Multi-Omic Biogeography of the Gastrointestinal Microbiota of a Pre-Weaned Lamb

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Description and Sample Collection
2.2. DNA Sample Preparation and Sequencing
2.3. Protein Sample Preparation and Metaproteome Analysis
2.4. Data Analysis and Graph Generation
3. Results and Discussion
3.1. Experimental Design and General Metrics of Omic Data
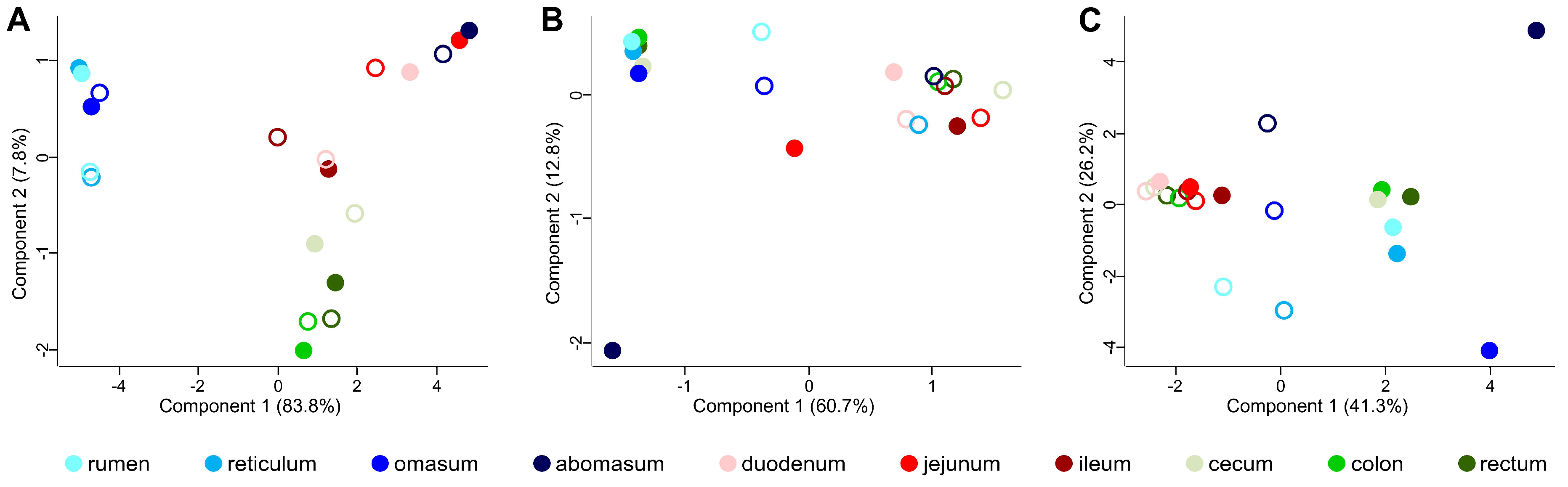
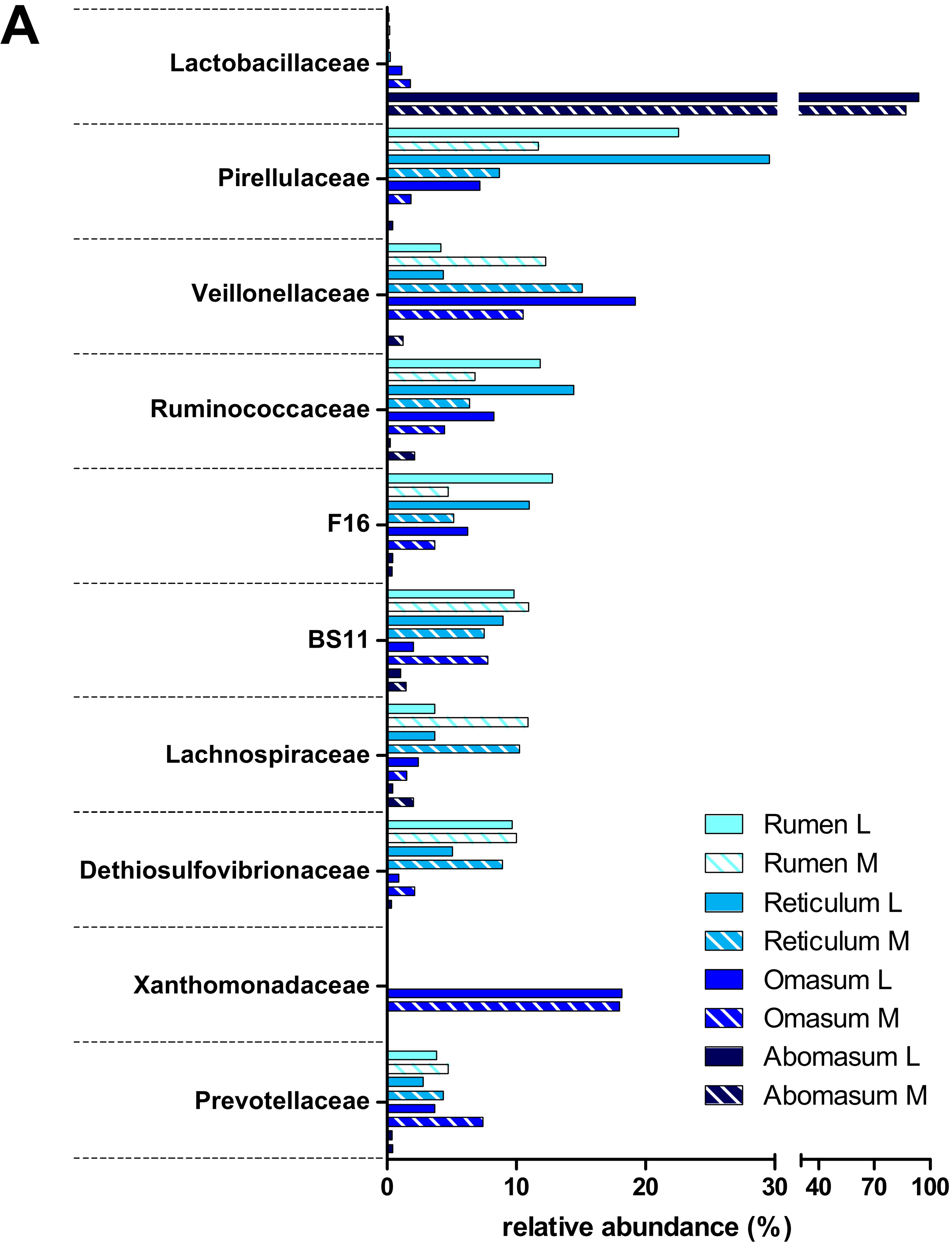
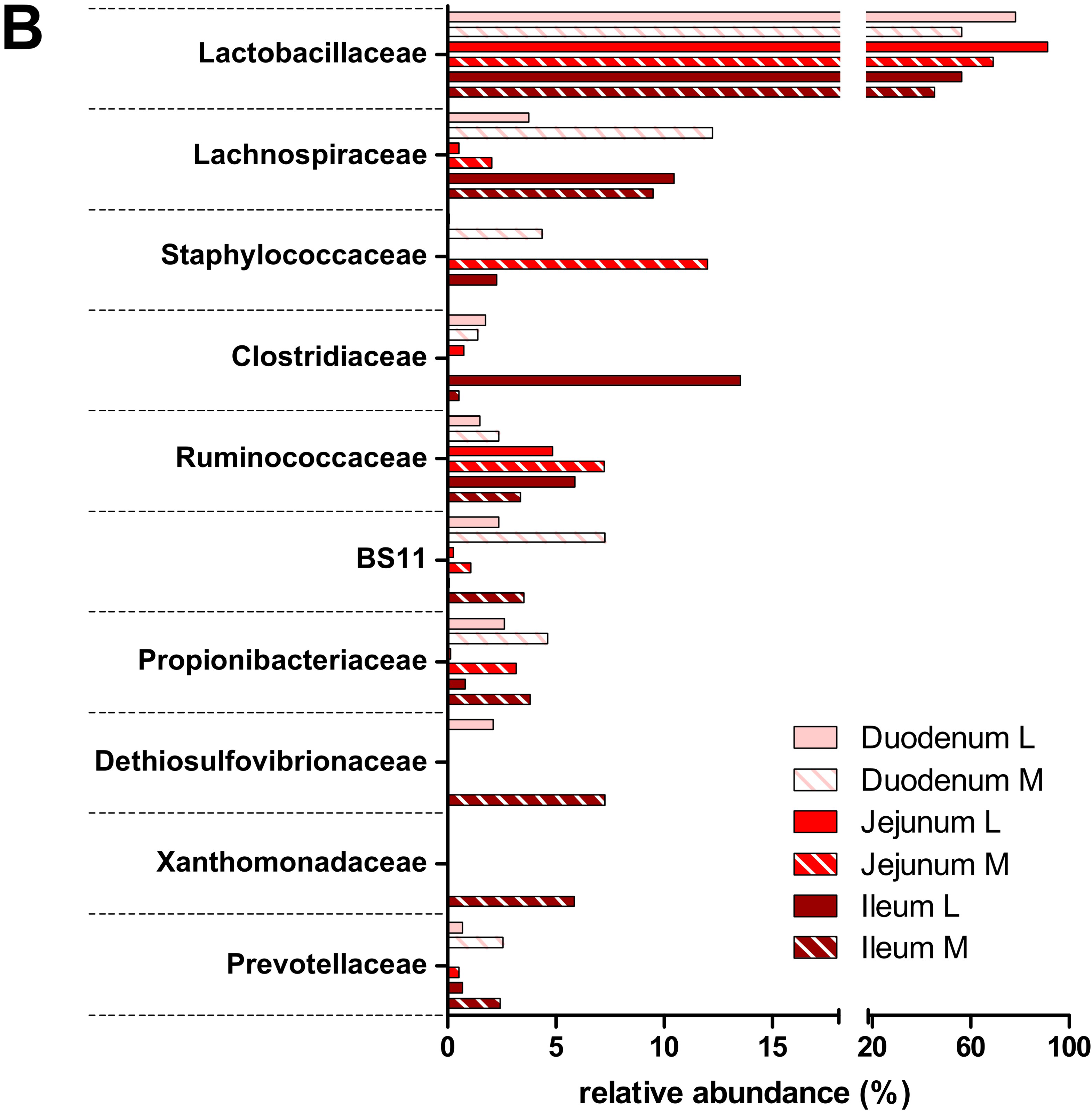
3.2. Taxonomic Distribution of the Microbiota along the Gastrointestinal Tract of a Pre-Weaned Lamb
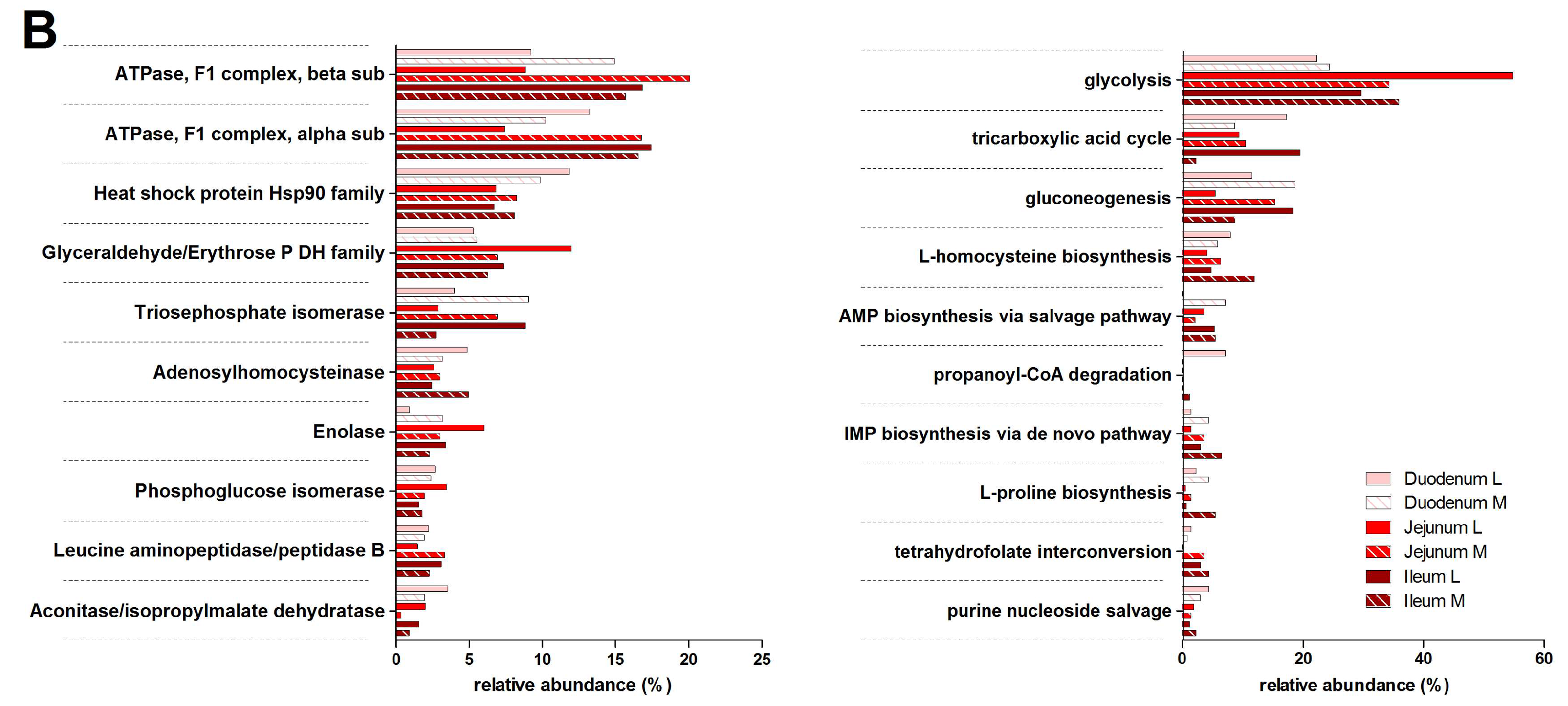
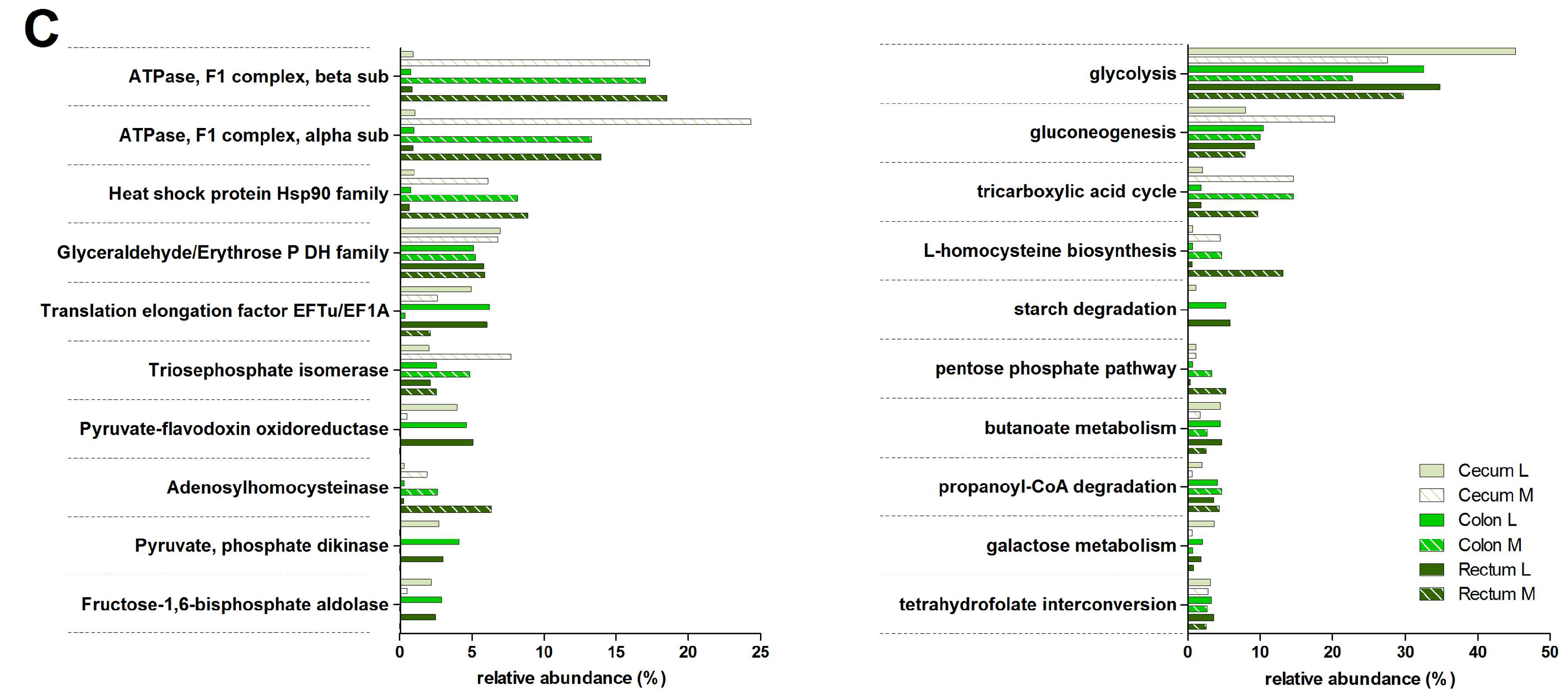
3.3. Functions and Metabolic Pathways of the Pre-Weaned Lamb Gastrointestinal Microbiota
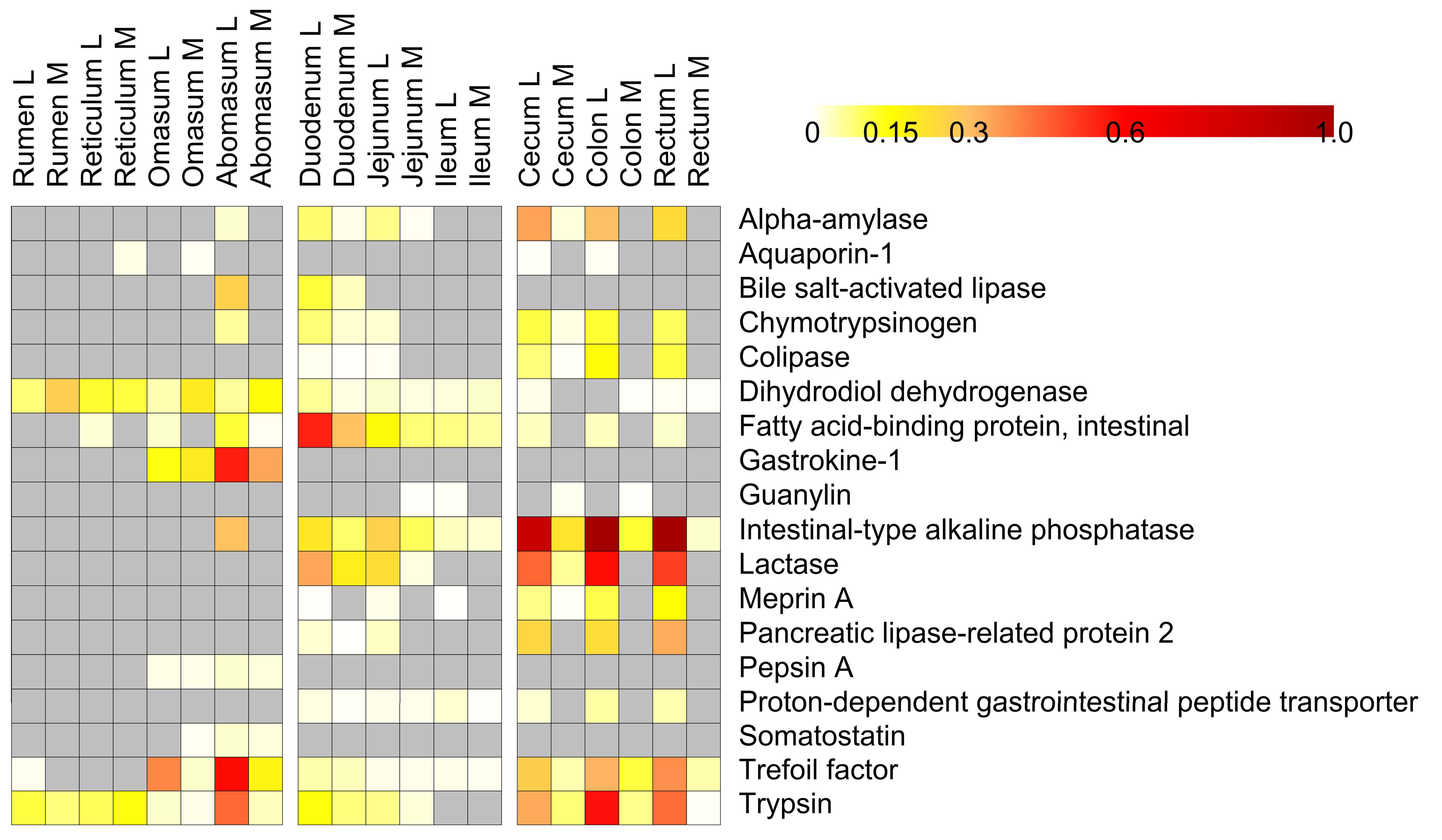
3.4. Host Protein Functions along the Pre-Weaned Lamb Gastrointestinal System: Focus on Digestion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| FASP | filter-aided sample preparation |
| F/B | Firmicutes/Bacteroidetes |
| FDR | false-discovery rate |
| GH | glycoside hydrolase |
| GIT | gastrointestinal tract |
| OTU | operational taxonomic unit |
| PCA | principal component analysis |
| PSM | peptide-spectrum matches |
| VFA | volatile fatty acid |
References
- Warner, A.C. Proteolysis by rumen micro-organisms. J. Gen. Microbiol. 1956, 14, 749–762. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meale, S.J.; Chaucheyras-Durand, F.; Berends, H.; Guan, L.L.; Steele, M.A. From pre- to postweaning: Transformation of the young calf’s gastrointestinal tract. J. Dairy Sci. 2017, 100, 5984–5995. