A Proteomic Approach to Investigate the Drought Response in the Orphan Crop Eragrostis tef

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Growth Conditions
2.2. Sampling and Dehydration Stress Treatment
Absolute Water Content (AWC) and Relative Water Content (RWC) Determination
2.3. Electrolyte Leakage
2.4. Chlorophyll Fluorescence
2.5. Transmission Electron Microscopy (TEM)
2.6. Plant Protein Material and iTRAQ Experimental Design
2.7. Protein Extraction and Quantification
2.8. Protein Preparation and Tryptic Digests
2.9. iTRAQ Labelling
2.10. Peptide Purification and OFFGEL Fractionation
2.11. Mass Spectrometry Settings
2.12. Mass Spectra Data Preparation
2.13. Database Selection and Searching
2.14. iTRAQ Data Processing—Protein Quantitation and Statistical Analysis
2.14.1. Data Refinement
2.14.2. protViz: For Visualising and Analysis of Proteomic Mass Spectrometry Data
2.15. Western Blot Analyses
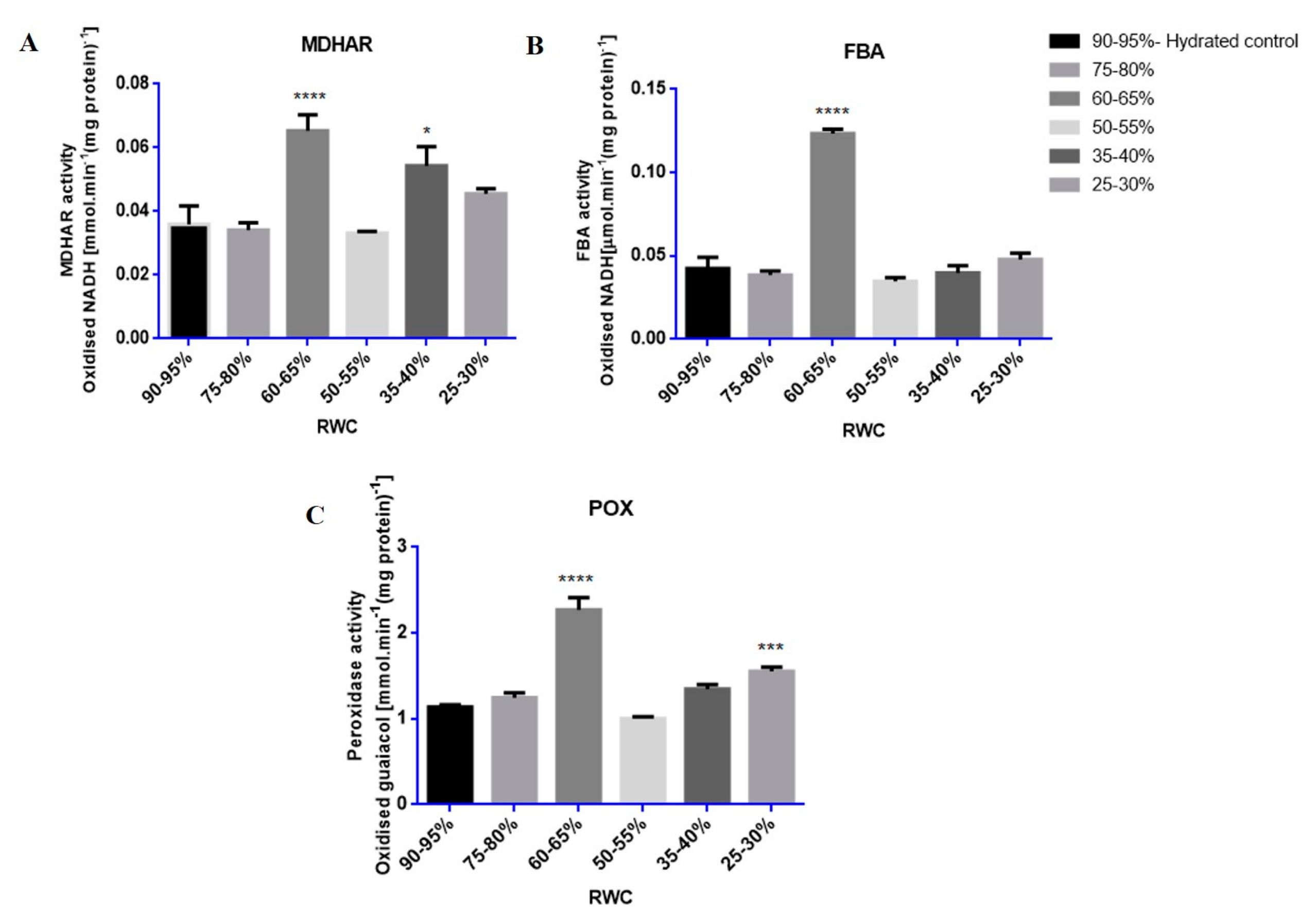
2.16. Enzyme Assays
2.16.1. Monodehydroascorbate Reductase (MDHAR, EC: 1.6.5.4)
2.16.2. Fructose-Bisphosphate Aldolase (FBA, EC: 4.1.2.13)
2.16.3. Peroxidase (POX, EC: 1.11.1.7)
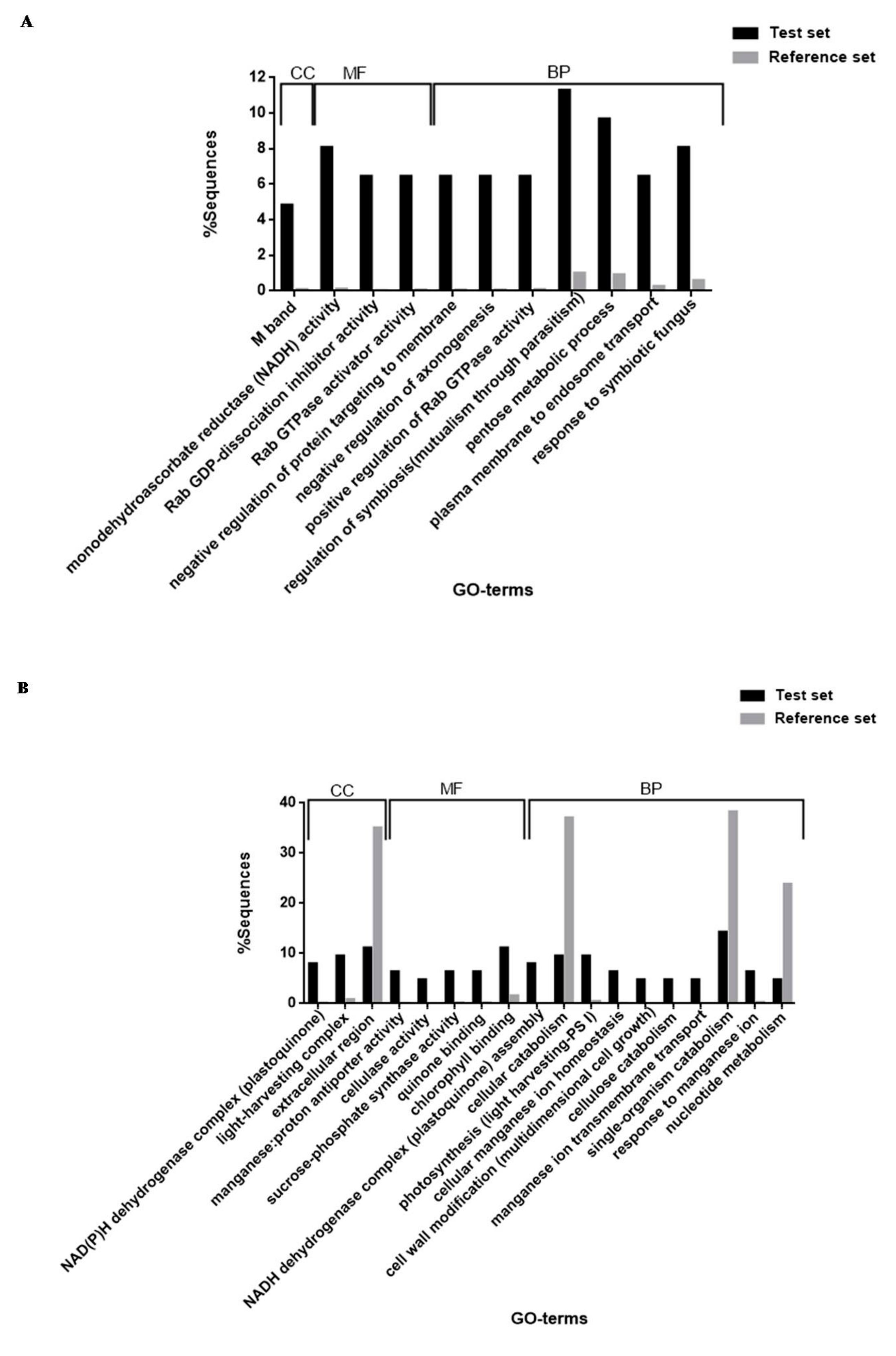
2.17. Blast2GO for Protein Identification, Annotation and Functional Enrichment Analysis
2.17.1. Protein Identification, Annotation and GO-Term Retrieval
2.17.2. Functional Enrichment Analysis
3. Results
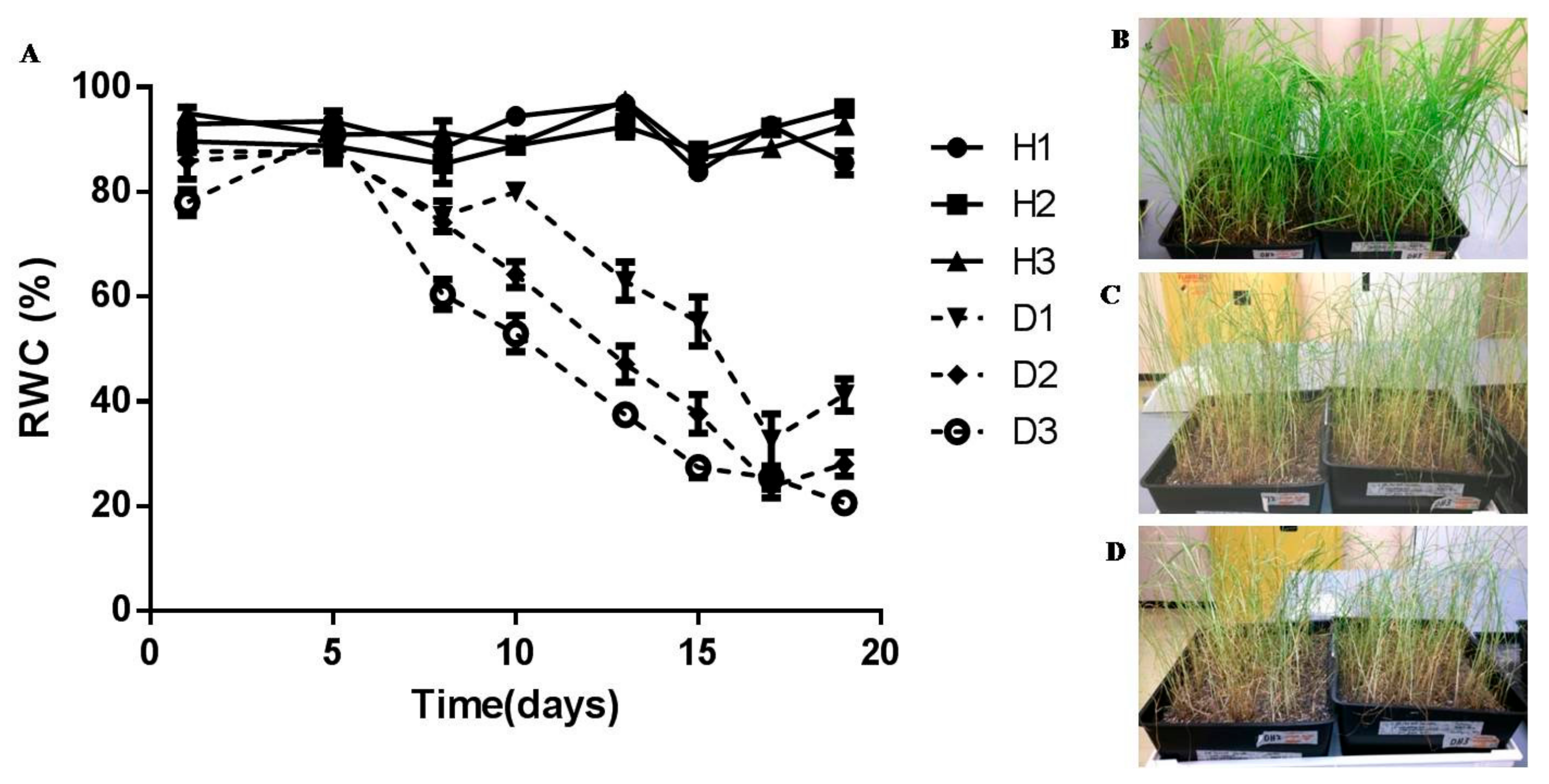
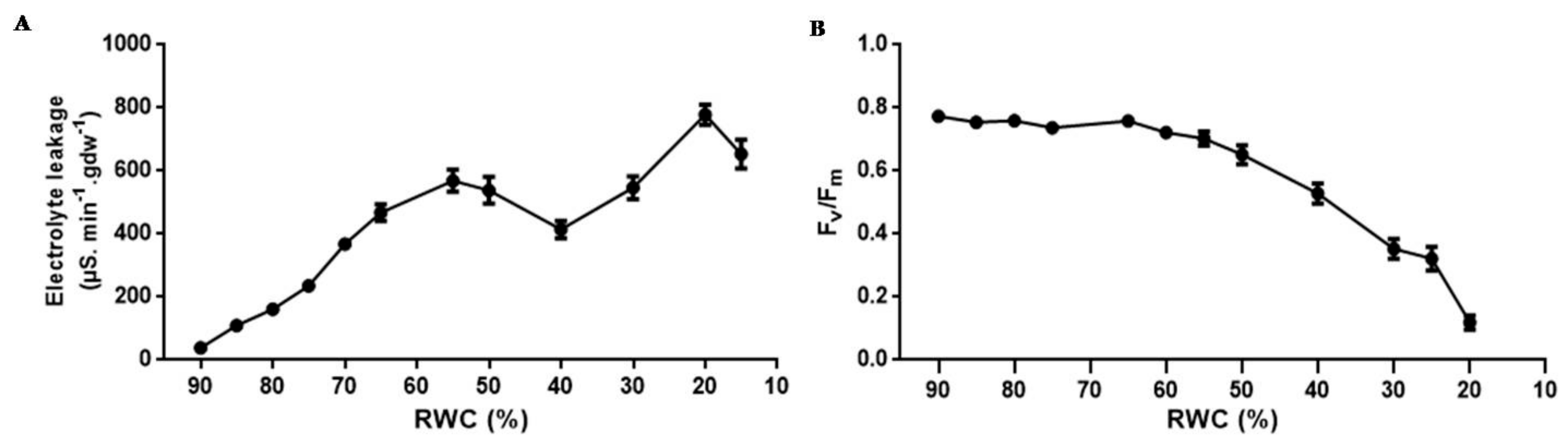
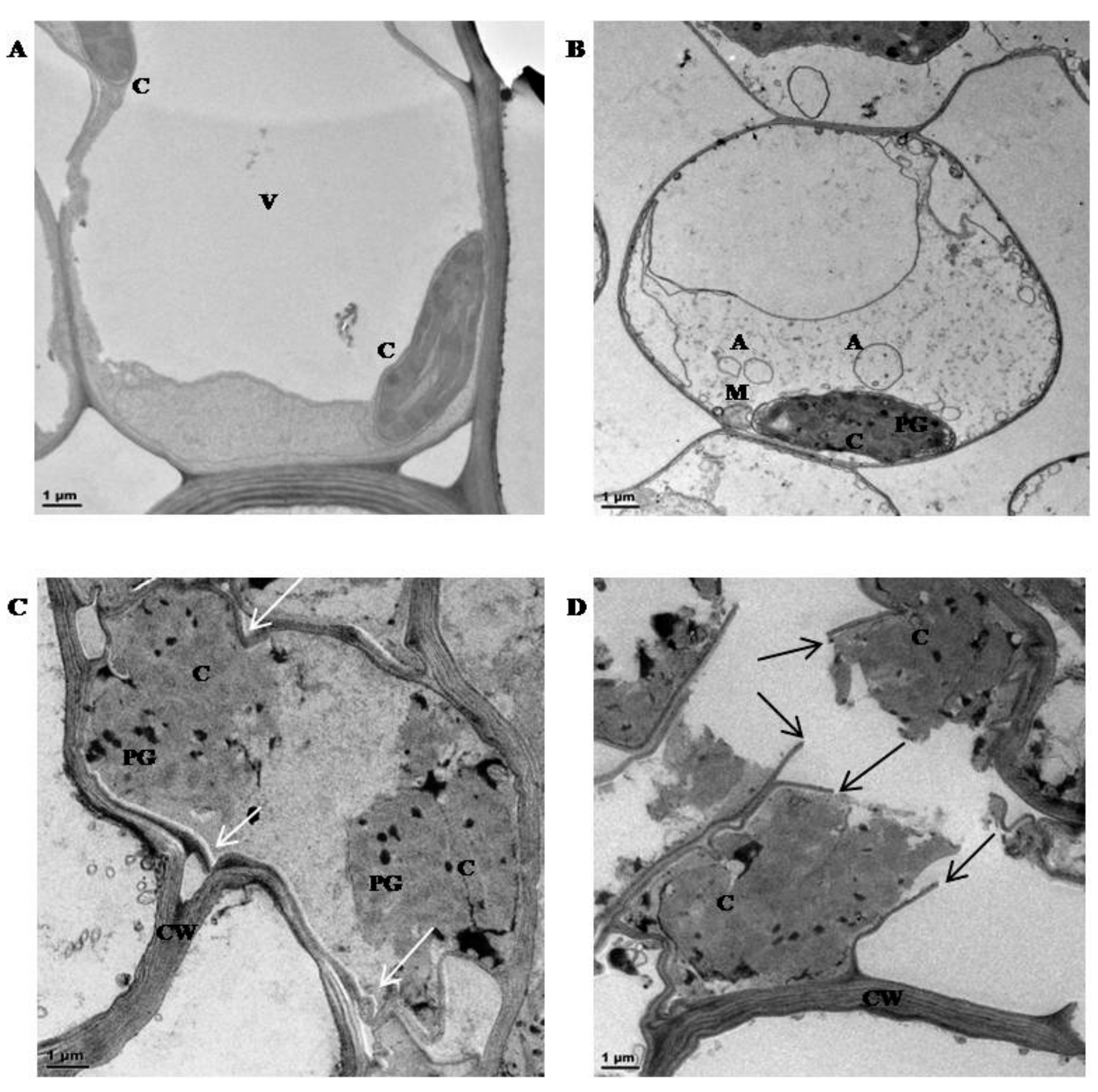
3.1. Physiological Characterisation
3.2. Tef Proteomic Analysis
Identification of Differentially Regulated Proteins
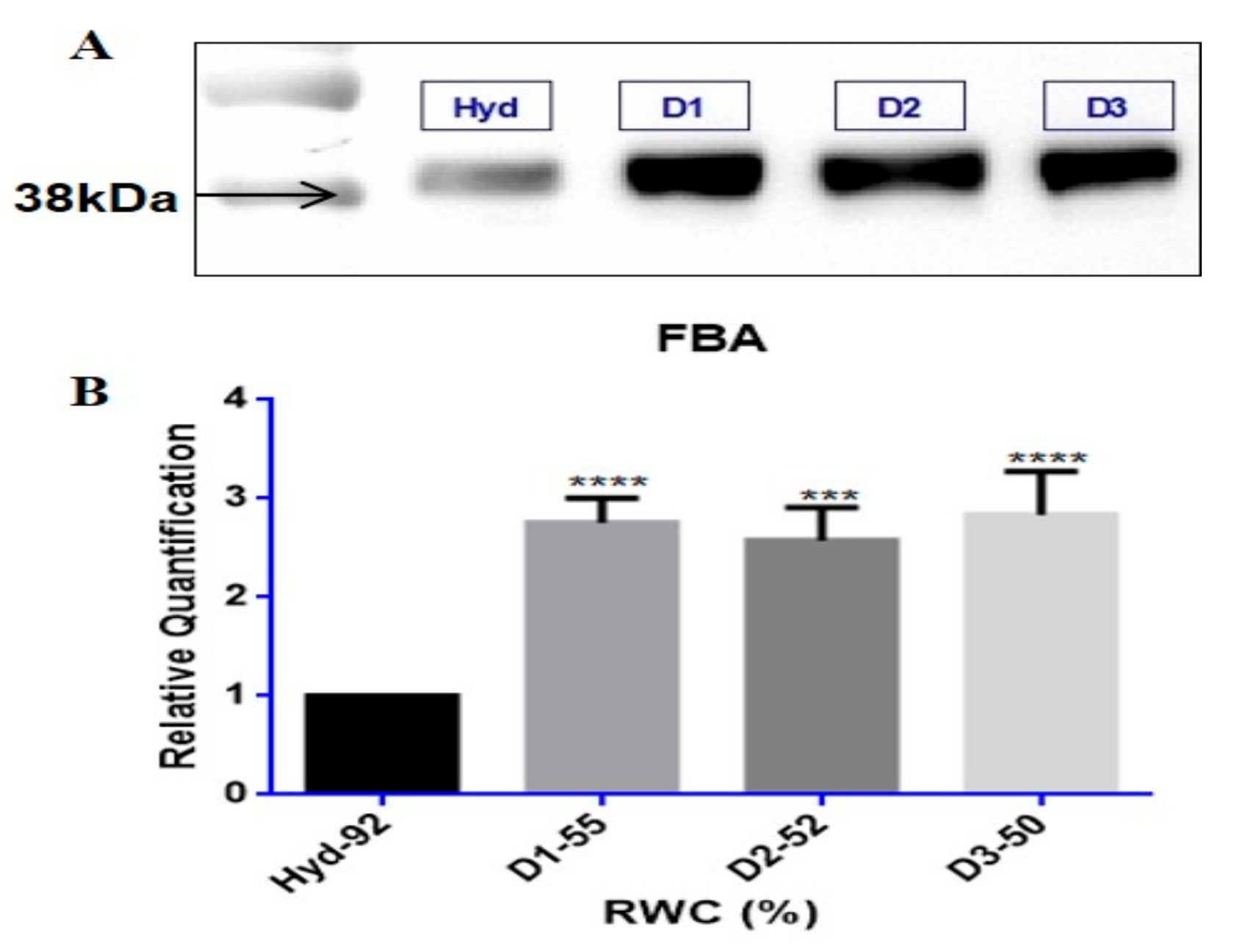
3.3. Tef Biological Validation
Western Blots and Enzyme Assays
3.4. Functional Enrichment Analysis (Fisher’s Exact Test)
4. Discussion
4.1. Physiological Characterisation
4.2. Tef Proteomics
4.3. Tef Protein Validation
4.4. Tef Bioinformatic Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AWC | Absolute water content |
| FASP | Filter assisted sample preparation procedure |
| FBA | Fructose bisphosphate aldolase |
| FDR | False discovery rate |
| GO | Gene ontology |
| GTP iTRAQ | Guanosine triphosphate Isobaric tag for relative and absolute quantitation |
| MDHAR | Monodehydroascorbate reductase |
| MS | Mass spectrometry |
| MS/MS | Tandem mass spectrometry |
| MU | Monocot unique |
| NAD POX | Nicotinamide adenine dinucleotide Peroxidase |
| ROS | Reactive oxygen species |
| RWC | Relative water content |
| TE | Tef Extended |
| TEM | Transmission electron microscopy |
| TEU | Tef Extended unique |
References
- Benešová, M.; Holá, D.; Fischer, L.; Jedelský, P.L.; Hnilička, F.; Wilhelmová, N.; Rothová, O.; Kočová, M.; Procházková, D.; Honnerová, J.; et al. The physiology and proteomics of drought tolerance in maize: Early stomatal closure as a cause of lower tolerance to short-term dehydration? PLoS ONE 2012, 7, e38017. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.R.; Chaitanya, K.V.; Vivekanandan, M. Drought-induced responses of photosynthesis and antioxidant metabolism in higher plants. J. Plant Physiol. 2004, 161, 1189–1202. [Google Scholar] [CrossRef]
- Dai, A. Increasing drought under global warming in observations and models. Nat. Clim. Chang. 2013, 3, 52–58. [Google Scholar] [CrossRef]
- Thornton, P.K.; Ericksen, P.J.; Herrero, M.; Challinor, A.J. Climate variability and vulnerability to climate change: A review. Glob. Chang. Biol. 2014, 20, 3313–3328. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.C. T’ef (Eragrostis tef) in ancient agricultural systems of highland ethiopia. Econ. Bot. 2008, 62, 547–566. [Google Scholar] [CrossRef]
- Tadele, Z.; Assefa, K. Increasing food production in Africa by boosting the productivity of understudied crops. Agronomy 2012, 2, 240–283. [Google Scholar] [CrossRef]
- Ayele, M. Genetic Diversity in tef [Eragrostis tef (Zucc.) Trotter] for Osmotic Adjustment, Root Traits, and Amplified Fragment Length Polymorphism; Texas Tech University: Lubbock, TX, USA, 1999. [Google Scholar]
- Costanza, S.H.; Dewet, J.M.J.; Harlan, J.R. Literature review and numerical taxonomy of Eragrostis tef (tef). Econ. Bot. 1979, 33, 413–424. [Google Scholar] [CrossRef]
- Tefara, H.; Belay, G. Eragrostis tef (Zuccagni) Trotter. In Plant Resources of Tropical Africa 1: Cereals and Pulses; Brink, M., Belay, G., Eds.; PROTA Foundation: Wageningen, The Netherlands, 2006; pp. 68–72. [Google Scholar]
- Tadele, Z.; Esfeld, K.; Plaza, S. Applications of high-throughput techniques to the understudied crops of Africa. Asp. Appl. Biol. 2010, 96, 233–240. [Google Scholar]
- Asfaw, K.G.; Dano, F.I. Effects of salinity on yield and yield components of tef [Eragrostis tef (Zucc.) Trotter] accessions and varieties. Curr. Res. J. Biol. Sci. 2011, 3, 289–299. [Google Scholar]
- Abate, E.; Hussein, S.; Laing, M.; Mengistu, F. Quantitative responses of tef [Eragrostis tef (Zucc.) Trotter] and weeping love grass [Eragrostis curvula (Schrad.) Nees] varieties to acid soil. Aust. J. Crop Sci. 2013, 7, 1854–1860. [Google Scholar]
- Degu, H.D.; Fujimura, T. Mapping QTLs related to plant height and root development of Eragrostis tef under drought. J. Agric. Sci. 2010, 2, 62–72. [Google Scholar] [CrossRef]
- Degu, H.D.; Ohta, M.; Fujimura, T. Drought tolerance of Eragrostis tef and development of roots. Int. J. Plant Sci. 2008, 169, 768–775. [Google Scholar] [CrossRef]
- Ginbot, Z.; Farrant, J.M. Physiological response of selected Eragrostis species to water-deficit stress. Afr. J. Biotechnol. 2011, 10, 10405–10417. [Google Scholar]
- Mengistu, D.K. The influence of soil water deficit imposed during various developmental phases on physiological processes of tef (Eragrostis tef). Agric. Ecosyst. Environ. 2009, 132, 283–289. [Google Scholar] [CrossRef]
