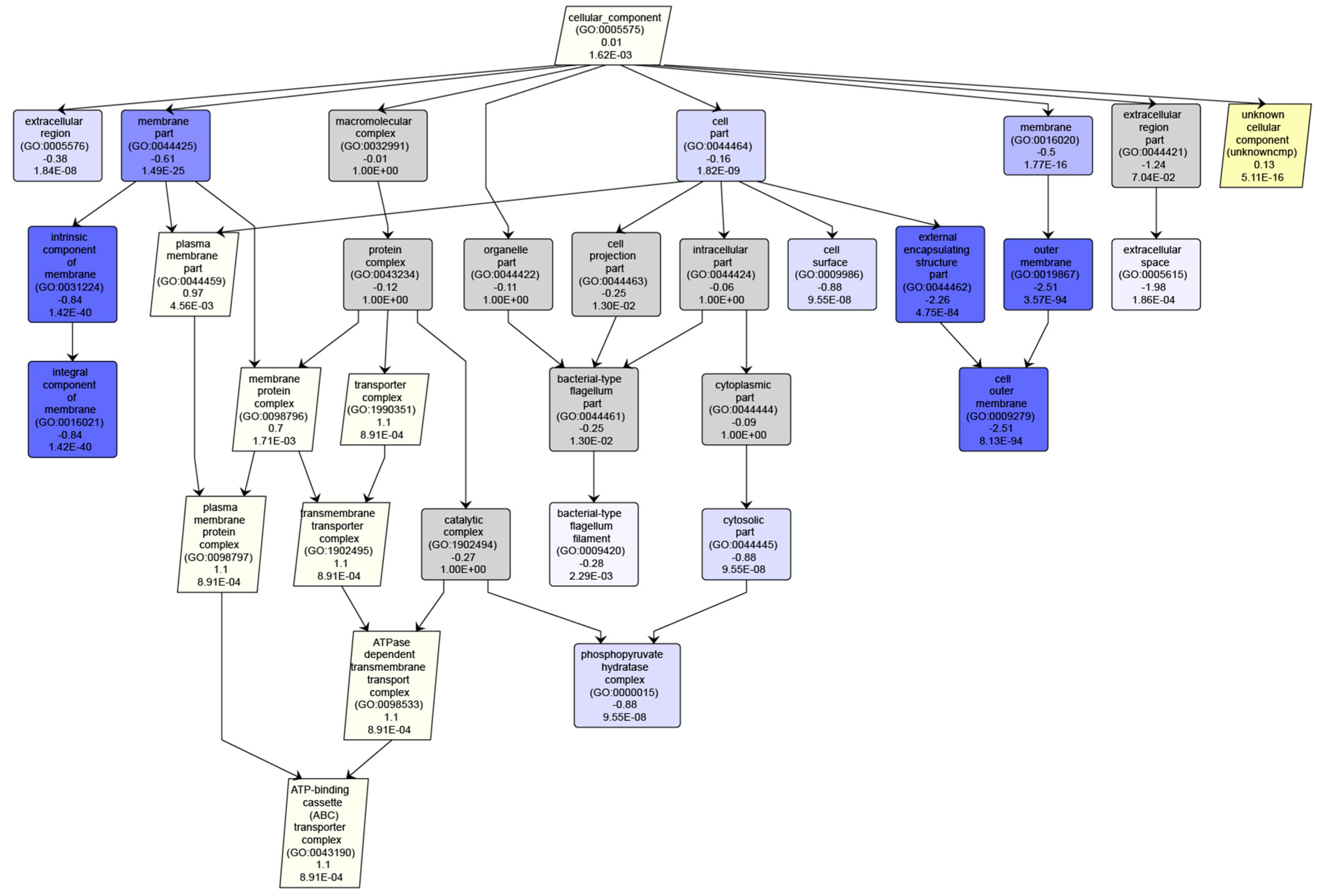
3.3. Interpreting Results With Many “Unknown” GO Annotations
3.4. Interpreting Taxonomic Changes
3.5. Current Usage
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed]
- Group, N.H.W.; Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; et al. The NIH human microbiome project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.M.; Nunn, B.L.; Frazar, C.; Goodlett, D.R.; Ting, Y.S.; Rocap, G. Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction. ISME J. 2010, 4, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Oulas, A.; Pavloudi, C.; Polymenakou, P.; Pavlopoulos, G.A.; Papanikolaou, N.; Kotoulas, G.; Arvanitidis, C.; Iliopoulos, I. Metagenomics: Tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform. Biol. Insights 2015, 9, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Jones, O.A.H.; Maguire, M.L.; Griffin, J.L.; Dias, D.A.; Spurgeon, D.J.; Svendsen, C. Metabolomics and its use in ecology. Austral Ecol. 2013, 38, 713–720. [Google Scholar] [CrossRef]
- Bashiardes, S.; Zilberman-Schapira, G.; Elinav, E. Use of metatranscriptomics in microbiome research. Bioinform. Biol. Insights 2016, 10, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Pal, R. Integrated analysis of transcriptomic and proteomic data. Curr. Genom. 2013, 14, 91–110. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mrna and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Petriz, B.A.; Franco, O.L. Metaproteomics as a complementary approach to gut microbiota in health and disease. Front. Chem. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; Jahan, T.A.; Hoopmann, M.R. Comet: An open-source ms/ms sequence database search tool. Proteomics 2013, 13, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef]
- Perkins, D.N.; Pappin, D.J.C.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Craig, R.; Beavis, R.C. Tandem: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar]
- Tanca, A.; Palomba, A.; Fraumene, C.; Pagnozzi, D.; Manghina, V.; Deligios, M.; Muth, T.; Rapp, E.; Martens, L.; Addis, M.F.; et al. The impact of sequence database choice on metaproteomic results in gut microbiota studies. Microbiome 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Timmins-Schiffman, E.; May, D.H.; Mikan, M.; Riffle, M.; Frazar, C.; Harvey, H.R.; Noble, W.S.; Nunn, B.L. Critical decisions in metaproteomics: Achieving high confidence protein annotations in a sea of unknowns. ISME J. 2017, 11, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Aebersold, R. Interpretation of shotgun proteomic data: The protein inference problem. Mol. Cell. Proteom. 2005, 4, 1419–1440. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M. What does it mean to identify a protein in proteomics? Trends Biochem. Sci. 2002, 27, 74–78. [Google Scholar] [CrossRef]