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bian, G.; Sun, D.; Zhu, W.; Mao, S. Starter feeding altered ruminal epithelial bacterial communities and some key immune-related genes’ expression before weaning in lambs. J. Anim. Sci. 2017, 95, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Perea, K.; Perz, K.; Olivo, S.K.; Williams, A.; Lachman, M.; Ishaq, S.L.; Thomson, J.; Yeoman, C.J. Feed efficiency phenotypes in lambs involve changes in ruminal, colonic, and small-intestine-located microbiota. J. Anim. Sci. 2017, 95, 2585–2592. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.; Boland, T.; Storey, S.; Doyle, E. Linseed oil supplementation of lambs’ diet in early life leads to persistent changes in rumen microbiome structure. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Rey, M.; Enjalbert, F.; Combes, S.; Cauquil, L.; Bouchez, O.; Monteils, V. Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 2014, 116, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Guzman, C.E.; Bereza-Malcolm, L.T.; De Groef, B.; Franks, A.E. Presence of selected methanogens, fibrolytic bacteria, and proteobacteria in the gastrointestinal tract of neonatal dairy calves from birth to 72 hours. PLoS ONE 2015, 10, e0133048. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.L.V.; McLeod, K.R.; Klotz, J.L.; Heitmann, R.N. Rumen development, intestinal growth and hepatic metabolism in the pre- and postweaning ruminant. J. Dairy Sci. 2004, 87, E55–E65. [Google Scholar] [CrossRef]
- Lane, M.A.; Baldwin, R.L., VI; Jesse, B.W. Sheep rumen metabolic development in response to age and dietary treatments. J. Anim. Sci. 2000, 78, 1990–1996. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.Z.; Guan, L.L.; Tan, Z.L.; Han, X.F.; Tang, S.X.; Zhou, C.S. Postnatal bacterial succession and functional establishment of hindgut in supplemental feeding and grazing goats. J. Anim. Sci. 2015, 93, 3528–3538. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P.; Nielsen, M.O. Impacts of prenatal nutrition on animal production and performance: A focus on growth and metabolic and endocrine function in sheep. J. Anim. Sci. Biotechnol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, C.; Li, F.; Wang, X.; Zhang, X.; Liu, T.; Nian, F.; Yue, X.; Pan, X.; La, Y.; et al. Effects of early feeding on the host rumen transcriptome and bacterial diversity in lambs. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Malmuthuge, N.; Guan, L.L. Understanding the gut microbiome of dairy calves: Opportunities to improve early-life gut health. J. Dairy Sci. 2017, 100, 5996–6005. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Weimer, P.J. Host-rumen microbe interactions may be leveraged to improve the productivity of dairy cows. J. Dairy Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. Qiime allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Fraumene, C.; Manghina, V.; Palomba, A.; Abbondio, M.; Deligios, M.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Diversity and functions of the sheep faecal microbiota: A multi-omic characterization. Microb. Biotechnol. 2017, 10, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Pisanu, S.; Deligios, M.; Fraumene, C.; Manghina, V.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. A straightforward and efficient analytical pipeline for metaproteome characterization. Microbiome 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Biosa, G.