Tissue Specific Labeling in Proteomics



Abstract
1. Introduction
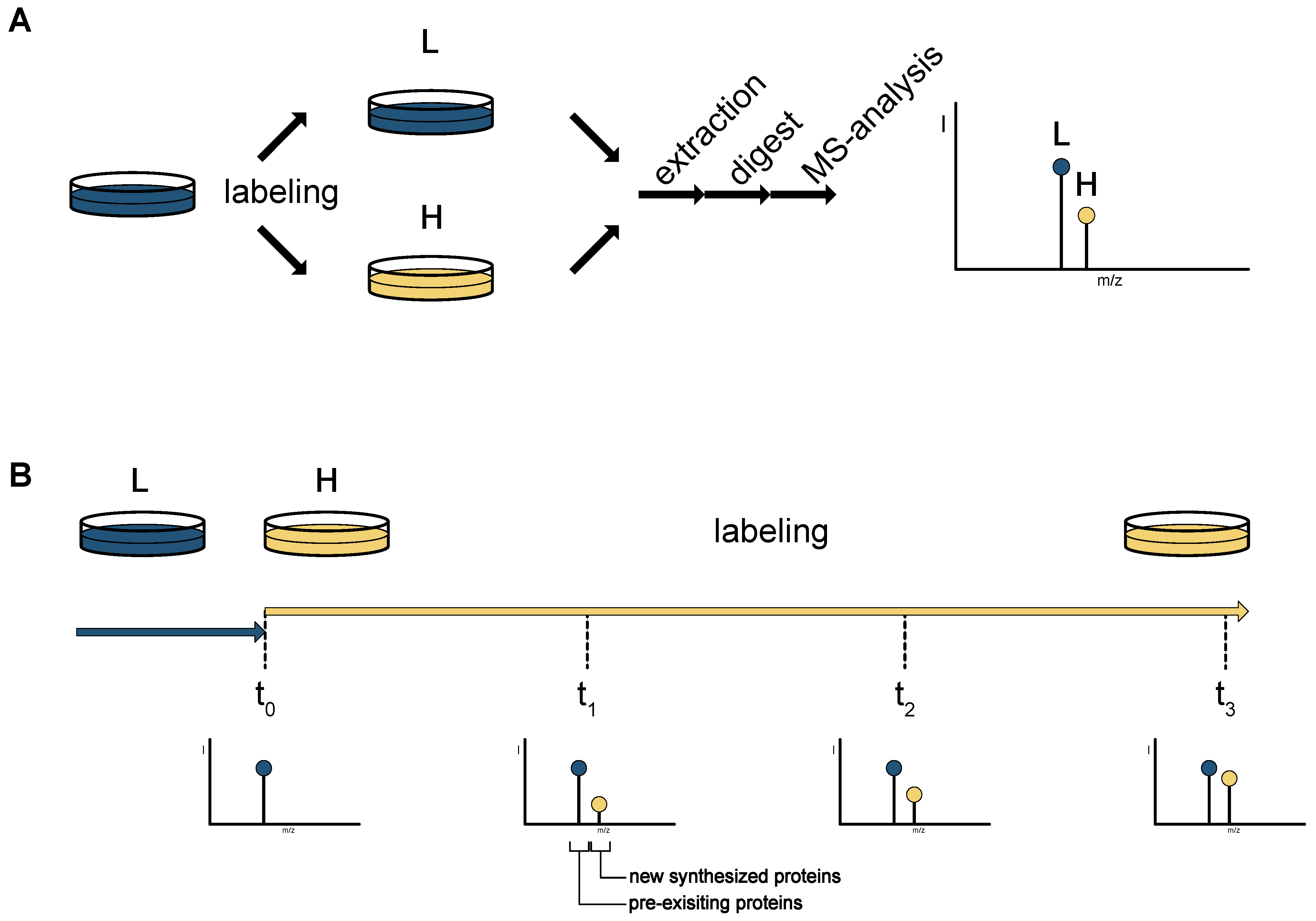
2. Mass Spectrometry-Based Proteomics
3. Bioorthogonal Amino Acids
4. Enrichment Using High Affinity or Covalent Binding
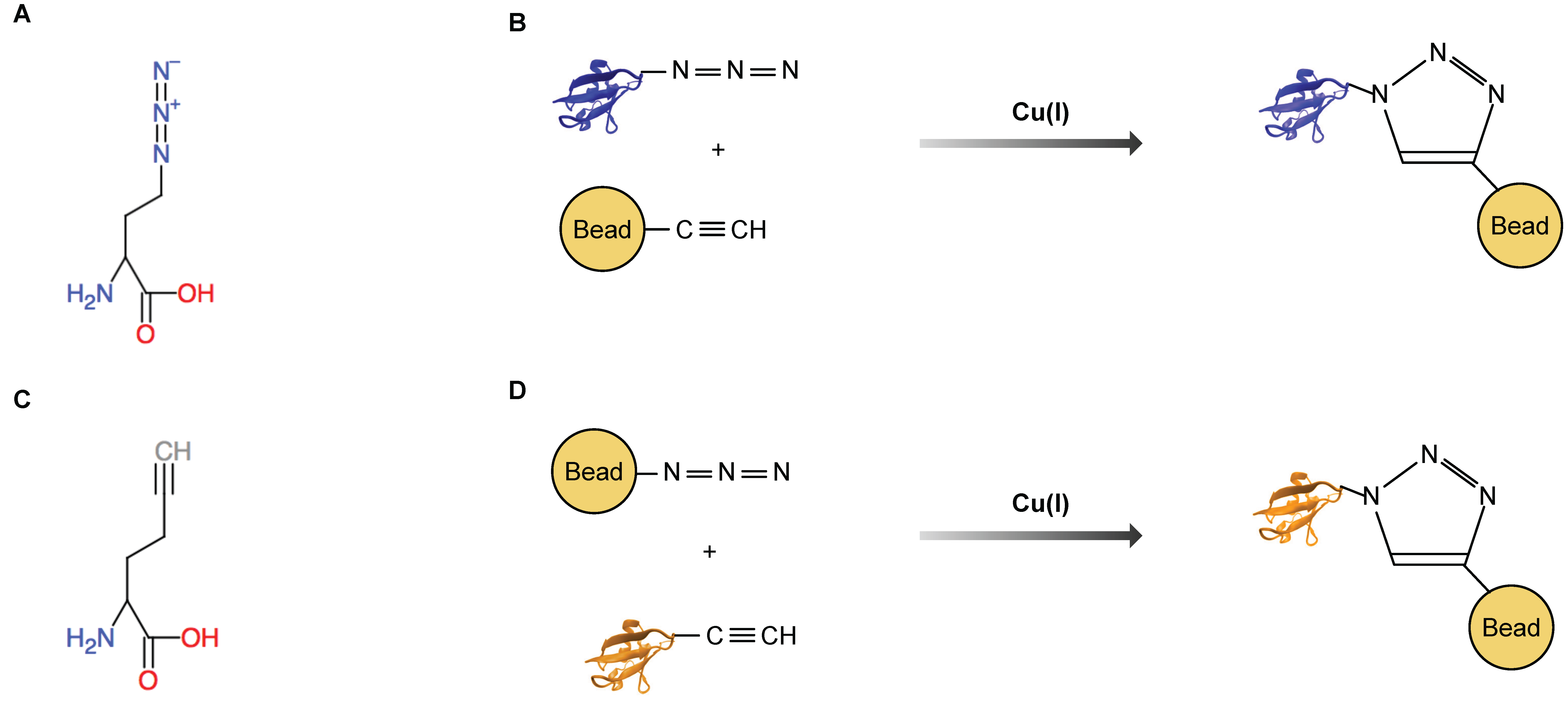
5. Azidohomoalanine and Homopropargylglycine
6. Modified Aminoacyl tRNA Synthetases
7. Isotopic Labeling of Amino Acid Precursors (CTAP and NANCAT)
8. Genetic Labeling
8.1. O-Propargyl-Purocmycin Labeling (OP-Puro)
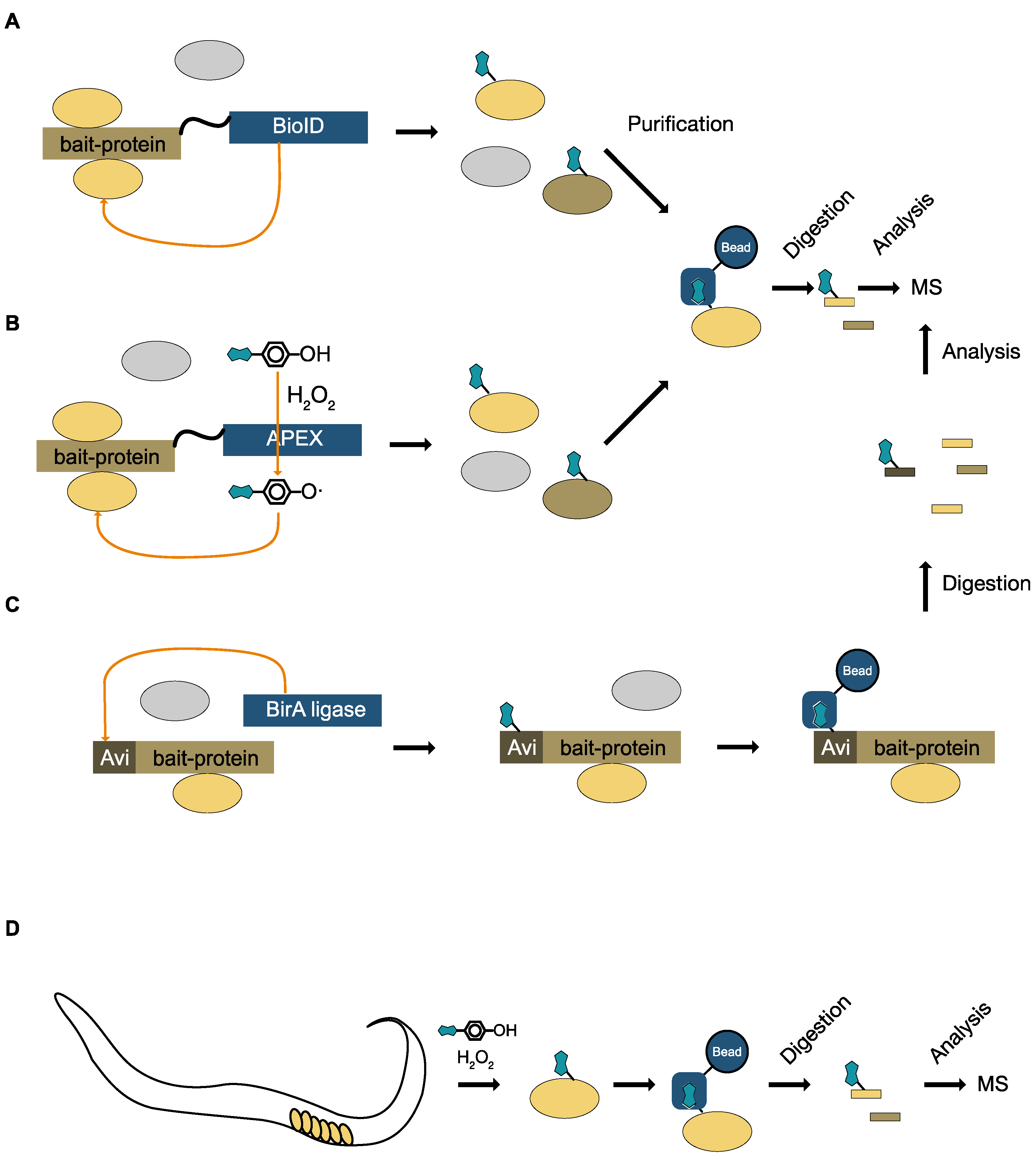
8.2. Specific Labeling with Biotin
8.3. Unspecific Labeling with Biotin
8.4. GFP-Labeling of a Subpopulation of Cells Combined with FACS
9. Analysis of Cell and Tissue Specific Signaling
9.1. Tyrosine Signaling
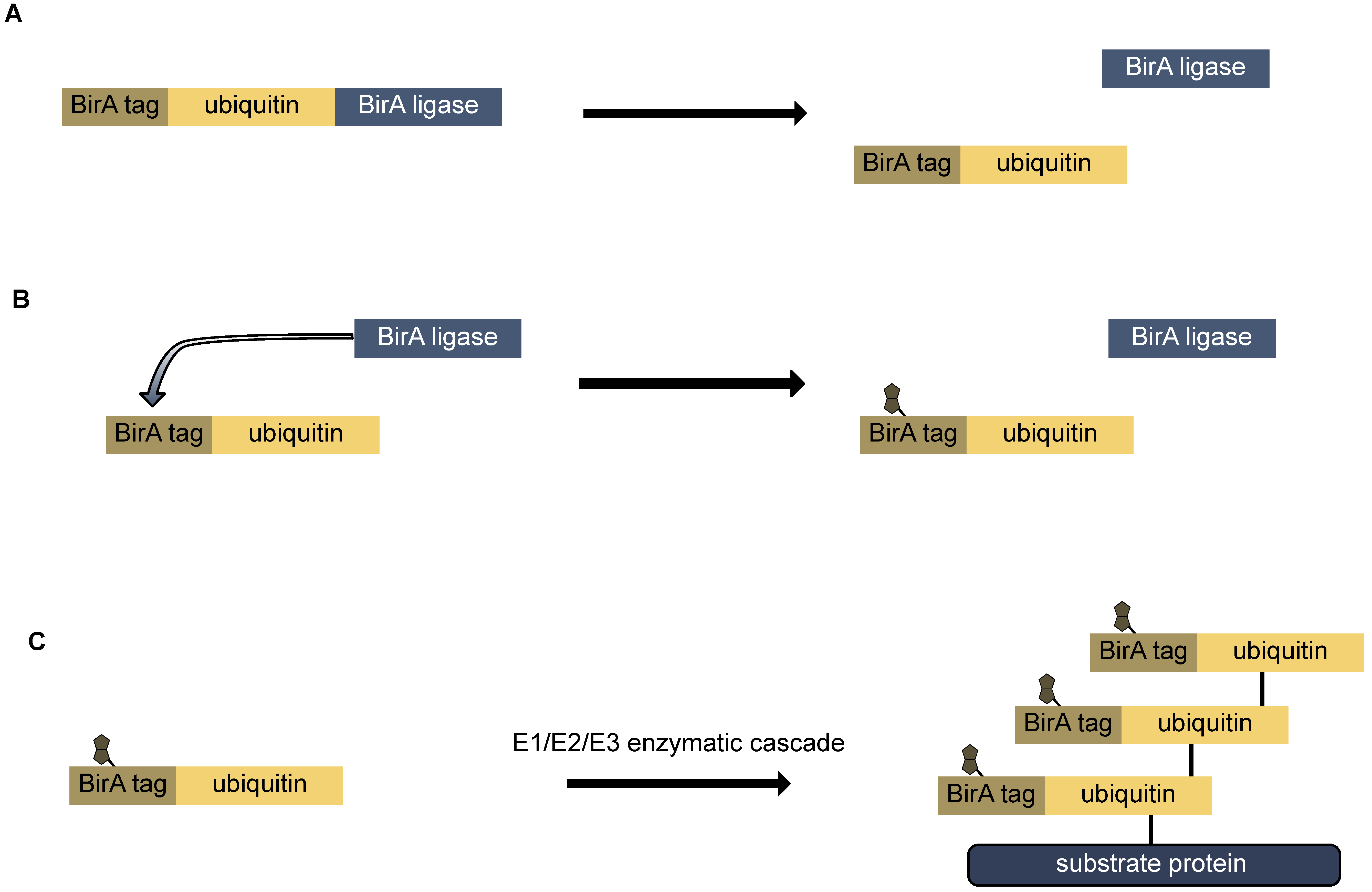
9.2. Bio-Ubiquitin
10. Monitoring Signaling for Specific Enzymes
11. Summary and Outlook
Acknowledgments
Conflicts of Interest
References
- Nielsen, M.L.; Hubner, N.C.; Fro, F.; De Godoy, L.M.F.; Olsen, J.V.; Walther, T.C.; Mann, M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 2008, 455. [Google Scholar] [CrossRef]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The One Hour Yeast Proteome. Mol. Cell. Proteom. 2014, 13, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Moser, M.; Ussar, S.; Thievessen, I.; Luber, C.A.; Forner, F.; Schmidt, S.; Zanivan, S.; Fässler, R.; Mann, M. SILAC Mouse for Quantitative Proteomics Uncovers Kindlin-3 as an Essential Factor for Red Blood Cell Function. Cell 2008, 134, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Sury, M.D.; Chen, J.; Selbach, M. In Vivo Stable Isotope Labeling by Amino Acids in Drosophila melanogaster. Methods Mol. Biol. 2014, 1188, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, C.; Sherman, A.; Chen, G.I.; Pasculescu, A.; Poliakov, A.; Hsiung, M.; Larsen, B.; Wilkinson, D.G.; Linding, R.; Pawson, T. Cell-Specific Information Processing in Segregating Populations of Eph Receptor Ephrin-Expressing Cells. Science 2009, 326, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, O.; Kalman, M.; Fang, Y.; Vernitsky, H.; Jacob-Hirsch, J.; Foster, L.J.; Kloog, Y.; Goldstein, I. Trans-SILAC: Sorting out the non-cell-autonomous proteome. Nat. Methods 2010, 7, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Schwanhäusser, B.; Gossen, M.; Dittmar, G.; Selbach, M. Global analysis of cellular protein translation by pulsed SILAC. Proteomics 2009, 9, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Tuorto, F.; Herbst, F.; Alerasool, N.; Bender, S.; Popp, O.; Federico, G.; Reitter, S.; Liebers, R.; Stoecklin, G.; Gröne, H.