- Shiferaw, W.; Balcha, A.; Mohammed, H. Evaluation of drought tolerance indices in tef [Eragrostis tef (Zucc.) Trotter]. Afr. J. Agric. Res. 2012, 7, 3433–3438. [Google Scholar]
- Cannarozzi, G.; Plaza-Wuthrich, S.; Esfeld, K.; Larti, S.; Wilson, Y.S.; Girma, D.; de Castro, E.; Chanyalew, S.; Blosch, R.; Farinelli, L.; et al. Genome and transcriptome sequencing identifies breeding targets in the orphan crop tef (Eragrostis tef). BMC Genom. 2014, 15, 581. [Google Scholar] [CrossRef] [PubMed]
- Ketema, S. Tef, Eragrostis tef (Zucc.) Trotter; Institute of Plant Genetics and Crop Plant Research, Gatersleben/International Plant Genetic Resources Institute: Rome, Italy, 1997; p. 52. [Google Scholar]
- Kreitschitz, A.; Tadele, Z.; Gola, E.M. Slime cells on the surface of Eragrostis seeds maintain a level of moisture around the grain to enhance germination. Seed Sci. Res. 2009, 19, 27–35. [Google Scholar] [CrossRef]
- Ingram, A.L.; Doyle, J.J. The origin and evolution of Eragrostis tef (Poaceae) and related polyploids: Evidence from nuclear waxy and plastid RPS16. Am. J. Bot. 2003, 90, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Assefa, K.; Merker, A.; Tefera, H. Inter simple sequence repeat (ISSR) analysis of genetic diversity in tef [Eragrostis tef (Zucc.) Trotter]. Hereditas 2003, 139, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Assefa, K.; Tefera, H.; Merker, A.; Kefyalew, T.; Hundera, F. Quantitative trait diversity in tef [Eragrostis tef (Zucc.) Trotter] germplasm from central and northern Ethiopia. Genet. Resour. Crop Evol. 2001, 48, 53–61. [Google Scholar] [CrossRef]
- Bai, G.; Ayele, M.; Tefera, H.; Nguyen, H.T. Genetic diversity in tef [Eragrostis tef (Zucc) Trotter] and its relatives as revealed by Random Amplified Polymorphic DNAs. Euphytica 2000, 112, 15–22. [Google Scholar] [CrossRef]
- Yu, J.K.; Graznak, E.; Breseghello, F.; Tefera, H.; Sorrells, M.E. QTL mapping of agronomic traits in tef [Eragrostis tef (Zucc) Trotter]. BMC Plant Biol. 2007, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.K.; Kantety, R.V.; Graznak, E.; Benscher, D.; Tefera, H.; Sorrells, M.E. A genetic linkage map for tef [Eragrostis tef (Zucc.) Trotter]. Theor. Appl. Genet. 2006, 113, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Zeid, M.; Assefa, K.; Haddis, A.; Chanyalew, S.; Sorrells, M.E. Genetic diversity in tef (Eragrostis tef) germplasm using SSR markers. Field Crops Res. 2012, 127, 64–70. [Google Scholar] [CrossRef]
- Zeid, M.; Belay, G.; Mulkey, S.; Poland, J.; Sorrells, M.E. QTL mapping for yield and lodging resistance in an enhanced SSR-based map for tef. Theor. Appl. Genet. 2011, 122, 77–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ayele, M.; Tefera, H.; Nguyen, H.T. RFLP linkage map of the Ethiopian cereal tef [Eragrostis tef (Zucc) Trotter]. Theor. Appl. Genet. 2001, 102, 957–964. [Google Scholar] [CrossRef]
- Assefa, K.; Yu, J.K.; Zeid, M.; Belay, G.; Tefera, H.; Sorrells, M.E. Breeding tef [Eragrostis tef (Zucc.) trotter]: Conventional and molecular approaches. Plant Breed. 2011, 130, 1–9. [Google Scholar] [CrossRef]
- Girma, D.; Assefa, K.; Chanyalew, S.; Cannarozzi, G.; Kuhlemeier, C.; Tadele, Z. The origins and progress of genomics research on tef (Eragrostis tef). Plant Biotechnol. J. 2014, 12, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Assefa, K.; Cannarozzi, G.; Girma, D.; Kamies, R.; Chanyalew, S.; Plaza-Wüthrich, S.; Blösch, R.; Rindisbacher, A.; Rafudeen, S.; Tadele, Z. Genetic diversity in tef [Eragrostis tef (Zucc.) Trotter]. Front. Plant Sci. 2015, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nanjo, Y.; Nouri, M.Z.; Komatsu, S. Quantitative proteomic analyses of crop seedlings subjected to stress conditions: A commentary. Phytochemistry 2011, 72, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Balbuena, T.S.; Dias, L.L.C.; Martins, M.L.B.; Chiquieri, T.B.; Santa-Catarina, C.; Floh, E.I.S.; Silveira, V. Challenges in proteome analyses of tropical plants. Braz. J. Plant Physiol. 2011, 23, 91–104. [Google Scholar] [CrossRef]
- Cañas, B.; López-Ferrer, D.; Ramos-Fernández, A.; Camafeita, E.; Calvo, E. Mass spectrometry technologies for proteomics. Brief. Funct. Genom. Proteom. 2006, 4, 295–320. [Google Scholar] [CrossRef] [PubMed]
- Neilson, K.A.; Gammulla, C.G.; Mirzaei, M.; Imin, N.; Haynes, P.A. Proteomic analysis of temperature stress in plants. Proteomics 2010, 10, 828–845. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.N.; Bekele, E. Amino acid composition of the cereal tef and related species of Eragrostis (Gramineae). Cereal Chem. 1981, 58, 113–115. [Google Scholar]
- Bekele, E.; Fido, R.J.; Tatham, A.S.; Shewry, P.R. Heterogeneity and polymorphism of seed proteins in tef (Eragrotis tef). Hereditas 1995, 122, 67–72. [Google Scholar] [CrossRef]
- Tatham, A.S.; Fido, R.J.; Moore, C.M.; Kasarda, D.D.; Kuzmicky, D.D.; Keen, J.N.; Shewry, P.R. Characterisation of the major prolamins of tef (Eragrostis tef) and finger millet (Eleusine coracana). J. Cereal Sci. 1996, 24, 65–71. [Google Scholar] [CrossRef]
- Maxwell, K.; Johnson, G.N. Chlorophyll fluorescence—A practical guide. J. Exp. Bot. 2000, 51, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Sherwin, H.W.; Farrant, J.M. Differences in rehydration of three desiccation-tolerant angiosperm species. Ann. Bot. 1996, 78, 703–710. [Google Scholar] [CrossRef]
