
A Routine ‘Top-Down’ Approach to Analysis of the Human Serum Proteome



Abstract
:1. Introduction
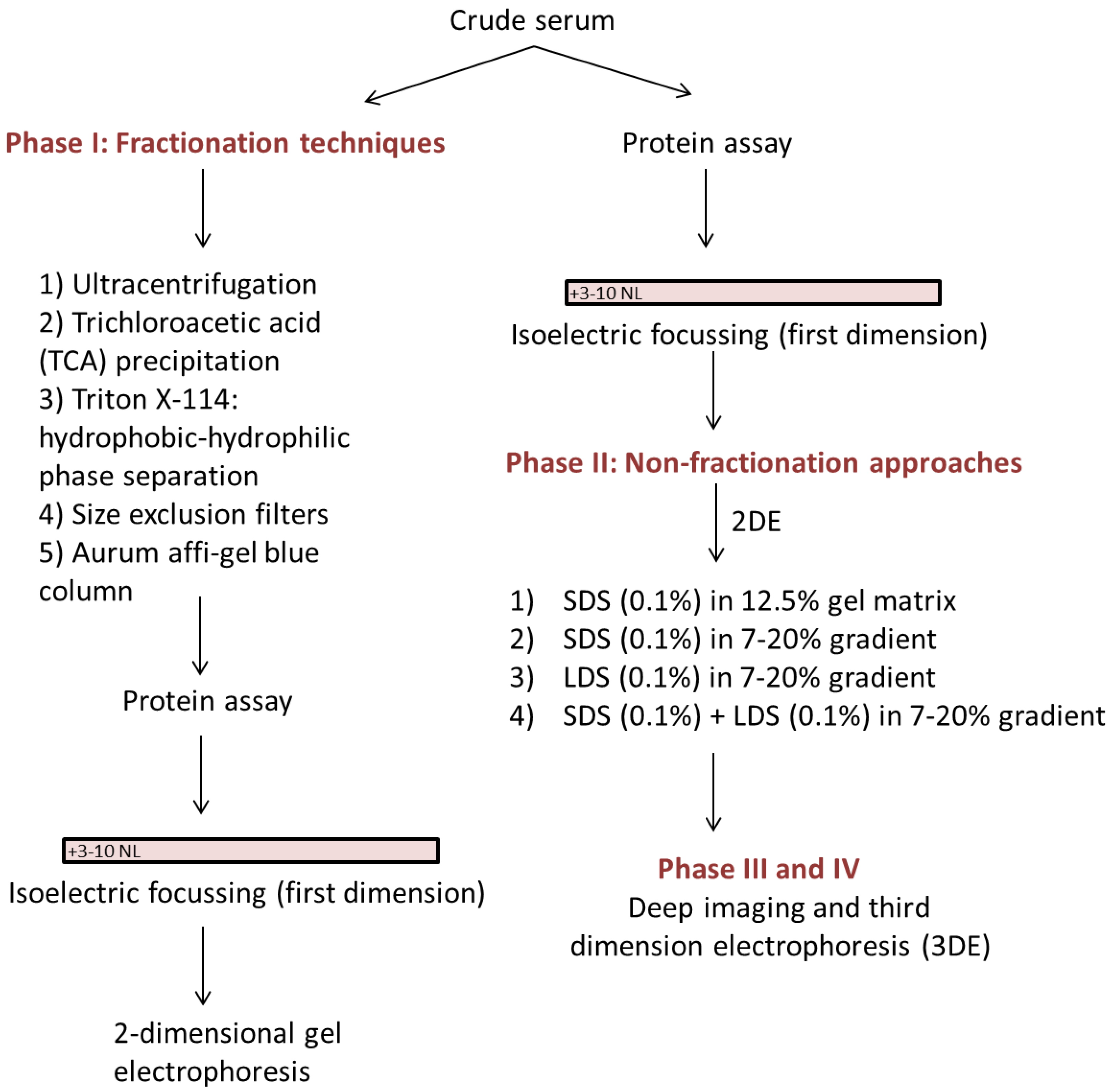
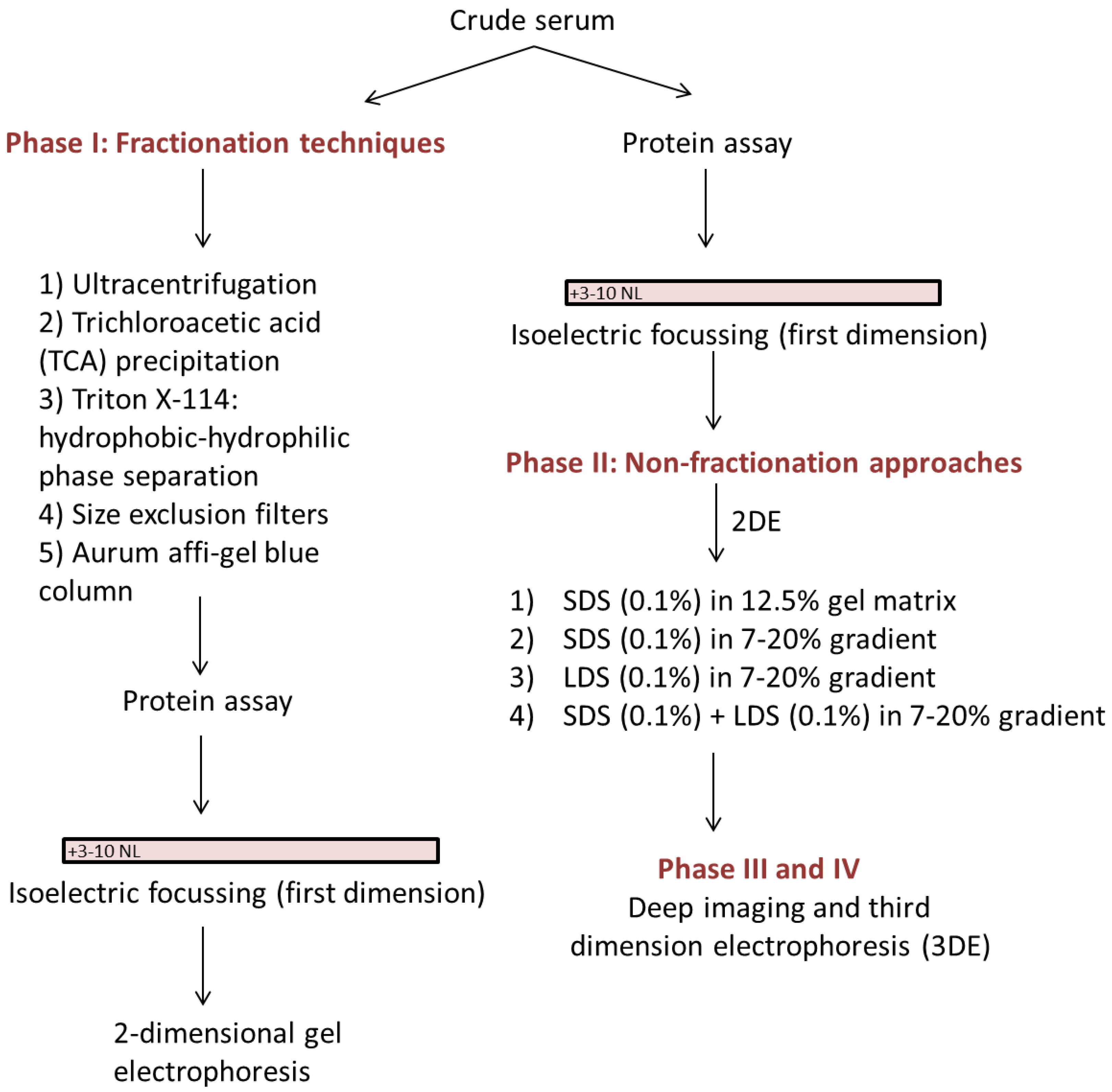