-J.; et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015, 34, 2350–2362. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.K.; Hammond, D.E.; Clague, M.J.; Gaskell, S.J.; Beynon, R.J. Turnover of the Human Proteome: Determination of Protein Intracellular Stability by Dynamic SILAC. J. Proteome Res. 2009, 8, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Sheean, M.E.; McShane, E.; Cheret, C.; Walcher, J.; Müller, T.; Wulf-Goldenberg, A.; Hoelper, S.; Garratt, A.N.; Krüger, M.; Rajewsky, K.; et al. Activation of MAPK overrides the termination of myelin growth and replaces Nrg1/ErbB3 signals during Schwann cell development and myelination. Genes Dev. 2014, 28, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, D.C.; Link, A.J.; Graumann, J.; Tirrell, D.A.; Schuman, E.M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl. Acad. Sci. USA 2006, 103, 9482–9487. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, D.C.; Hodas, J.J.L.; Gouzer, G.; Shadrin, I.Y.; Ngo, J.T.; Triller, A.; Tirrell, D.A.; Schuman, E.M. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 2010, 13, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Kiick, K.L.; Saxon, E.; Tirrell, D.A.; Bertozzi, C.R. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. USA 2002, 99, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Calve, S.; Witten, A.J.; Ocken, A.R.; Kinzer-Ursem, T.L. Incorporation of non-canonical amino acids into the developing murine proteome. Sci. Rep. 2016, 6, 32377. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.T.; Champion, J.A.; Mahdavi, A.; Tanrikulu, I.C.; Beatty, K.E.; Connor, R.E.; Yoo, T.H.; Dieterich, D.C.; Schuman, E.M.; Tirrell, D.A. Cell-selective metabolic labeling of proteins. Nat. Chem. Biol. 2009, 5, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Link, A.J.; Vink, M.K.S.; Agard, N.J.; Prescher, J.A.; Bertozzi, C.R.; Tirrell, D.A. Discovery of aminoacyl-tRNA synthetase activity through cell-surface display of noncanonical amino acids. Proc. Natl. Acad. Sci. USA 2006, 103, 10180–10185. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, I.; Marter, K.; Kobler, O.; Niehues, S.; Abele, J.; Müller, A.; Bussmann, J.; Storkebaum, E.; Ziv, T.; Thomas, U.; Dieterich, D.C. Cell-selective labelling of proteomes in Drosophila melanogaster. Nat. Commun. 2015, 6, 7521. [Google Scholar] [CrossRef] [PubMed]