- Reynolds, E.S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 1963, 17, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, T.; Damasceno, C.M.; Saravanan, R.S.; He, Y.; Catala, C.; Saladie, M.; Rose, J.K. Sample extraction techniques for enhanced proteomic analysis of plant tissues. Nat. Protoc. 2006, 1, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Stajich, J.E.; Block, D.; Boulez, K.; Brenner, S.E.; Chervitz, S.A.; Dagdigian, C.; Fuellen, G.; Gilbert, J.G.; Korf, I.; Lapp, H.; et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002, 12, 1611–1618. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Panse, C.; Grossmann, J. protViz: Visualizing and Analyzing Mass Spectrometry Related Data in Proteomics, version 0.1.26; University of Zurich: Zürich, Switzerland, 2012. [Google Scholar]
- Valyova, M.; Stoyanov, S.; Markovska, Y.; Ganeva, Y. Evaluation of in vitro antioxidant activity and free radical scavenging potential of variety of Tagetes erecta L. flowers growing in Bulgaria. Int. J. Appl. Res. Nat. Prod. 2012, 5, 19–25. [Google Scholar]
- Miyake, C.; Asada, K. Thylakoid-bound ascorbate peroxidase in spinach chloroplasts and photoreduction of its primary oxidation product monodehydroascorbate radicals in thylakoids. Plant Cell Physiol. 1992, 33, 541–553. [Google Scholar]
- Kingston-Smith, A.H.; Foyer, C.H. Overexpression of Mn-superoxide dismutase in maize leaves leads to increased monodehydroascorbate reductase, dehydroascorbate reductase and glutathione reductase activities. J. Exp. Bot. 2000, 51, 1867–1877. [Google Scholar] [CrossRef] [PubMed]
- Mundree, S.G.; Whittaker, A.; Thomson, J.A.; Farrant, J.M. An aldose reductase homolog from the resurrection plant Xerophyta viscosa Baker. Planta 2000, 211, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Dionisio-Sese, M.L.; Tobita, S. Antioxidant responses of rice seedlings to salinity stress. Plant Sci. 1998, 135, 1–9. [Google Scholar] [CrossRef]
- Chance, B.; Maehly, A.C. Assay of catalase and peroxidase. Methods Enzymol. 1955, 2, 764–775. [Google Scholar]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Bluthgen, N.; Brand, K.; Cajavec, B.; Swat, M.; Herzel, H.; Beule, D. Biological profiling of gene groups utilizing Gene Ontology. Genome Inform. 2005, 16, 106–115. [Google Scholar] [PubMed]
- Farrant, J.M.; Vander Willigen, C.; Loffell, D.A.; Bartsch, S.; Whittaker, A. An investigation into the role of light during desiccation of three angiosperm resurrection plants. Plant Cell Environ. 2003, 26, 1275–1286. [Google Scholar] [CrossRef]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef] [PubMed]
- Balsamo, R.A.; Willigen, C.V.; Bauer, A.M.; Farrant, J. Drought tolerance of selected Eragrostis species correlates with leaf tensile properties. Ann. Bot. 2006, 97, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Ahmadizadeh, M.; Valizadeh, M.; Zaefizadeh, M.; Shahbazi, H. Antioxidative protection and electrolyte leakage in Durum wheat under drought stress condition. J. Appl. Sci. Res. 2011, 73, 236–246. [Google Scholar]
- Navari-Izzo, F.; Meneguzzo, S.; Loggini, B.; Vazzana, C.; Sgherri, C.L.M. The role of the glutathione system during dehydration of Boea hygroscopica. Physiol. Plant. 1997, 99, 23–30. [Google Scholar] [CrossRef]
- Kranner, I.; Birtic, S.; Anderson, K.M.; Pritchard, H.W. Glutathione half-cell reduction potential: A universal stress marker and modulator of programmed cell death? Free Radic. Biol. Med. 2006, 40, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Smirnoff, N. The role of active oxygen in the response of plants to water deficit and desiccation. New Phytol. 1993, 125, 27–58. [Google Scholar] [CrossRef]
- Liu, Y.; Xiong, Y.; Bassham, D.C. Autophagy is required for tolerance of drought and salt stress in plants. Autophagy 2009, 5, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.; Njaci, I.; Moghaddam, L.; Long, H.; Dickman, M.B.; Zhang, X.; Mundree, S. Trehalose Accumulation Triggers Autophagy during Plant Desiccation. PLoS Genet. 2015, 11, e1005705. [Google Scholar] [CrossRef] [PubMed]
- Choe, L.; D’Ascenzo, M.; Relkin, N.R.; Pappin, D.; Ross, P.; Williamson, B.; Guertin, S.; Pribil, P.; Lee, K.H. 8-Plex Quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer’s disease. Proteomics 2007, 7, 3651–3660. [Google Scholar] [CrossRef] [PubMed]
- Karp, N.A.; Huber, W.; Sadowski, P.G.; Charles, P.D.; Hester, S.V.; Lilley, K.S. Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteom. 2010, 9, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Abu, M.; Hanger, D.P. Key issues in the acquisition and analysis of qualitative and quantitative mass spectrometry data for peptide-centric proteomic experiments. Amino Acids 2012, 43, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Cappadona, S.; Baker, P.R.; Cutillas, P.R.; Heck, A.J.R.; van Breukelen, B. Current challenges in software solutions for mass spectrometry-based quantitative proteomics. Amino Acids 2012, 43, 1087–1108. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Pevzner, P.A. False discovery rates of protein identifications: A strike against the two-peptide rule. J. Proteome Res. 2009, 8, 4173–4181. [Google Scholar] [CrossRef] [PubMed]
- Kazan, K. Alternative splicing and proteome diversity in plants: The tip of the iceberg has just emerged. Trends Plant Sci. 2003, 8, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S. Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu. Rev. Plant Biol. 2007, 58, 267–294. [Google Scholar] [CrossRef] [PubMed]
- Duque, P. A role for SR proteins in plant stress responses. Plant Signal. Behav. 2011, 6, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Staiger, D. Shaping the Arabidopsis transcriptome through alternative splicing. Adv. Bot. 2015, 2015, 13. [Google Scholar] [CrossRef]
- Carpentier, S.C.; Coemans, B.; Podevin, N.; Laukens, K.; Witters, E.; Matsumura, H.; Terauchi, R.; Swennen, R.; Panis, B. Functional genomics in a non-model crop: Transcriptomics or proteomics? Physiol. Plant. 2008, 133, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, S.C.; Panis, B.; Vertommen, A.; Swennen, R.; Sergeant, K.; Renaut, J.; Laukens, K.; Witters, E.; Samyn, B.; Devreese, B. Proteome analysis of non-model plants: A challenging but powerful approach. Mass Spectrom. Rev. 2008, 27, 354–377. [Google Scholar] [CrossRef] [PubMed]