- Griffin, N.M.; Yu, J.Y.; Long, F.; Oh, P.; Shore, S.; Li, Y.; Koziol, J.A.; Schnitzer, J.E. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat. Biotechnol. 2010, 28, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (empai) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, A.C.; Parmely, T.J.; Tomomori-Sato, C.; Sato, S.; Zhu, D.X.; Conaway, R.C.; Conaway, J.W.; Florens, L.; Washburn, M.P. Quantitative proteomic analysis of distinct mammalian mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. USA 2006, 103, 18928–18933. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, Z.H.; Washburn, M.P.; Florens, L. Refinements to label free proteome quantitation: How to deal with peptides shared by multiple proteins. Anal. Chem. 2010, 82, 2272–2281. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.F.G.; Arnold, R.J.; Li, Y.X.; Radivojac, P.; Sheng, Q.H.; Tang, H.X. A bayesian approach to protein inference problem in shotgun proteomics. J. Comput. Biol. 2009, 16, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chambers, M.C.; Tabb, D.L. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 2007, 6, 3549–3557. [Google Scholar] [CrossRef] [PubMed]
- Serang, O.; Moruz, L.; Hoopmann, M.R.; Kall, L. Recognizing uncertainty increases robustness and reproducibility of mass spectrometry-based protein inferences. J. Proteome Res. 2012, 11, 5586–5591. [Google Scholar] [CrossRef] [PubMed]
- Audain, E.; Uszkoreit, J.; Sachsenberg, T.; Pfeuffer, J.; Liang, X.; Hermjakob, H.; Sanchez, A.; Eisenacher, M.; Reinert, K.; Tabb, D.L.; et al. In-depth analysis of protein inference algorithms using multiple search engines and well-defined metrics. J. Proteom. 2017, 150, 1701–1782. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. Megan analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Behne, A.; Heyer, R.; Kohrs, F.; Benndorf, D.; Hoffmann, M.; Lehteva, M.; Reichl, U.; Martens, L.; Rapp, E. The metaproteomeanalyzer: A powerful open-source software suite for metaproteomics data analysis and interpretation. J. Proteome Res. 2015, 14, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Mesuere, B.; Devreese, B.; Debyser, G.; Aerts, M.; Vandamme, P.; Dawyndt, P. Unipept: Tryptic peptide-based biodiversity analysis of metaproteome samples. J. Proteome Res. 2012, 11, 5773–5780. [Google Scholar] [CrossRef] [PubMed]
- Jaschob, D.; Riffle, M. Jobcenter: An open source, cross-platform, and distributed job queue management system optimized for scalability and versatility. Source Code Biol. Med. 2012, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Chen, C.; Huang, H.; Wu, C.H. Protein bioinformatics databases and resources. Methods Mol. Biol. 2017, 1558, 33–39. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Methodol. 1995, 57, 289–300. [Google Scholar]