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Comparison of detergent-based sample preparation workflows for LTQ-Orbitrap analysis of the Escherichia coli proteome. Proteomics 2013, 13, 2597–2607. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Pisanu, S.; Addis, M.F.; Uzzau, S. Enrichment or depletion? The impact of stool pretreatment on metaproteomic characterization of the human gut microbiota. Proteomics 2015, 15, 3474–3485. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Flyvbjerg, H. Error filtering, pair assembly and error correction for next-generation sequencing reads. Bioinformatics 2015, 31, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Namiki, T.; Hachiya, T.; Tanaka, H.; Sakakibara, Y. Metavelvet: An extension of velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res. 2012, 40, e155. [Google Scholar] [CrossRef] [PubMed]
- Rho, M.; Tang, H.; Ye, Y. Fraggenescan: Predicting genes in short and error-prone reads. Nucleic Acids Res. 2010, 38, e191. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Gorska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.J.; Tappu, R. Megan community edition—Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Pundir, S.; Martin, M.J.; O’Donovan, C. Uniprot tools. Curr. Protoc. Bioinform. 2016, 53. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the pride database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.V.; Jimenez, C.R. An accurate paired sample test for count data. Bioinformatics 2012, 28, i596–i602. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.V.; Piersma, S.R.; Warmoes, M.; Jimenez, C.R. On the beta-binomial model for analysis of spectral count data in label-free tandem mass spectrometry-based proteomics. Bioinformatics 2010, 26, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar]