- Romero-Rodriguez, M.C.; Pascual, J.; Valledor, L.; Jorrin-Novo, J. Improving the quality of protein identification in non-model species. Characterization of Quercus ilex seed and Pinus radiata needle proteomes by using SEQUEST and custom databases. J. Proteom. 2014, 105, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, S.C.; Witters, E.; Laukens, K.; Van Onckelen, H.; Swennen, R.; Panis, B. Banana (Musa spp.) as a model to study the meristem proteome: Acclimation to osmotic stress. Proteomics 2007, 7, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Hajheidari, M.; Abdollahian-Noghabi, M.; Askari, H.; Heidari, M.; Sadeghian, S.Y.; Ober, E.S.; Salekdeh, G.H. Proteome analysis of sugar beet leaves under drought stress. Proteomics 2005, 5, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Graciet, E.; Lebreton, S.; Gontero, B. Emergence of new regulatory mechanisms in the Benson–Calvin pathway via protein–protein interactions: A glyceraldehyde-3-phosphate dehydrogenase/CP12/phosphoribulokinase complex. J. Exp. Bot. 2004, 55, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, K.; Suzuki, N.; Iwamae, T.; Inui, M.; Yukawa, H. Increased fructose 1,6-bisphosphate aldolase in plastids enhances growth and photosynthesis of tobacco plants. J. Exp. Bot. 2012, 63, 3001–3009. [Google Scholar] [CrossRef] [PubMed]
- Salekdeh, G.H.; Komatsu, S. Crop proteomics: Aim at sustainable agriculture of tomorrow. Proteomics 2007, 7, 2976–2996. [Google Scholar] [CrossRef] [PubMed]
- Salekdeh, G.H.; Siopongco, J.; Wade, L.J.; Ghareyazie, B.; Bennett, J. Proteomic analysis of rice leaves during drought stress and recovery. Proteomics 2002, 2, 1131–1145. [Google Scholar] [CrossRef]
- Kamal, A.H.M.; Cho, K.; Kim, D.E.; Uozumi, N.; Chung, K.Y.; Lee, S.Y.; Choi, J.S.; Cho, S.W.; Shin, C.S.; Woo, S.H. Changes in physiology and protein abundance in salt-stressed wheat chloroplasts. Mol. Biol. Rep. 2012, 39, 9059–9074. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Kamal, A.H.M.; Hossain, Z. Wheat proteomics: Proteome modulation and abiotic stress acclimation. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.W.; Roy, S.K.; Kamal, A.H.; Cho, K.; Cho, S.W.; Park, C.S.; Choi, J.S.; Komatsu, S.; Woo, S.H. Proteome analysis of roots of wheat seedlings under aluminum stress. Mol. Biol. Rep. 2014, 41, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Berla, B.M.; Sheffield, J.; Cahoon, R.E.; Jez, J.M.; Hicks, L.M. Comprehensive analysis of the Brassica juncea root proteome in response to cadmium exposure by complementary proteomic approaches. Proteomics 2009, 9, 2419–2431. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.J.; Deng, J.S.; Chen, H.J.; Huang, S.S.; Shih, C.C.; Lin, Y.H. Dehydroascorbate reductase and monodehydroascorbate reductase activities of two metallothionein-like proteins from sweet potato (Ipomoea batatas [L.] Lam. ‘Tainong 57’) storage roots. Bot. Stud. 2013, 54, 7. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Kumar, R.G.; Verma, S.; Dubey, R.S. Effect of cadmium on lipid peroxidation, superoxide anion generation and activities of antioxidant enzymes in growing rice seedlings. Plant Sci. 2001, 161, 1135–1144. [Google Scholar] [CrossRef]
- Zhang, J.; Kirkham, M.B. Drought-stress-induced changes in activities of superoxide dismutase, catalase, and peroxidase in wheat species. Plant Cell Physiol. 1994, 35, 785–791. [Google Scholar] [CrossRef]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Van Breusegem, F. Reactive oxygen gene network of plants. Trends Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Sreenivasulu, N.; Ramanjulu, S.; Ramachandra-Kini, K.; Prakash, H.S.; Shekar-Shetty, H.; Savithri, H.S.; Sudhakar, C. Total peroxidase activity and peroxidase isoforms as modified by salt stress in two cultivars of fox-tail millet with differential salt tolerance. Plant Sci. 1999, 141, 1–9. [Google Scholar] [CrossRef]
- Chakraborty, U.; Pradhan, B. Oxidative stress in five wheat varieties (Triticum aestivum L.) exposed to water stress and study of their antioxidant enzyme defense system, water stress responsive metabolites and H2O2 accumulation. Braz. J. Plant Physiol. 2012, 24, 117–130. [Google Scholar] [CrossRef]
- Abedi, T.; Pakniyat, H. Antioxidant enzyme changes in response to drought stress in ten cultivars of oilseed Rape (Brassica napus L.). Czech J. Genet. Plant Breed. 2010, 46, 27–34. [Google Scholar]
- Nazarli, H.; Zardashti, M.R.; Darvishzadeh, R.; Mohammadi, M. Change in activity of antioxidative enzymes in young leaves of sunflower (Helianthus annuus L.) by application of super absorbent synthetic polymers under drought stress condition. Aust. J. Crop Sci. 2011, 5, 1334–1338. [Google Scholar]
- Murthy, S.M.; Devaraj, V.R.; Anitha, P.; Tejavathi, D.H. Studies on the activities of antioxidant enzymes under induced drought stress in in vivo and in vitro plants of Macrotyloma uniflorum (Lam.) Verdc. Recent Res. Sci. Technol. 2012, 4, 34–37. [Google Scholar]
- Quan, L.J.; Zhang, B.; Shi, W.W.; Li, H.Y. Hydrogen peroxide in plants: A versatile molecule of the reactive oxygen species network. J. Integr. Plant Biol. 2008, 50, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Morell, S.; Follmann, H.; De Tullio, M.; Häberlein, I. Dehydroascorbate and dehydroascorbate reductase are phantom indicators of oxidative stress in plants. FEBS Lett. 1997, 414, 567–570. [Google Scholar] [CrossRef]