- Yekutieli, D.; Benjamini, Y. Resampling-based false discovery rate controlling multiple test procedures for correlated test statistics. J. Stat. Plan. Inference 1999, 82, 171–196. [Google Scholar] [CrossRef]
- O’Donovan, C.; Martin, M.J.; Gattiker, A.; Gasteiger, E.; Bairoch, A.; Apweiler, R. High-quality protein knowledge resource: Swiss-prot and trembl. Brief. Bioinform. 2002, 3, 275–284. [Google Scholar] [CrossRef] [PubMed]
- May, D.H.; Timmins-Schiffman, E.; Mikan, M.P.; Harvey, H.R.; Borenstein, E.; Nunn, B.L.; Noble, W.S. An alignment-free “metapeptide” strategy for metaproteomic characterization of microbiome samples using shotgun metagenomic sequencing. J. Proteome Res. 2016, 15, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Kall, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Accession String | GO Aspect | GO Name | Spectral Count | Ratio |
|---|---|---|---|---|
| GO:0005575 | cellular_component | cellular_component | 12,217 | 1 |
| GO:0008150 | biological_process | biological_process | 12,217 | 1 |
| GO:0003674 | molecular_function | molecular_function | 12,217 | 1 |
| unknownprc | biological_process | unknown biological process | 5472 | 0.45 |
| GO:0005488 | molecular_function | binding | 4185 | 0.34 |
| GO:0097159 | molecular_function | organic cyclic compound binding | 3579 | 0.29 |
| GO:1901363 | molecular_function | heterocyclic compound binding | 3579 | 0.29 |
| GO:0005524 | molecular_function | ATP binding | 1712 | 0.14 |
| GO:1901566 | biological_process | organonitrogen compound biosynthetic process | 1353 | 0.11 |
| GO:0042026 | biological_process | protein refolding | 1145 | 0.09 |
| GO:1990351 | cellular_component | transporter complex | 200 | 0.02 |
| Taxon Name | Taxonomy Rank | Spectral Count | Ratio of GO | Ratio of Experiment |
|---|---|---|---|---|
| Bacteria | superkingdom | 240 | 0.88 | 3.50 × 10−2 |
| Bacteroidia | class | 141 | 0.52 | 2.05 × 10−2 |
| Bacteroidetes | phylum | 141 | 0.52 | 2.05 × 10−2 |
| Bacteroidales | order | 141 | 0.52 | 2.05 × 10−2 |
| Prevotella | genus | 81 | 0.3 | 1.18 × 10−2 |
| Prevotellaceae | family | 81 | 0.3 | 1.18 × 10−2 |
| Firmicutes | phylum | 41 | 0.15 | 5.97 × 10−3 |
| Lactobacillales | order | 33 | 0.12 | 4.81 × 10−3 |
| Lactobacillaceae | family | 33 | 0.12 | 4.81 × 10−3 |
| Lactobacillus | genus | 33 | 0.12 | 4.81 × 10−3 |
| Bacilli | class | 33 | 0.12 | 4.81 × 10−3 |
| Prevotella sp. CAG:873 | species | 23 | 0.08 | 3.35 × 10−3 |
| Clostridiales | order | 6 | 0.02 | 8.74 × 10−4 |