- Tanca, A.; Palomba, A.; Deligios, M.; Cubeddu, T.; Fraumene, C.; Biosa, G.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Evaluating the impact of different sequence databases on metaproteome analysis: Insights from a lab-assembled microbial mixture. PLoS ONE 2013, 8, e82981. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Fraumene, C.; Pagnozzi, D.; Manghina, V.; Deligios, M.; Muth, T.; Rapp, E.; Martens, L.; Addis, M.F.; et al. The impact of sequence database choice on metaproteomic results in gut microbiota studies. Microbiome 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Kolmeder, C.A.; Salojarvi, J.; Keskitalo, S.; Varjosalo, M.; Verdam, F.J.; Rensen, S.S.; Reichl, U.; de Vos, W.M.; Rapp, E.; et al. Navigating through metaproteomics data: A logbook of database searching. Proteomics 2015, 15, 3439–3453. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Goslinga, J.; Kooren, J.A.; McGowan, T.; Wroblewski, M.S.; Seymour, S.L.; Griffin, T.J. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics 2013, 13, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Zeng, D.; Ni, X.; Zhu, H.; Jian, P.; Zhou, Y.; Xu, S.; Lin, Y.; Li, Y.; Yin, Z.; et al. Microbial community compositions in the gastrointestinal tract of Chinese Mongolian sheep using Illumina MiSeq sequencing revealed high microbial diversity. AMB Express 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Van Niftrik, L.; Devos, D.P. Editorial: Planctomycetes-verrucomicrobia-chlamydiae bacterial superphylum: New model organisms for evolutionary cell biology. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Al-Masaudi, S.; El Kaoutari, A.; Drula, E.; Al-Mehdar, H.; Redwan, E.M.; Lombard, V.; Henrissat, B. A metagenomics investigation of carbohydrate-active enzymes along the gastrointestinal tract of Saudi sheep. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Popova, M.; McGovern, E.; McCabe, M.S.; Martin, C.; Doreau, M.; Arbre, M.; Meale, S.J.; Morgavi, D.P.; Waters, S.M. The structural and functional capacity of ruminal and cecal microbiota in growing cattle was unaffected by dietary supplementation of linseed oil and nitrate. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Jewell, K.A.; McCormick, C.A.; Odt, C.L.; Weimer, P.J.; Suen, G. Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency. Appl. Environ. Microbiol. 2015, 81, 4697–4710. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Elshahed, M.S.; Youssef, N.H.; Luo, Q.; Najar, F.Z.; Roe, B.A.; Sisk, T.M.; Buhring, S.I.; Hinrichs, K.U.; Krumholz, L.R. Phylogenetic and metabolic diversity of Planctomycetes from anaerobic, sulfide- and sulfur-rich Zodletone Spring, Oklahoma. Appl. Environ. Microbiol. 2007, 73, 4707–4716. [Google Scholar] [CrossRef] [PubMed]
- Solden, L.M.; Hoyt, D.W.; Collins, W.B.; Plank, J.E.; Daly, R.A.; Hildebrand, E.; Beavers, T.J.; Wolfe, R.; Nicora, C.D.; Purvine, S.O.; et al. New roles in hemicellulosic sugar fermentation for the uncultivated Bacteroidetes family BS11. ISME J. 2017, 11, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Guan, L.L. Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Abecia, L.; Jimenez, E.; Martinez-Fernandez, G.; Martin-Garcia, A.I.; Ramos-Morales, E.; Pinloche, E.; Denman, S.E.; Newbold, C.J.; Yanez-Ruiz, D.R. Natural and artificial feeding management before weaning promote different rumen microbial colonization but not differences in gene expression levels at the rumen epithelium of newborn goats. PLoS ONE 2017, 12, e0182235. [Google Scholar] [CrossRef] [PubMed]