- Elliott, T.S.; Townsley, F.M.; Bianco, A.; Ernst, R.J.; Sachdeva, A.; Elsässer, S.J.; Davis, L.; Lang, K.; Pisa, R.; Greiss, S.; et al. Proteome labeling and protein identification in specific tissues and at specific developmental stages in an animal. Nat. Biotechnol. 2014, 32, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Dieck, S.; Anke, M.; Nehring, A.; Hinz, F.I. Metabolic Labeling with Noncanonical Amino Acids and Visualization by Chemoselective Fluorescent Tagging. Curr. Protoc. Cell Biol. 2012, 1–29. [Google Scholar] [CrossRef]
- Bagert, J.D.; Xie, Y.J.; Sweredoski, M.J.; Qi, Y.; Hess, S.; Schuman, E.M.; Tirrell, D.A. Quantitative, Time-Resolved Proteomic Analysis by Combining Bioorthogonal Noncanonical Amino Acid Tagging and Pulsed Stable Isotope Labeling by Amino Acids in Cell Culture. Mol. Cell. Proteom. 2014, 13, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Hinz, F.I.; Dieterich, D.C.; Tirrell, D.A.; Schuman, E.M. Noncanonical Amino Acid Labeling in Vivo to Visualize and Affinity Purify Newly Synthesized Proteins in Larval Zebrafish. ACS Chem. Neurosci. 2012, 3, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Liu, H.H.; Schiapparelli, L.; McClatchy, D.; He, H.Y.; Yates, J.R.; Cline, H.T. Acute Synthesis of CPEB Is Required for Plasticity of Visual Avoidance Behavior in Xenopus. Cell Rep. 2014, 6, 737–747. [Google Scholar] [CrossRef] [PubMed]
- McClatchy, D.B.; Ma, Y.; Liu, C.; Stein, B.D.; Martínez-Bartolomé, S.; Vasquez, D.; Hellberg, K.; Shaw, R.J.; Yates, J.R. Pulsed azidohomoalanine labeling in mammals (PALM) detects changes in liver-specific LKB1 knockout mice. J. Proteome Res. 2015, 14, 4815–4822. [Google Scholar] [CrossRef] [PubMed]
- McShane, E.; Sin, C.; Zauber, H.; Wells, J.N.; Donnelly, N.; Wang, X.; Hou, J.; Chen, W.; Storchova, Z.; Marsh, J.A.; et al. Kinetic Analysis of Protein Stability Reveals Age-Dependent Degradation. Cell 2016, 167, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Howden, A.J.M.; Geoghegan, V.; Katsch, K.; Efstathiou, G.; Bhushan, B.; Boutureira, O.; Thomas, B.; Trudgian, D.C.; Kessler, B.M.; Dieterich, D.C.; et al. QuaNCAT: Quantitating proteome dynamics in primary cells. Nat. Methods 2013, 10, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Kramer, G.; Sprenger, R.R.; Back, J.; Dekker, H.L.; Nessen, M.A.; van Maarseveen, J.H.; de Koning, L.J.; Hellingwerf, K.J.; de Jong, L.; de Koster, C.G. Identification and Quantitation of Newly Synthesized Proteins in Escherichia coli by Enrichment of Azidohomoalanine-labeled Peptides with Diagonal Chromatography. Mol. Cell. Proteom. 2009, 8, 1599–1611. [Google Scholar] [CrossRef] [PubMed]