- Zarsky, V.; Cvrckova, F.; Bischoff, F.; Palme, K. At-GDI1 from Arabidopsis thaliana encodes a rab-specific GDP dissociation inhibitor that complements the sec19 mutation of Saccharomyces cerevisiae. FEBS Lett. 1997, 403, 303–308. [Google Scholar] [CrossRef]
- Chrispeels, M.J.; Crawford, N.M.; Schroeder, J.I. Proteins for Transport of Water and Mineral Nutrients across the Membranes of Plant Cells. Plant Cell 1999, 11, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.Y.; De Vries, S.C. Membrane trafficking: Intracellular highways and country roads. Plant Physiol. 2008, 147, 1451–1453. [Google Scholar] [CrossRef] [PubMed]
- Vernoud, V.; Horton, A.C.; Yang, Z.; Nielsen, E. Analysis of the small GTPase gene superfamily of Arabidopsis. Plant Physiol. 2003, 131, 1191–1208. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K.; Agarwal, P.; Jain, P.; Jha, B.; Reddy, M.K.; Sopory, S.K. Constitutive overexpression of a stress-inducible small GTP-binding protein PgRab7 from Pennisetum glaucum enhances abiotic stress tolerance in transgenic tobacco. Plant Cell Rep. 2008, 27, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Guo, J.; Bai, P.; Duan, Y.; Wang, X.; Cheng, Y.; Feng, H.; Huang, L.; Kang, Z. Wheat TaRab7 GTPase is part of the signaling pathway in responses to stripe rust and abiotic stimuli. PLoS ONE 2012, 7, e37146. [Google Scholar] [CrossRef] [PubMed]
- Stallkneecht, G.F.; Gilberson, K.M.; Eckhoff, J.L. Tef: Food crop for humans and animals. In New Crops; Janick, J., Simon, J.E., Eds.; Wiley: New York, NY, USA, 1993; pp. 231–234. [Google Scholar]
- Couee, I.; Sulmon, C.; Gouesbet, G.; El Amrani, A. Involvement of soluble sugars in reactive oxygen species balance and responses to oxidative stress in plants. J. Exp. Bot. 2006, 57, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Jain, M. Emerging role of metabolic pathways in abiotic stress tolerance. J. Plant Biochem. Physiol. 2013, 1, 108. [Google Scholar] [CrossRef]
- Jacoby, R.P.; Li, L.; Huang, S.; Pong Lee, C.; Millar, A.H.; Taylor, N.L. Mitochondrial composition, function and stress response in plants. J. Integr. Plant Biol. 2012, 54, 887–906. [Google Scholar] [CrossRef] [PubMed]
- Keunen, E.; Remans, T.; Bohler, S.; Vangronsveld, J.; Cuypers, A. Metal-induced oxidative stress and plant mitochondria. Int. J. Mol. Sci. 2011, 12, 6894–6918. [Google Scholar] [CrossRef] [PubMed]
- Peltier, G.; Cournac, L. Chlororespiration. Annu. Rev. Plant Biol. 2002, 53, 523–550. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Yamamoto, H.; Shikanai, T. Structure and biogenesis of the chloroplast NAD(P)H dehydrogenase complex. (BBA) Bioenergetics 2011, 1807, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Rumeau, D.; Becuwe-Linka, N.; Beyly, A.; Louwagie, M.; Garin, J.; Peltier, G. New subunits NDH-M, -N, and -O, encoded by nuclear genes, are essential for plastid Ndh complex functioning in higher plants. Plant Cell 2005, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Huda, K.M.K.; Banu, M.S.A.; Tuteja, R.; Tuteja, N. Global calcium transducer P-type Ca2+-ATPases open new avenues for agriculture by regulating stress signalling. J. Exp. Bot. 2013, 64, 3099–3109. [Google Scholar] [CrossRef] [PubMed]
- Peiter, E.; Montanini, B.; Gobert, A.; Pedas, P.; Husted, S.; Maathuis, F.J.M.; Blaudez, D.; Chalot, M.; Sanders, D. A secretory pathway-localized cation diffusion facilitator confers plant manganese tolerance. Proc. Natl. Acad. Sci. USA 2007, 104, 8532–8537. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, H.; Philippe, F.; Domon, J.-M.; Gillet, F.; Pelloux, J.; Rayon, C. Cell wall metabolism in response to abiotic stress. Plants 2015, 4, 112. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.G.; Dumbroff, E.B. Turgor regulation via cell wall adjustment in white spruce. Plant Physiol. 1999, 119, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.P.; Nguema-Ona, E.; Chevalier, L.; Lindsey, G.G.; Brandt, W.F.; Lerouge, P.; Farrant, J.M.; Driouich, A. Response of the leaf cell wall to desiccation in the resurrection plant Myrothamnus flabellifolius. Plant Physiol. 2006, 141, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cosgrove, D.J. Adaptation of roots to low water potentials by changes in cell wall extensibility and cell wall proteins. J. Exp. Bot. 2000, 51, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Pelleschi, S.; Rocher, J.P.; Prioul, J.L. Effect of water restriction on carbohydrate metabolism and photosynthesis in mature maize leaves. Plant Cell Environ. 1997, 20, 493–503. [Google Scholar] [CrossRef]
- Du, Y.C.; Nose, A.; Wasano, K. Effects of chilling temperature on photosynthetic rates, photosynthetic enzyme activities and metabolite levels in leaves of three sugarcane species. Plant Cell Environ. 1999, 22, 317–324. [Google Scholar] [CrossRef]
- Whittaker, A.; Martinelli, T.; Farrant, J.M.; Bochicchio, A.; Vazzana, C. Sucrose phosphate synthase activity and the co-ordination of carbon partitioning during sucrose and amino acid accumulation in desiccation-tolerant leaf material of the C4 resurrection plant Sporobolus stapfianus during dehydration. J. Exp. Bot. 2007, 58, 3775–3787. [Google Scholar] [CrossRef] [PubMed]
- Abreu, I.A.; Farinha, A.P.; Negrão, S.; Gonçalves, N.; Fonseca, C.; Rodrigues, M.; Batista, R.; Saibo, N.J.M.; Oliveira, M.M. Coping with abiotic stress: Proteome changes for crop improvement. J. Proteom. 2013, 93, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, N.A. Alternative splicing confers a dual role in polar auxin transport and drought stress tolerance to the major facilitator superfamily transporter ZIFL1. Plant Cell 2013, 25, 779. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, N.J.; Urwin, P.E. The interaction of plant biotic and abiotic stresses: From genes to the field. J. Exp. Bot. 2012, 63, 3523–3543. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein ID | Protein Description | Hydrated (Group 1) | Dehydrated (Group 2) | Fold Change | p-Value |