- Koropatkin, N.M.; Cameron, E.A.; Martens, E.C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012, 10, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Waldo, D. Extent and partition of cereal grain starch digestion in ruminants. J. Anim. Sci. 1973, 37, 1062–1074. [Google Scholar] [CrossRef]
- Li, S.; Khafipour, E.; Krause, D.O.; Kroeker, A.; Rodriguez-Lecompte, J.C.; Gozho, G.N.; Plaizier, J.C. Effects of subacute ruminal acidosis challenges on fermentation and endotoxins in the rumen and hindgut of dairy cows. J. Dairy Sci. 2012, 95, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.A.N.; France, J.; Dijkstra, J. A review of starch digestion in the lactating dairy cow and proposals for a mechanistic model: (1) dietary starch characterisation and ruminal starch digestion. J. Anim. Feed Sci. 1999, 8, 291–340. [Google Scholar] [CrossRef]
- Larsen, M.; Lund, P.; Weisbjerg, M.R.; Hvelplund, T. Digestion site of starch from cereals and legumes in lactating dairy cows. Anim. Feed Sci. Technol. 2009, 153, 236–248. [Google Scholar] [CrossRef]
- Van Soest, P.J. Nutritional Ecology of the Ruminant; Cornell University Press: Ithaca, NY, USA, 1994. [Google Scholar]
- Fonty, G.; Joblin, K.; Chavarot, M.; Roux, R.; Naylor, G.; Michallon, F. Establishment and development of ruminal hydrogenotrophs in methanogen-free lambs. Appl. Environ. Microbiol. 2007, 73, 6391–6403. [Google Scholar] [CrossRef] [PubMed]