- Eichelbaum, K.; Winter, M.; Diaz, M.B.; Herzig, S.; Krijgsveld, J. Selective enrichment of newly synthesized proteins for quantitative secretome analysis. Nat. Biotechnol. 2012, 30, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Bowling, H.; Bhattacharya, A.; Zhang, G.; Lebowitz, J.Z.; Smith, P.T.; Kirshenbaum, K.; Neubert, T.A.; Vogel, C.; Chao, V.; Klann, E. BONLAC: A Combinatorial Proteomic Technique to Measure Stimulus-induced Translational Profiles in Brain Slices. Neuropharmacology 2016, 100, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Grammel, M.; Zhang, M.M.; Hang, H.C. Orthogonal alkynyl-amino acid reporter for selective labeling of bacetrial proteomes during infection. Angew. Chem. Int. Ed. 2010, 49, 5970–5974. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, A.; Szychowski, J.; Ngo, J.T.; Sweredoski, M.J.; Graham, R.L.J.; Hess, S.; Schneewind, O.; Mazmanian, S.K.; Tirrell, D.A. Identification of secreted bacterial proteins by noncanonical amino acid tagging. Proc. Natl. Acad. Sci. USA 2014, 111, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, A.; Segall-shapiro, T.H.; Kou, S.; Jindal, G.A.; Ho, K.G.; Liu, S.; Chitsaz, M.; Ismagilov, R.F.; Silberg, J.J.; Tirrell, D.A. A Genetically Encoded AND Gate for Cell-Targeted Metabolic Labeling of Proteins. J. Am. Chem. Soc. 2013, 135, 2979–2982. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, A.; Hamblin, G.D.; Jindal, G.A.; Bagert, J.D.; Dong, C.; Sweredoski, M.J.; Hess, S.; Schuman, E.M.; Tirrell, D.A. Engineered Aminoacyl-tRNA Synthetase for Cell-Selective Analysis of Mammalian Protein Synthesis. J. Am. Chem. Soc. 2016, 138, 4278–4281. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Stellmacher, A.; Freitag, C.E.; Landgraf, P.; Dieterich, D.C. Monitoring Astrocytic Proteome Dynamics by Cell Type-Specific Protein Labeling. PLoS ONE 2015, 10, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Yuet, K.P.; Doma, M.K.; Ngo, J.T.; Sweredoski, M.J.; Graham, R.L.J.; Moradian, A.; Hess, S.; Schuman, E.M.; Sternberg, P.W.; Tirrell, D.A. Cell-specific proteomic analysis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2015, 112, 2705–2710. [Google Scholar] [CrossRef] [PubMed]
- Elliott, T.S.; Bianco, A.; Townsley, F.M.; Fried, S.D.; Chin, J.W. Tagging and Enriching Proteins Enables Cell-Specific Proteomics. Cell Chem. Biol. 2016, 23, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Ernst, R.J.; Krogager, T.P.; Maywood, E.S.; Zanchi, R.; Beránek, V.; Elliott, T.S.; Barry, N.P.; Hastings, M.H.; Chin, J.W. Genetic code expansion in the mouse brain. Nat. Chem. Biol. 2016, 12, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yang, A.; Lee, S.; Lee, H.-W.; Park, C.B.; Park, H.