2. Materials and Methods
2.1. Protein Assay
2.1.1. 2-Dimensional Gel Electrophoresis (2DE)
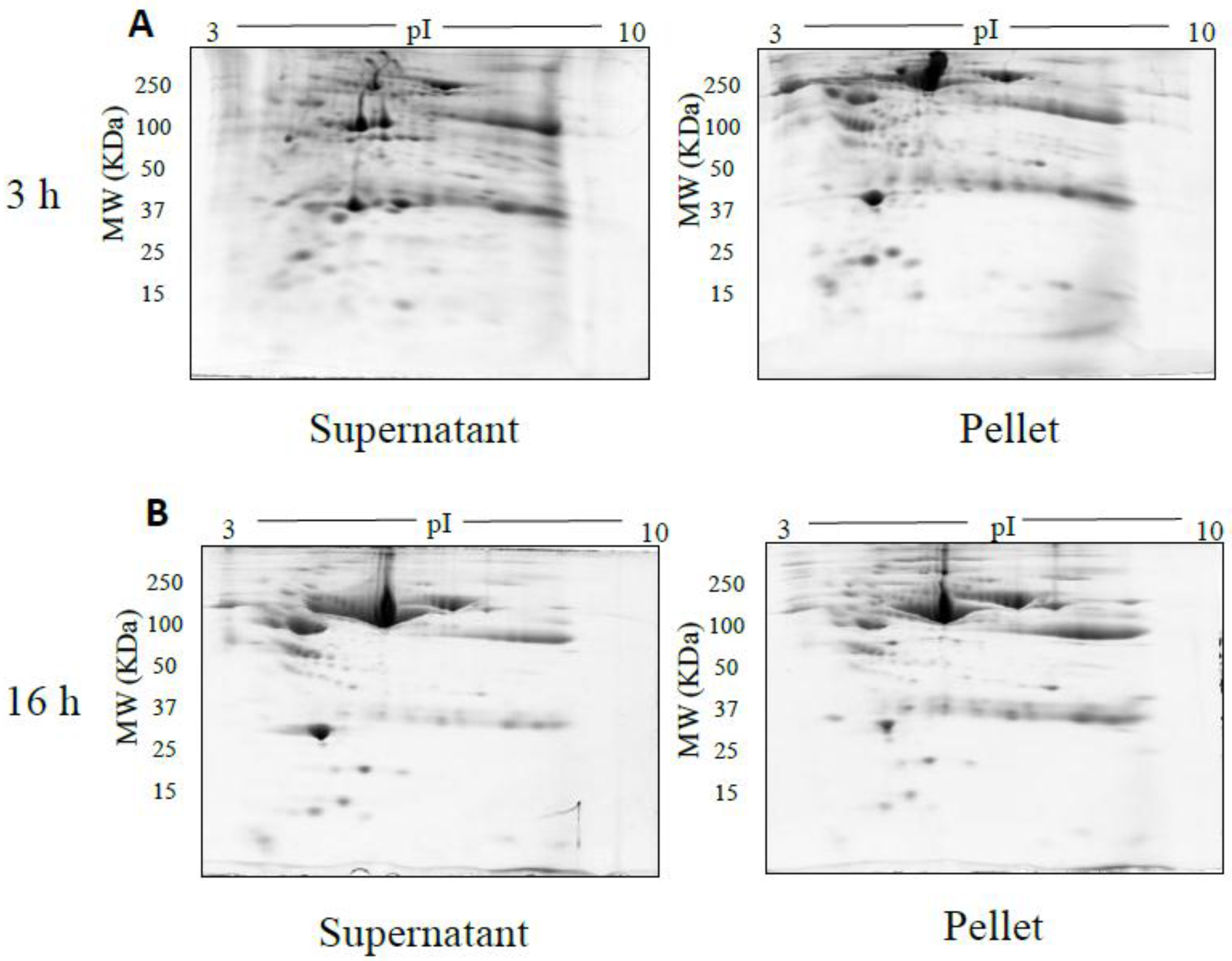
2.1.2. Ultracentrifugation
2.1.3. Trichloroacetic Acid (TCA) Precipitation