|---|---|---|---|---|---|
| CL1Contig10009 | ---NA--- | 7.22 | 8.09 | 1.12 | 0.001 |
| CL5492Contig2 | fructose-bisphosphate aldolase cytoplasmic isozyme | 10.70 | 10.86 | 1.02 | 0.001 |
| CL856Contig3 | 40s ribosomal protein s28 | 9.17 | 9.33 | 1.02 | 0.001 |
| CL856Contig4 | 40s ribosomal protein s28 | 9.17 | 9.33 | 1.02 | 0.001 |
| CL68Contig25 | peroxidase 3-rare cold-inducible protein | 7.14 | 7.73 | 1.08 | 0.002 |
| CL4104Contig2 | gras family protein 2 | 0.69 | 4.68 | 6.75 | 0.003 |
| CL3156Contig1 | ---NA--- | 0.69 | 6.05 | 8.73 | 0.004 |
| CL6974Contig4 | ---NA--- | 0.69 | 6.05 | 8.73 | 0.004 |
| CL715Contig2 | protease includes: reverse transcriptase inlcudes: endonuclease | 9.03 | 9.43 | 1.04 | 0.004 |
| Locus_2136_1_5 | ENO2_ERATE, Enolase 2 | 11.01 | 11.18 | 1.02 | 0.004 |
| CL8690Contig2 | ---NA--- | 0.69 | 5.34 | 7.70 | 0.005 |
| CL1942Contig1 | ---NA--- | 0.69 | 4.53 | 6.54 | 0.006 |
| CL2699Contig6 | monodehydroascorbate cytoplasmic isoform 2 | 0.69 | 4.53 | 6.54 | 0.006 |
| isotig02308 | probable monodehydroascorbate cytoplasmic isoform 2 | 0.69 | 4.53 | 6.54 | 0.006 |
| CL2976Contig3 | ---NA--- | 0.69 | 4.94 | 7.12 | 0.007 |
| CL68Contig6 | peroxidase 3-rare cold-inducible protein | 0.66 | 4.49 | 6.82 | 0.008 |
| CL799Contig2 | hua2-like protein 2 | 9.35 | 9.58 | 1.02 | 0.008 |
| CL8983Contig3 | ---NA--- | 0.66 | 4.49 | 6.82 | 0.008 |
| CL2991Contig2 | hydroxyphenylpyruvate reductase | 10.22 | 10.29 | 1.01 | 0.009 |
| CL3629Contig1 | poly polymerase i | 0.69 | 5.13 | 7.41 | 0.013 |
| CL3629Contig2 | poly polymerase i | 0.69 | 5.13 | 7.41 | 0.013 |
| CL7746Contig3 | ---NA--- | 0.69 | 5.04 | 7.28 | 0.013 |
| CL873Contig3 | cyclin-p4-1 | 0.69 | 5.04 | 7.28 | 0.013 |
| CL2289Contig1 | ---NA--- | 6.60 | 7.55 | 1.14 | 0.014 |
| CL2761Contig4 | red chlorophyll catabolite reductase | 8.78 | 9.12 | 1.04 | 0.014 |
| CL5492Contig1 | fructose-bisphosphate aldolase cytoplasmic isozyme | 10.66 | 10.84 | 1.02 | 0.014 |
| CL1Contig421 | calcium-dependent protein kinase 5 | 7.83 | 8.40 | 1.07 | 0.015 |
| CL1Contig7756 | serine carboxypeptidase-like 51 | 9.51 | 9.77 | 1.03 | 0.015 |
| CL3374Contig5 | ---NA--- | 0.69 | 5.41 | 7.80 | 0.016 |
| CL57Contig23 | ---NA--- | 0.69 | 5.41 | 7.80 | 0.016 |
| CL4404Contig1 | ---NA--- | 4.75 | 6.49 | 1.37 | 0.017 |
| CL8759Contig1 | proteasome subunit beta type-3 | 4.75 | 6.49 | 1.37 | 0.017 |
| CL8759Contig2 | proteasome subunit beta type-3 | 4.75 | 6.49 | 1.37 | 0.017 |
| CL1Contig7029 | protease includes: reverse transcriptase includes:endonuclease | 6.94 | 7.76 | 1.12 | 0.018 |
| CL1Contig4553 | chlorophyll a-b binding protein chloroplastic | 7.16 | 7.82 | 1.09 | 0.019 |
| CL2228Contig1 | s phase cyclin a-associated protein in the endoplasmic reticulum | 9.04 | 9.19 | 1.02 | 0.019 |
| CL124Contig7 | ---NA--- | 8.82 | 9.24 | 1.05 | 0.02 |
| CL415Contig1 | glutathione hydrolase 3 | 6.58 | 7.51 | 1.14 | 0.02 |
| CL124Contig2 | ---NA--- | 8.30 | 8.72 | 1.05 | 0.021 |
| CL3894Contig4 | leucoanthocyanidin dioxygenase | 0.69 | 5.49 | 7.92 | 0.021 |
| CL1498Contig7 | guanosine nucleotide diphosphate dissociation inhibitor 2 | 5.56 | 6.77 | 1.22 | 0.022 |
| CL413Contig11 | u-box domain-containing protein 4 | 7.18 | 7.96 | 1.11 | 0.022 |
| CL8890Contig3 | ENO3_ERATE, Enolase 3 | 10.96 | 11.08 | 1.01 | 0.022 |
| CL1888Contig1 | glycerophosphodiester phosphodiesterase gdpdl3 | 11.40 | 11.61 | 1.02 | 0.023 |
| CL1888Contig2 | glycerophosphodiester phosphodiesterase gdpdl3 | 11.40 | 11.61 | 1.02 | 0.023 |
| CL1498Contig4 | guanosine nucleotide diphosphate dissociation inhibitor 1 | 6.53 | 7.06 | 1.08 | 0.024 |
| CL1498Contig5 | guanosine nucleotide diphosphate dissociation inhibitor 2 | 6.53 | 7.06 | 1.08 | 0.024 |
| CL5028Contig3 | plant intracellular ras-group-related lrr protein 6 | 8.98 | 9.21 | 1.03 | 0.024 |
| isotig02787 | guanosine nucleotide diphosphate dissociation inhibitor 2 | 6.53 | 7.06 | 1.08 | 0.024 |
| CL3527Contig4 | nucleolar complex protein 2 homolog | 6.24 | 7.62 | 1.22 | 0.027 |
| CL1Contig6763 | ---NA--- | 5.57 | 6.63 | 1.19 | 0.028 |
| CL1224Contig6 | gtp-binding protein sar1a | 7.63 | 8.20 | 1.07 | 0.029 |
| CL90Contig16 | ---NA--- | 7.59 | 8.02 | 1.06 | 0.029 |
| CL18849Contig1 | ---NA--- | 5.79 | 6.54 | 1.13 | 0.031 |
| CL4737Contig2 | acetohydroxy-acid reductoisomerase | 9.65 | 9.89 | 1.03 | 0.032 |
| CL577Contig14 | ubiquinol oxidase chloroplastic chromoplastic | 9.39 | 9.56 | 1.02 | 0.032 |
| CL24657Contig1 | fructokinase-1 | 7.48 | 7.92 | 1.06 | 0.036 |
| CL546Contig2 | f-box protein skip24 | 5.26 | 6.16 | 1.17 | 0.036 |
| CL7996Contig1 | fructokinase-1 | 7.48 | 7.92 | 1.06 | 0.036 |
| CL1073Contig1 | peptidyl-prolyl cis-trans isomerase d | 7.15 | 7.79 | 1.09 | 0.037 |
| CL3347Contig4 | delta-aminolevulinic acid chloroplastic | 8.68 | 9.00 | 1.04 | 0.037 |
| CL5Contig21 | probable wrky transcription factor 19 | 7.72 | 8.19 | 1.06 | 0.037 |
| CL7405Contig3 | chlorophyll a-b binding protein cp24 chloroplastic s | 10.71 | 10.85 | 1.01 | 0.038 |
| CL136Contig17 | ---NA--- | 7.58 | 8.24 | 1.09 | 0.04 |
| CL1Contig5054 | f-box only protein 8 | 7.30 | 7.68 | 1.05 | 0.04 |
| CL326Contig6 | ---NA--- | 4.92 | 6.56 | 1.33 | 0.04 |
| CL3687Contig5 | ---NA--- | 4.92 | 6.56 | 1.33 | 0.04 |
| CL4000Contig1 | monodehydroascorbate reductase | 9.99 | 10.26 | 1.03 | 0.04 |
| CL4000Contig2 | monodehydroascorbate reductase | 9.99 | 10.26 | 1.03 | 0.04 |
| CL4000Contig3 | monodehydroascorbate reductase | 9.99 | 10.26 | 1.03 | 0.04 |
| CL4207Contig1 | ---NA--- | 4.92 | 6.56 | 1.33 | 0.04 |
| CL445Contig4 | elongator complex protein 6 | 4.92 | 6.56 | 1.33 | 0.04 |