-S. Expanding the genetic code of Mus musculus. Nat. Commun. 2017, 8, 14568. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the Genetic Code of Escherichia coli. Science 2001, 292, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Smits, A.H.; Borrmann, A.; Roosjen, M.; Van Hest, J.C.M.; Vermeulen, M. Click-MS: Tagless Protein Enrichment Using Bioorthogonal Chemistry for Quantitative Proteomics. ACS Chem. Biol. 2016, 11, 3245–3250. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, N.P.; Soufi, B.; Walkowicz, W.E.; Pedicord, V.A.; Mavrakis, K.J.; Macek, B.; Gin, D.Y.; Sander, C.; Martin, L. Cell-selective labeling with amino acid precursors for proteomic studies of Studies of multicellular environments. Nat. Methods 2014, 10, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Norrie, I.C.; Worboys, J.D.; Lim, L.; Lauffenburger, D.A.; Jorgensen, C. Cell-specific Labeling Enzymes for Analysis of Cell-Cell Communication in Continuous Co-culture. Mol. Cell. Proteom. 2014, 13, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhu, Y.; Sun, Y.; Qin, K.; Liu, W.; Zhou, W.; Chen, X. Nitrilase-Activatable Noncanonical Amino Acid Precursors for Cell-Selective Metabolic Labeling of Proteomes. ACS Chem. Biol. 2016, 11, 3273–3277. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xu, Y.; Stoleru, D.; Salic, A. Imaging protein synthesis in cells and tissues with an alkyne analog of puromycin. Proc. Natl. Acad. Sci. USA 2012, 109, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.M.; Liu, H.W.; Jin, H.; Goodman, R.H.; Cohen, M.S. Cell-specific Profiling of Nascent Proteomes Using Orthogonal Enzyme-mediated Puromycin Incorporation. ACS Chem. Biol. 2016, 11, 1532–1536. [Google Scholar] [CrossRef] [PubMed]
- Waaijers, S.; Muñoz, J.; Berends, C.; Ramalho, J.J.; Goerdayal, S.S.; Low, T.Y.; Zoumaro-Djayoon, A.D.; Hoffmann, M.; Koorman, T.; Tas, R.P.; et al. A tissue-specific protein purification approach in Caenorhabditis elegans identifies novel interaction partners of DLG-1/Discs large. BMC Biol. 2016, 14, 66. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Schiapparelli, L.M.; McClatchy, D.B.; Liu, H.H.; Sharma, P.; Yates, J.R.; Cline, H.T. Direct detection of biotinylated proteins by mass spectrometry. J. Proteome Res. 2014, 13, 3966–3978. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Vamsi, K.M.; Carr, S.A.; Ting, A.Y. Proteomic Mapping of Mitochondria in Living Cells via Spatially-Restricted Enzymatic Tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Martell, J.D.; Deerinck, T.J.; Sancak, Y.; Poulos, T.L.; Mootha, V.K.; Sosinsky, G.E.; Ellisman, M.H.; Ting, A.Y. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012, 30, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2014, 12, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Hu, Y.; Udeshi, N.D.; Lau, T.Y.; Wirtz-Peitz, F.; He, L.; Ting, A.Y.; Carr, S.A.; Perrimon, N. Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proc. Natl. Acad. Sci. USA 2015, 112, 12093–12098. [Google Scholar] [CrossRef] [PubMed]
- Reinke, A.W.; Mak, R.; Troemel, E.R.; Bennett, E.J. In vivo mapping of tissue- and subcellular-specific proteomes in Caenorhabditis elegans. Sci. Adv. 2017, 3, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Di Palma, S.; Stange, D.; Van De Wetering, M.; Clevers, H.; Heck, A.J.R.; Mohammed, S. Highly sensitive proteome analysis of FACS-sorted adult colon stem cells. J. Proteome Res. 2011, 10, 3814–3819. [Google Scholar] [CrossRef] [PubMed]
- Jahn, M.; Seifert, J.; von Bergen, M.; Schmid, A.; Bühler, B.; Müller, S. Subpopulation-proteomics in prokaryotic populations. Curr. Opin. Biotechnol. 2013, 24, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Beaudette, P.; Popp, O.; Dittmar, G. Proteomic techniques to probe the ubiquitin landscape. Proteomics 2016, 16, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Lectez, B.; Migotti, R.; Lee, S.Y.; Ramirez, J.; Beraza, N.; Mansfield, B.; Sutherland, J.D.; Martinez-Chantar, M.L.; Dittmar, G.; Mayor, U. Ubiquitin Profiling in Liver Using a Transgenic Mouse with Biotinylated Ubiquitin. J. Proteome Res. 2014, 13, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Martinez, A.; Lectez, B.; Lee, S.Y.; Franco, M.; Barrio, R.; Dittmar, G.; Mayor, U. Proteomic analysis of the ubiquitin landscape in the Drosophila embryonic nervous system and the adult photoreceptor cells. PLoS ONE 2015, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Lectez, B.; Ramirez, J.; Popp, O.; Sutherland, J.D.; Urbé, S.; Dittmar, G.; Clague, M.J.; Mayor, U. Quantitative proteomic analysis of Parkin substrates in Drosophila neurons. Mol. Neurodegener. 2017, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.; Seyfried, N.T.; Brand, A.H.; Peng, J.; Mayor, U. A Novel Strategy to Isolate Ubiquitin Conjugates Reveals Wide Role for Ubiquitination during Neural Development. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Akimov, V.; Henningsen, J.; Hallenborg, P.; Rigbolt, K.T.G.; Jensen, S.S.; Nielsen, M.M.; Kratchmarova, I.; Blagoev, B. StUbEx: Stable tagged ubiquitin exchange system for the global investigation of cellular ubiquitination. J. Proteome Res. 2014, 13, 4192–4204. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Welcker, M.; Hizli, A.A.; Posakony, J.J.; Aebersold, R.; Clurman, B.E. Identification of CDK2 substrates in human cell lysates. Genome Biol. 2008, 9, R149. [Google Scholar] [CrossRef] [PubMed]