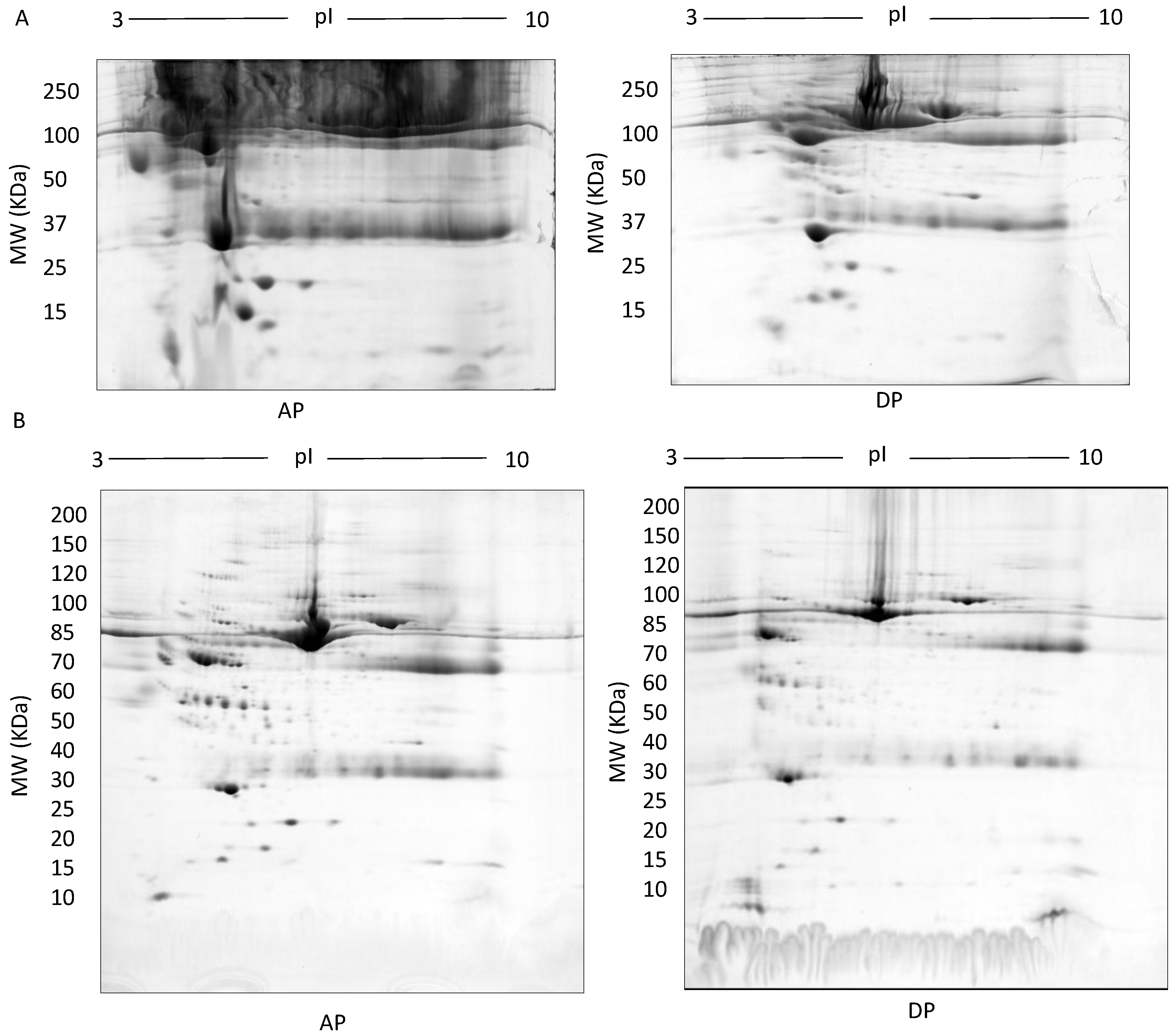
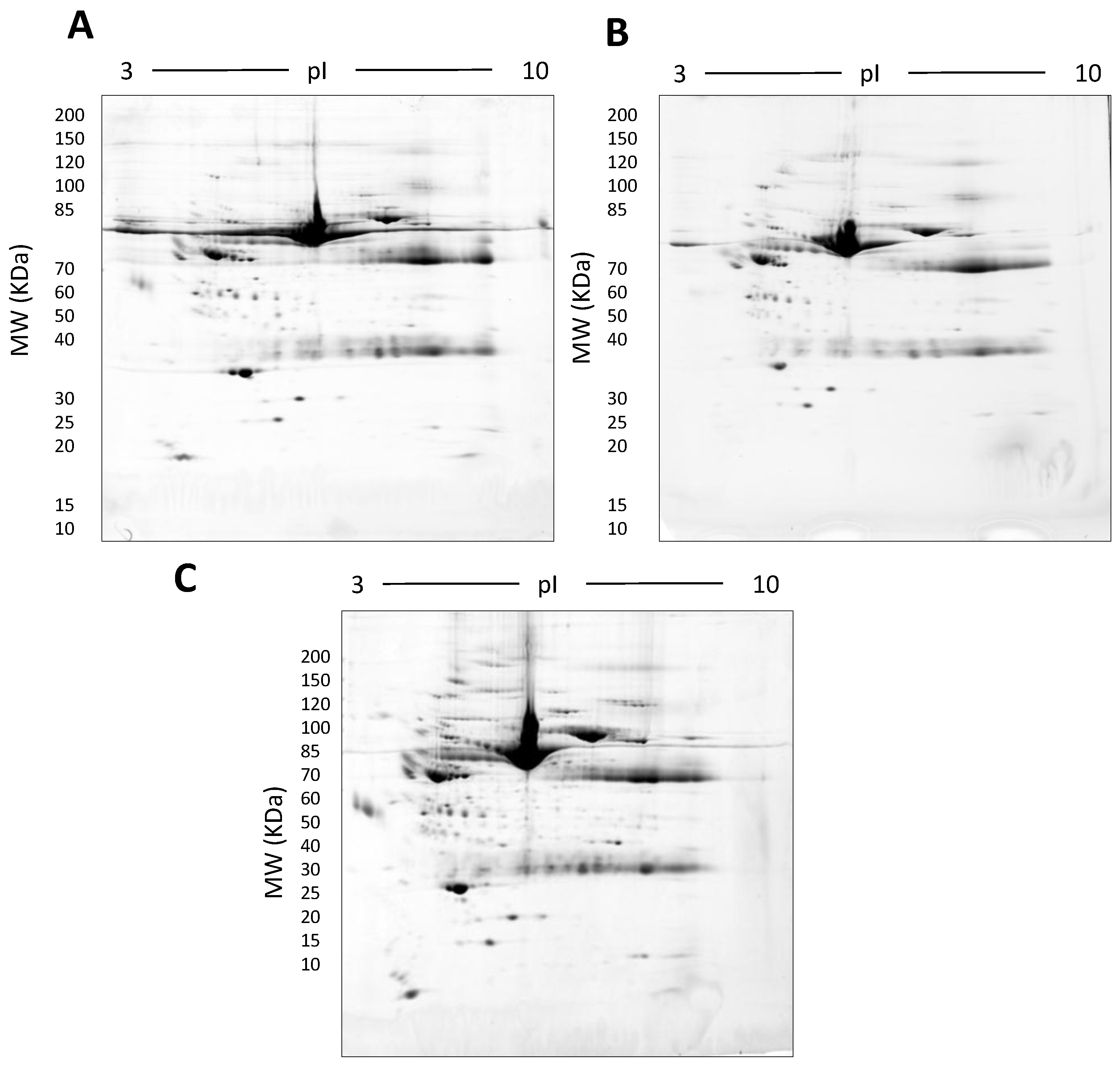