- Šimůnek, J.; Skřivanová, V.; Hoza, I.; Březina, P.; Marounek, M. Ontogenesis of enzymatic activities in the gastrointestinal tract of young goats. Small Rumin. Res. 1995, 17, 207–211. [Google Scholar] [CrossRef]
- Rey, M.; Enjalbert, F.; Monteils, V. Establishment of ruminal enzyme activities and fermentation capacity in dairy calves from birth through weaning. J. Dairy Sci. 2012, 95, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.L.; Lang, X.; Wu, P.J.; Casper, D.P.; Li, F.D. Development of small intestinal enzyme activities and their relationship with some gut regulatory peptides in grazing sheep. J. Anim. Sci. 2017, 95, 3783–3791. [Google Scholar] [CrossRef] [PubMed][Green Version]









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomba, A.; Tanca, A.; Fraumene, C.; Abbondio, M.; Fancello, F.; Atzori, A.S.; Uzzau, S. Multi-Omic Biogeography of the Gastrointestinal Microbiota of a Pre-Weaned Lamb. Proteomes 2017, 5, 36. https://doi.org/10.3390/proteomes5040036
Palomba A, Tanca A, Fraumene C, Abbondio M, Fancello F, Atzori AS, Uzzau S. Multi-Omic Biogeography of the Gastrointestinal Microbiota of a Pre-Weaned Lamb. Proteomes. 2017; 5(4):36. https://doi.org/10.3390/proteomes5040036
Chicago/Turabian StylePalomba, Antonio, Alessandro Tanca, Cristina Fraumene, Marcello Abbondio, Francesco Fancello, Alberto Stanislao Atzori, and Sergio Uzzau. 2017. "Multi-Omic Biogeography of the Gastrointestinal Microbiota of a Pre-Weaned Lamb" Proteomes 5, no. 4: 36. https://doi.org/10.3390/proteomes5040036
APA StylePalomba, A., Tanca, A., Fraumene, C., Abbondio, M., Fancello, F., Atzori, A. S., & Uzzau, S. (2017). Multi-Omic Biogeography of the Gastrointestinal Microbiota of a Pre-Weaned Lamb. Proteomes, 5(4), 36. https://doi.org/10.3390/proteomes5040036