- Blethrow, J.D.; Glavy, J.S.; Morgan, D.O.; Shokat, K.M. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. USA 2008, 105, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.; Bothwell, I.; Chen, Y.; Sengelaub, C.; Wang, R.; Deng, H.; Luo, M. Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-L-methionine. J. Am. Chem. Soc. 2012, 134, 5909–5915. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, R.; Zheng, W.; Chen, Y.; Blum, G.; Deng, H.; Luo, M. Profiling substrates of protein arginine N-methyltransferase 3 with S-adenosyl-L-methionine analogues. ACS Chem. Biol. 2014, 9, 476–484. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Label | Cell Specific? | Genetic Modification Necessary | Already Applied in | Enrichment of Labeled Proteins Possible | Time Scale and Applications | References |
|---|---|---|---|---|---|---|---|
| Stable isotope labeling of amino acids in cell culture (SILAC) | heavy isotope containing amino acids | no | no | wide range of cell lines and model organisms | no | 5 doubling times to achieve complete labeling of a proteome. Pulsed labeling possible but number of identifications is compromised | [1,4,5,6,7,8,9,10,11,12,13] |
| Bioorthogonal labeling of amino acids in cell culture (BONCAT) | methione analogues AHA or HPG | no | no | wide range of cell lines and model organisms | yes (covalent capture with click chemistry) | short pulses (down to minutes) and subsequent enrichment of newly synthesized proteins. Prolonged labeling possible | [14,15,16,17,23,24,25,26,27,28,29,30,31,32] |
| Cell specific BONCAT | biorthogonal amino acids that require a modified tRNA sythetase (azidonorleucine or p-azido-l-phenylalanine) | yes | yes (mutated tRNA synthetase) | cell lines, worm, fly | yes (covalent capture with click chemistry) | short pulses (down to minutes) and subsequent enrichment of newly synthesized proteins. Prolonged labeling is possible but dependent on the system side effects are possible | [18,19,20,33,34,35,36,37] |
| Stochastic orthogonal recoding of translation (SORT) | bioorthogonal amino acid in combination with an orthogonal tRNA and tRNA synthetase | yes | yes (mutated tRNA synthetase and tRNA) | cell lines, fly, mouse brain | yes (covalent capture with click chemistry) | short pulses (down to minutes) and subsequent enrichment of newly synthesized proteins. Prolonged labeling is possible. Many codons can be tagged | [21,39,40,41] |