| CL445Contig6 | elongator complex protein 7 | 4.92 | 6.56 | 1.33 | 0.04 |
| CL4771Contig3 | ---NA--- | 4.92 | 6.56 | 1.33 | 0.04 |
| CL4956Contig4 | premnaspirodiene oxygenase | 4.92 | 6.56 | 1.33 | 0.04 |
| CL522Contig8 | ---NA--- | 4.92 | 6.56 | 1.33 | 0.04 |
| CL6050Contig1 | ---NA--- | 4.92 | 6.56 | 1.33 | 0.04 |
| CL7668Contig1 | nadh dehydrogenase complex assembly factor 6 | 6.77 | 7.66 | 1.13 | 0.04 |
| CL7668Contig2 | nadh dehydrogenase complex assembly factor 6 | 6.77 | 7.66 | 1.13 | 0.04 |
| CL837Contig7 | cell division cycle protein 48 homolog | 10.78 | 10.96 | 1.02 | 0.04 |
| CL102Contig20 | ---NA--- | 6.34 | 7.08 | 1.12 | 0.041 |
| CL5577Contig3 | ---NA--- | 7.34 | 7.84 | 1.07 | 0.041 |
| CL1Contig7889 | ---NA--- | 8.42 | 9.03 | 1.07 | 0.042 |
| CL2761Contig3 | red chlorophyll catabolite reductase | 8.59 | 8.97 | 1.04 | 0.042 |
| CL836Contig11 | probable polyamine transporter | 7.78 | 8.38 | 1.08 | 0.042 |
| CL4591Contig2 | phosphatidylinositol n-acetylglucosaminyltransferase subunit a | 8.95 | 9.18 | 1.03 | 0.043 |
| CL61Contig20 | probable ufm1-specific protease | 8.15 | 8.64 | 1.06 | 0.043 |
| CL61Contig7 | probable ufm1-specific protease | 8.15 | 8.64 | 1.06 | 0.043 |
| CL4852Contig2 | ---NA--- | 9.18 | 9.68 | 1.05 | 0.046 |
| CL2637Contig1 | peroxisome biogenesis protein 6 (PEX6) | 9.85 | 10.06 | 1.02 | 0.047 |
| CL680Contig10 | elongation factor tu gtp-binding domain-containing protein 2 | 7.43 | 8.05 | 1.08 | 0.048 |
| CL680Contig5 | elongation factor tu gtp-binding domain-containing protein 2 | 7.43 | 8.05 | 1.08 | 0.048 |
| CL7065Contig1 | ---NA--- | 5.58 | 6.49 | 1.16 | 0.048 |
| CL7065Contig2 | ---NA--- | 5.58 | 6.49 | 1.16 | 0.048 |
| CL140Contig10 | npk1-activating kinesin-1 | 8.98 | 9.27 | 1.03 | 0.049 |
| CL4289Contig6 | ---NA--- | 8.29 | 8.58 | 1.03 | 0.049 |
| isotig08284 | protein disulfide isomerase-like 1-1 | 10.76 | 10.98 | 1.02 | 0.049 |
| Protein ID | Protein Description | Hydrated (Group 1) | Dehydrated (Group 2) | Fold Change | p-Value |
|---|---|---|---|---|---|
| CL5604Contig1 | 2-methyl-6-phytyl-hydroquinone methyltransferase chloroplastic | 9.36 | 8.94 | 0.96 | 0.001 |
| CL977Contig4 | ---NA--- | 7.70 | 7.08 | 0.92 | 0.001 |
| CL2349Contig3 | protein dek | 9.77 | 9.35 | 0.96 | 0.002 |
| CL36Contig35 | nad-dependent malic enzyme 59 kda mitochondrial | 10.52 | 10.36 | 0.99 | 0.002 |
| CL5457Contig2 | ---NA--- | 9.09 | 8.91 | 0.98 | 0.002 |
| CL11972Contig1 | metal tolerance protein 5 | 6.23 | 1.19 | 0.19 | 0.004 |
| CL700Contig3 | nad-dependent malic enzyme 62 kda mitochondrial | 9.37 | 9.12 | 0.97 | 0.005 |
| CL236Contig5 | probable sucrose-phosphate synthase 2 | 9.46 | 9.21 | 0.97 | 0.006 |
| CL236Contig6 | probable sucrose-phosphate synthase 2 | 9.44 | 9.18 | 0.97 | 0.006 |
| CL1456Contig11 | s-adenosylmethionine decarboxylase proenzyme | 5.33 | 0.69 | 0.13 | 0.007 |
| CL1456Contig8 | s-adenosylmethionine decarboxylase proenzyme | 5.33 | 0.69 | 0.13 | 0.007 |
| CL2948Contig2 | haloalkane dehalogenase | 9.85 | 9.63 | 0.98 | 0.007 |
| isotig10649 | s-adenosylmethionine decarboxylase proenzyme | 5.33 | 0.69 | 0.13 | 0.007 |
| CL13Contig40 | rhodanese-like domain-containing protein 10 | 8.66 | 7.80 | 0.90 | 0.009 |
| CL1595Contig2 | ---NA--- | 4.99 | 0.69 | 0.14 | 0.009 |
| CL1042Contig2 | alpha-glucan water chloroplastic | 9.41 | 9.13 | 0.97 | 0.01 |
| CL19Contig25 | ---NA--- | 8.92 | 8.29 | 0.93 | 0.01 |
| CL7534Contig1 | cellulose synthase-like protein a9 | 7.36 | 6.55 | 0.89 | 0.01 |
| CL7716Contig2 | ---NA--- | 7.78 | 7.25 | 0.93 | 0.013 |
| CL7716Contig3 | Putative uncharacterized protein CysX | 7.78 | 7.25 | 0.93 | 0.013 |
| CL7582Contig1 | ribosomal rna processing protein 36 homolog | 8.47 | 7.75 | 0.92 | 0.014 |
| CL14686Contig1 | alliin lyase 1 | 5.51 | 0.69 | 0.13 | 0.015 |
| CL2382Contig6 | chlorophyll a-b binding chloroplastic | 9.39 | 8.99 | 0.96 | 0.015 |
| Locus_49_75_82 | ---NA--- | 11.12 | 10.92 | 0.98 | 0.015 |
| CL1759Contig3 | ---NA--- | 7.61 | 7.19 | 0.94 | 0.016 |
| CL1Contig8969 | ---NA--- | 10.54 | 10.40 | 0.99 | 0.016 |
| CL14672Contig1 | ---NA--- | 8.33 | 7.87 | 0.94 | 0.017 |
| CL16131Contig1 | ---NA--- | 8.33 | 7.87 | 0.94 | 0.017 |
| CL456Contig16 | ---NA--- | 10.12 | 9.95 | 0.98 | 0.017 |
| CL7059Contig2 | cytochrome b561 and domon domain-containing protein | 8.00 | 7.36 | 0.92 | 0.017 |
| CL9348Contig2 | ras-related protein raba5c; ras-related protein ara-4; ras-related protein rab11f | 9.07 | 8.73 | 0.96 | 0.017 |
| CL1630Contig1 | nad h azoreductase | 9.33 | 9.12 | 0.98 | 0.018 |
| CL3227Contig1 | ---NA--- | 5.69 | 0.69 | 0.12 | 0.018 |
| CL3227Contig2 | ---NA--- | 5.69 | 0.69 | 0.12 | 0.018 |
| CL58Contig14 | ---NA--- | 5.69 | 0.69 | 0.12 | 0.018 |
| CL58Contig2 | ---NA--- | 5.69 | 0.69 | 0.12 | 0.018 |
| CL467Contig12 | ---NA--- | 9.78 | 9.57 | 0.98 | 0.02 |
| isotig23406 | photosystem ii protein d1 | 10.59 | 10.33 | 0.98 | 0.02 |
| CL1Contig3562 | ---NA--- | 8.20 | 7.53 | 0.92 | 0.022 |
| CL3294Contig3 | nad h-quinone oxidoreductase subunit chloroplastic | 9.37 | 9.17 | 0.98 | 0.022 |
| CL3294Contig4 | nad h-quinone oxidoreductase subunit chloroplastic | 9.37 | 9.17 | 0.98 | 0.022 |
| CL3294Contig5 | nad h-quinone oxidoreductase subunit chloroplastic | 9.37 | 9.17 | 0.98 | 0.022 |
| CL3294Contig6 | nad h-quinone oxidoreductase subunit chloroplastic | 9.37 | 9.17 | 0.98 | 0.022 |
| CL6495Contig2 | polyamine oxidase | 8.91 | 8.42 | 0.95 | 0.022 |
| comp294_c0_seq1 | nad h-quinone oxidoreductase subunit chloroplastic | 9.37 | 9.17 | 0.98 | 0.022 |
| CL5963Contig1 | 60s ribosomal protein l5-1 | 10.95 | 10.86 | 0.99 | 0.023 |
| CL8805Contig2 | ---NA--- | 9.60 | 9.14 | 0.95 | 0.023 |
| CL5672Contig2 | ---NA--- | 8.49 | 8.03 | 0.95 | 0.024 |
| CL6932Contig1 | ---NA--- | 9.87 | 9.60 | 0.97 | 0.024 |