| Clostridia | class | 6 | 0.02 | 8.74 × 10−4 |
| Actinobacteria | phylum | 5 | 0.02 | 7.28 × 10−4 |
| GO Name | Fold Change | q-Value |
|---|---|---|
| outer membrane | 1.55 | 5.27 × 10−106 |
| cell outer membrane | 1.55 | 5.61 × 10−106 |
| external encapsulating structure part | 1.5 | 5.64 × 10−102 |
| membrane | 1.14 | 3.00 × 10−101 |
| receptor activity | 1.47 | 5.03 × 10−93 |
| intrinsic component of membrane | 1.44 | 6.37 × 10−88 |
| integral component of membrane | 1.44 | 6.37 × 10−88 |
| molecular transducer activity | 1.35 | 4.14 × 10−81 |
| membrane part | 1.01 | 4.69 × 10−53 |
| carbohydrate derivative binding | −2.03 | 1.25 × 10−49 |
| ribonucleotide binding | −2.03 | 1.25 × 10−49 |
| purine ribonucleoside binding | −2.04 | 5.36 × 10−47 |
| ribonucleoside binding | −2.04 | 5.36 × 10−47 |
| purine ribonucleoside triphosphate binding | −2.04 | 5.36 × 10−47 |
| Biological Process | |||||
| Higher in Ocean Surface Water | Higher in Ocean Bottom Water | ||||
| GO Term | log Change | q-Value | GO Term | log Change | q-Value |
| d-xylose transport | −6.09 | 3.49 × 10−73 | protein refolding | 1.66 | 1.01 × 10−167 |
| translation | −0.77 | 2.43 × 10−57 | chromosome condensation | 1.52 | 3.69 × 10−50 |
| translational elongation | −1.11 | 9.60 × 10−26 | DNA repair | 1.61 | 1.82 × 10−7 |
| transcription anti-termination | −2.79 | 5.66 × 10−8 | dephosphorylation | 2.04 | 6.62 × 10−7 |
| fatty acid biosynthetic process | −1.21 | 6.42 × 10−8 | de novo’ pyrimidine nucleobase biosynthetic process | 3.74 | 4.53 × 10−5 |
| GTP biosynthetic process | −5.12 | 7.30 × 10−8 | RNA phosphodiester bond hydrolysis, exonucleolytic | 1 | 5.52 × 10−5 |
| UTP biosynthetic process | −5.12 | 7.30 × 10−8 | mRNA catabolic process | 0.93 | 1.69 × 10−4 |
| CTP biosynthetic process | −5.12 | 7.30 × 10−8 | 7,8-dihydroneopterin 3′-triphosphate biosynthetic process | 3.09 | 4.48 × 10−3 |
| tricarboxylic acid cycle | −3.84 | 4.41 × 10−8 | response to cadmium ion | 1.45 | 7.42 × 10−3 |
| cell division | −1.07 | 1.18 × 10−5 | |||
| Molecular Function | |||||
| Higher in Ocean Surface Water | Higher in Ocean Bottom Water | ||||
| GO Term | log Change | q-Value | GO Term | log Change | q-Value |
| monosaccharide binding | −6.07 | 6.71 × 10−72 | histidine ammonia-lyase activity | 4.54 | 1.93 × 10−113 |
| receptor activity | −1.03 | 5.77 × 10−65 | unfolded protein binding | 0.83 | 2.85 × 10−57 |
| structural constituent of ribosome | −0.68 | 1.12 × 10−34 | nitrate reductase activity | 7.35 | 4.13 × 10−35 |
| DNA-directed RNA polymerase activity | −1.68 | 4.06 × 10−34 | heme binding | 3.38 | 4.23 × 10−35 |
| translation elongation factor activity | −1.09 | 7.50 × 10−25 | ATP binding | 0.51 | 6.87 × 10−34 |