| O-propargyl-purocmycin labeling (OP-Puro) | Puromycin analogue (OP-Puro) binding to nascent polypeptides | yes | yes (penicillin G acylase ) | cell lines | yes (covalent capture with click chemistry) | very short labeling (minutes). Provides a snapshot of actively translated proteins in a cell | [48,49] |
| isotopic labeling of amino acid precursors (CTAP) | heavy isotope containing lysine | yes | yes (lysine synthesizing enzymes) | cell lines | no | labeling comparable to SILAC. Cell specific labeling of cells in co-culture | [44,45,46] |
| GFP -labeling and sorting | GFP | yes | yes (GFP) | cell lines, unicellular organisms, mouse | sorting of labeled cells with FACS | steady state proteome of a subpopulation of cells | [59,60] |
| proximity-dependent biotin identification with a promiscous biotin ligase (BioID) | biotin | yes | yes (promiscous biotin ligase fused to protein of interest) | cell lines, unicellular organisms | yes (affinity purification with streptavidin) | proximity labeling of interacting proteins | [51,52,53] |
| biotinylation with sequence specific biotin ligase BirA | biotin | yes | yes (BirA and Avi tagged protein of interest) | cell lines, wide range of model organisms | yes (affinity purification with streptavidin) | biotinylation of tagged proteins only in cells expressing BirA. Purification of interacting proteins | [50] |
| biotin-ubiquitin | yes | yes (Avi tagged Ubiquitin in fusion with BirA ligase) | cell lines, fly, mouse | yes (affinity purification with streptavidin) | biotinylation of ubiquitin and enrichment of ubiquitinated proteins | [62,63,64,65] | |
| labeling with an engineered ascorbate peroxidase (APEX) | biotin phenol | yes | yes (APEX) | cell lines, fly, worm | yes (affinity purification with streptavidin) | proximity labeling of interacting proteins or cellular compartment specific proteins | [54,55,56,57,58] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramberger, E.; Dittmar, G. Tissue Specific Labeling in Proteomics. Proteomes 2017, 5, 17. https://doi.org/10.3390/proteomes5030017
Ramberger E, Dittmar G. Tissue Specific Labeling in Proteomics. Proteomes. 2017; 5(3):17. https://doi.org/10.3390/proteomes5030017
Chicago/Turabian StyleRamberger, Evelyn, and Gunnar Dittmar. 2017. "Tissue Specific Labeling in Proteomics" Proteomes 5, no. 3: 17. https://doi.org/10.3390/proteomes5030017
APA StyleRamberger, E., & Dittmar, G. (2017). Tissue Specific Labeling in Proteomics. Proteomes, 5(3), 17. https://doi.org/10.3390/proteomes5030017