| Locus_954_4_4 | ---NA--- | 9.87 | 9.60 | 0.97 | 0.024 |
| CL4237Contig3 | SYN3_ERATE, Asparagine tRNA ligase 3 | 9.96 | 9.63 | 0.97 | 0.024 |
| CL4237Contig2 | SYN8_ERATE, Asparagine tRNA ligase 8 | 9.96 | 9.63 | 0.97 | 0.024 |
| CL1805Contig10 | protein dj-1 homolog b | 8.06 | 7.57 | 0.94 | 0.025 |
| CL1805Contig2 | protein dj-1 homolog b | 8.06 | 7.57 | 0.94 | 0.025 |
| CL1Contig3266 | ---NA--- | 7.90 | 7.43 | 0.94 | 0.025 |
| CL327Contig3 | cbs domain-containing protein cbsppr1 | 7.85 | 7.37 | 0.94 | 0.025 |
| Locus_2288_7_9 | ---NA--- | 9.38 | 9.05 | 0.97 | 0.025 |
| CL2320Contig2 | ---NA--- | 7.27 | 6.85 | 0.94 | 0.026 |
| CL977Contig1 | ---NA--- | 7.27 | 6.85 | 0.94 | 0.026 |
| CL10226Contig1 | ---NA--- | 8.21 | 7.20 | 0.88 | 0.027 |
| CL2336Contig7 | ---NA--- | 9.33 | 9.15 | 0.98 | 0.027 |
| CL1Contig5286 | metal tolerance protein 5 | 5.00 | 0.69 | 0.14 | 0.029 |
| CL1Contig5699 | metal tolerance protein 6 | 5.00 | 0.69 | 0.14 | 0.029 |
| CL1Contig8303 | metal tolerance protein 5 | 5.00 | 0.69 | 0.14 | 0.029 |
| CL73Contig10 | clathrin heavy chain 1 | 11.05 | 10.94 | 0.99 | 0.03 |
| CL3204Contig2 | ---NA--- | 6.50 | 5.77 | 0.89 | 0.031 |
| CL10162Contig3 | ---NA--- | 7.16 | 6.23 | 0.87 | 0.032 |
| CL349Contig4 | homeobox-leucine zipper protein roc6 | 7.24 | 6.70 | 0.93 | 0.033 |
| CL349Contig7 | homeobox-leucine zipper protein roc6 | 7.24 | 6.70 | 0.93 | 0.033 |
| CL5826Contig1 | long chain acyl- synthetase 4 | 10.35 | 10.18 | 0.98 | 0.033 |
| CL5826Contig2 | long chain acyl- synthetase 4 | 10.35 | 10.18 | 0.98 | 0.033 |
| CL5942Contig6 | ---NA--- | 9.19 | 8.87 | 0.97 | 0.033 |
| CL6511Contig2 | v-type proton atpase subunit g1 | 9.82 | 9.58 | 0.98 | 0.033 |
| CL7Contig43 | ---NA--- | 7.48 | 7.28 | 0.97 | 0.033 |
| comp13984_c0_se | ---NA--- | 8.33 | 7.85 | 0.94 | 0.035 |
| CL1Contig3395 | endoglucanase 7 | 8.19 | 7.50 | 0.92 | 0.036 |
| CL1Contig3396 | endoglucanase 7 | 8.19 | 7.50 | 0.92 | 0.036 |
| CL1Contig3397 | endoglucanase 7 | 8.19 | 7.50 | 0.92 | 0.036 |
| CL3496Contig11 | chlorophyll a-b binding protein 1b- chloroplastic -light-harvesting complex i | 10.21 | 9.97 | 0.98 | 0.036 |
| CL154Contig2 | ankyrin repeat domain-containing protein chloroplastic | 9.95 | 9.70 | 0.97 | 0.038 |
| CL1Contig4279 | ---NA--- | 9.23 | 8.89 | 0.96 | 0.038 |
| CL4622Contig2 | rubredoxin | 11.01 | 10.88 | 0.99 | 0.038 |
| CL1Contig4299 | histone-lysine n-methyltransferase setd3 | 8.28 | 7.84 | 0.95 | 0.039 |
| CL1Contig4635 | protease do-like 14 | 6.90 | 5.87 | 0.85 | 0.039 |
| CL8953Contig2 | ---NA--- | 9.05 | 8.80 | 0.97 | 0.039 |
| CL5563Contig3 | ---NA--- | 9.08 | 8.84 | 0.97 | 0.04 |
| CL1Contig242 | bax inhibitor 1 | 9.79 | 9.67 | 0.99 | 0.041 |
| CL2736Contig1 | ubiquinol-cytochrome-c reductase subunit ii | 9.80 | 9.66 | 0.99 | 0.041 |
| CL3528Contig3 | golgin candidate 4 | 9.24 | 8.95 | 0.97 | 0.041 |
| CL5380Contig1 | glutathione s-transferase t3 | 8.68 | 8.50 | 0.98 | 0.041 |
| CL94Contig6 | ---NA--- | 9.49 | 9.33 | 0.98 | 0.041 |
| CL5774Contig2 | r60s acidic ribosomal protein p0 | 9.92 | 9.78 | 0.99 | 0.042 |
| CL3496Contig14 | chlorophyll a-b binding protein 1b- chloroplastic -light-harvesting complex i | 10.11 | 9.95 | 0.98 | 0.043 |
| CL3496Contig15 | chlorophyll a-b binding protein 1b- chloroplastic -light-harvesting complex i | 10.11 | 9.95 | 0.98 | 0.043 |
| CL811Contig3 | myb-like transcription factor 1 | 7.47 | 6.96 | 0.93 | 0.043 |
| CL3168Contig2 | ---NA--- | 8.47 | 8.06 | 0.95 | 0.044 |
| Locus_393_4_9 | ---NA--- | 10.96 | 10.81 | 0.99 | 0.044 |
| CL1Contig492 | ---NA--- | 9.24 | 8.99 | 0.97 | 0.045 |
| CL1Contig6871 | chlorophyll a-b binding protein chloroplastic | 11.19 | 10.99 | 0.98 | 0.045 |
| CL1Contig7112 | hexose carrier protein hex6 | 8.75 | 8.57 | 0.98 | 0.045 |
| CL185Contig19 | probable disease resistance protein rf45 | 9.19 | 8.93 | 0.97 | 0.046 |
| CL885Contig1 | formin-like protein 3 | 9.72 | 9.54 | 0.98 | 0.046 |
| Locus_61_5_6 | ---NA--- | 10.11 | 9.92 | 0.98 | 0.046 |
| CL3496Contig13 | chlorophyll a-b binding protein 1b- chloroplastic -light-harvesting complex i | 10.10 | 9.88 | 0.98 | 0.048 |
| CL3496Contig17 | chlorophyll a-b binding protein 1b- chloroplastic -light-harvesting complex i | 10.10 | 9.88 | 0.98 | 0.048 |
| CL321Contig12 | ---NA--- | 8.27 | 7.66 | 0.93 | 0.049 |
| CL785Contig5 | ---NA--- | 7.16 | 6.68 | 0.93 | 0.049 |
| CL94Contig5 | choline chloroplastic | 9.49 | 9.35 | 0.99 | 0.049 |
| CL131Contig9 | ---NA--- | 7.77 | 7.25 | 0.93 | 0.05 |
| CL19309Contig1 | ---NA--- | 8.53 | 8.27 | 0.97 | 0.05 |
| CL236Contig2 | ---NA--- | 8.87 | 8.56 | 0.97 | 0.05 |
| CL236Contig9 | ---NA--- | 8.87 | 8.56 | 0.97 | 0.05 |
| CL305Contig27 | ---NA--- | 8.53 | 8.27 | 0.97 | 0.05 |
| CL7612Contig2 | ---NA--- | 8.53 | 8.27 | 0.97 | 0.05 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamies, R.; Farrant, J.M.; Tadele, Z.; Cannarozzi, G.; Rafudeen, M.S. A Proteomic Approach to Investigate the Drought Response in the Orphan Crop Eragrostis tef. Proteomes 2017, 5, 32. https://doi.org/10.3390/proteomes5040032
Kamies R, Farrant JM, Tadele Z, Cannarozzi G, Rafudeen MS. A Proteomic Approach to Investigate the Drought Response in the Orphan Crop Eragrostis tef. Proteomes. 2017; 5(4):32. https://doi.org/10.3390/proteomes5040032
Chicago/Turabian StyleKamies, Rizqah, Jill M. Farrant, Zerihun Tadele, Gina Cannarozzi, and Mohammed Suhail Rafudeen. 2017. "A Proteomic Approach to Investigate the Drought Response in the Orphan Crop Eragrostis tef" Proteomes 5, no. 4: 32. https://doi.org/10.3390/proteomes5040032
APA StyleKamies, R., Farrant, J. M., Tadele, Z., Cannarozzi, G., & Rafudeen, M. S. (2017). A Proteomic Approach to Investigate the Drought Response in the Orphan Crop Eragrostis tef. Proteomes, 5(4), 32. https://doi.org/10.3390/proteomes5040032