| GTP binding | −0.92 | 6.40 × 10−17 | 4 iron, 4 sulfur cluster binding | 2.33 | 3.28 × 10−27 |
| GTPase activity | −0.92 | 8.64 × 10−7 | prephenate dehydratase activity | 3.94 | 4.38 × 10−13 |
| nucleoside diphosphate kinase activity | −5.11 | 1.28 × 10−7 | selenium binding | 2 | 8.97 × 10−13 |
| tRNA binding | −0.87 | 3.00 × 10−6 | 4-phytase activity | 3.45 | 7.83 × 10−73 |
| acetyl-CoA carboxylase activity | −3.91 | 3.46 × 10−6 | formate dehydrogenase (NAD+) activity | 1.48 | 9.79 × 10−6 |
| Cellular Component | |||||
| Higher in Ocean Surface Water | Higher in Ocean Bottom Water | ||||
| GO Term | log Change | q-Value | GO Term | log Change | q-Value |
| cell outer membrane | −0.98 | 1.17 × 10−43 | cytoplasm | 0.74 | 8.53 × 10−84 |
| intracellular | −0.71 | 1.09 × 10−36 | bacterial-type flagellum filament | 3.49 | 1.47 × 10−15 |
| ribosome | −0.64 | 1.78 × 10−33 | bacterial-type flagellum | 2.01 | 1.53 × 10−8 |
| integral component of membrane | −0.98 | 9.77 × 10−24 | unknown cellular component | 0.07 | 1.04 × 10−6 |
| thylakoid | −2.08 | 1.07 × 10−11 | ATP-binding cassette (ABC) transporter complex | 0.6 | 1.04 × 10−5 |
| large ribosomal subunit | −1.21 | 1.54 × 10−11 | cytosolic small ribosomal subunit | 3.66 | 9.19 × 10−3 |
| acetyl-CoA carboxylase complex | −3.84 | 4.22 × 10−6 | |||
| plasma membrane | −0.29 | 3.37 × 10−4 | |||
| pyruvate dehydrogenase complex | −3.99 | 7.77 × 10−4 | |||
| proton-transporting ATP synthase complex, catalytic core F(1) | −0.37 | 1.36 × 10−3 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riffle, M.; May, D.H.; Timmins-Schiffman, E.; Mikan, M.P.; Jaschob, D.; Noble, W.S.; Nunn, B.L. MetaGOmics: A Web-Based Tool for Peptide-Centric Functional and Taxonomic Analysis of Metaproteomics Data. Proteomes 2018, 6, 2. https://doi.org/10.3390/proteomes6010002
Riffle M, May DH, Timmins-Schiffman E, Mikan MP, Jaschob D, Noble WS, Nunn BL. MetaGOmics: A Web-Based Tool for Peptide-Centric Functional and Taxonomic Analysis of Metaproteomics Data. Proteomes. 2018; 6(1):2. https://doi.org/10.3390/proteomes6010002
Chicago/Turabian StyleRiffle, Michael, Damon H. May, Emma Timmins-Schiffman, Molly P. Mikan, Daniel Jaschob, William Stafford Noble, and Brook L. Nunn. 2018. "MetaGOmics: A Web-Based Tool for Peptide-Centric Functional and Taxonomic Analysis of Metaproteomics Data" Proteomes 6, no. 1: 2. https://doi.org/10.3390/proteomes6010002
APA StyleRiffle, M., May, D. H., Timmins-Schiffman, E., Mikan, M. P., Jaschob, D., Noble, W. S., & Nunn, B. L. (2018). MetaGOmics: A Web-Based Tool for Peptide-Centric Functional and Taxonomic Analysis of Metaproteomics Data. Proteomes, 6(1), 2. https://doi.org/10.3390/proteomes6010002