2.1.4. Triton X-114: Hydrophobic-Hydrophilic Phase Separation
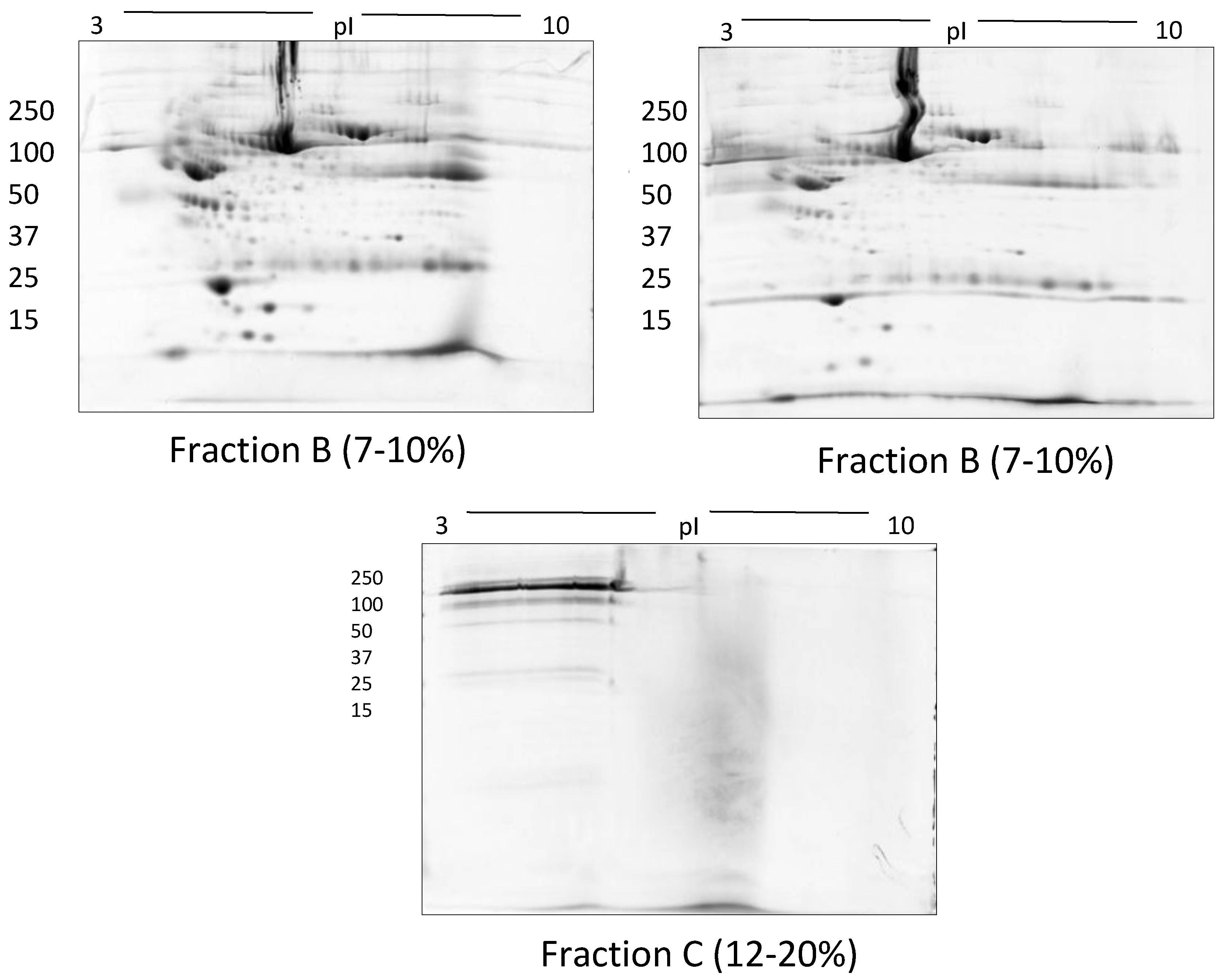
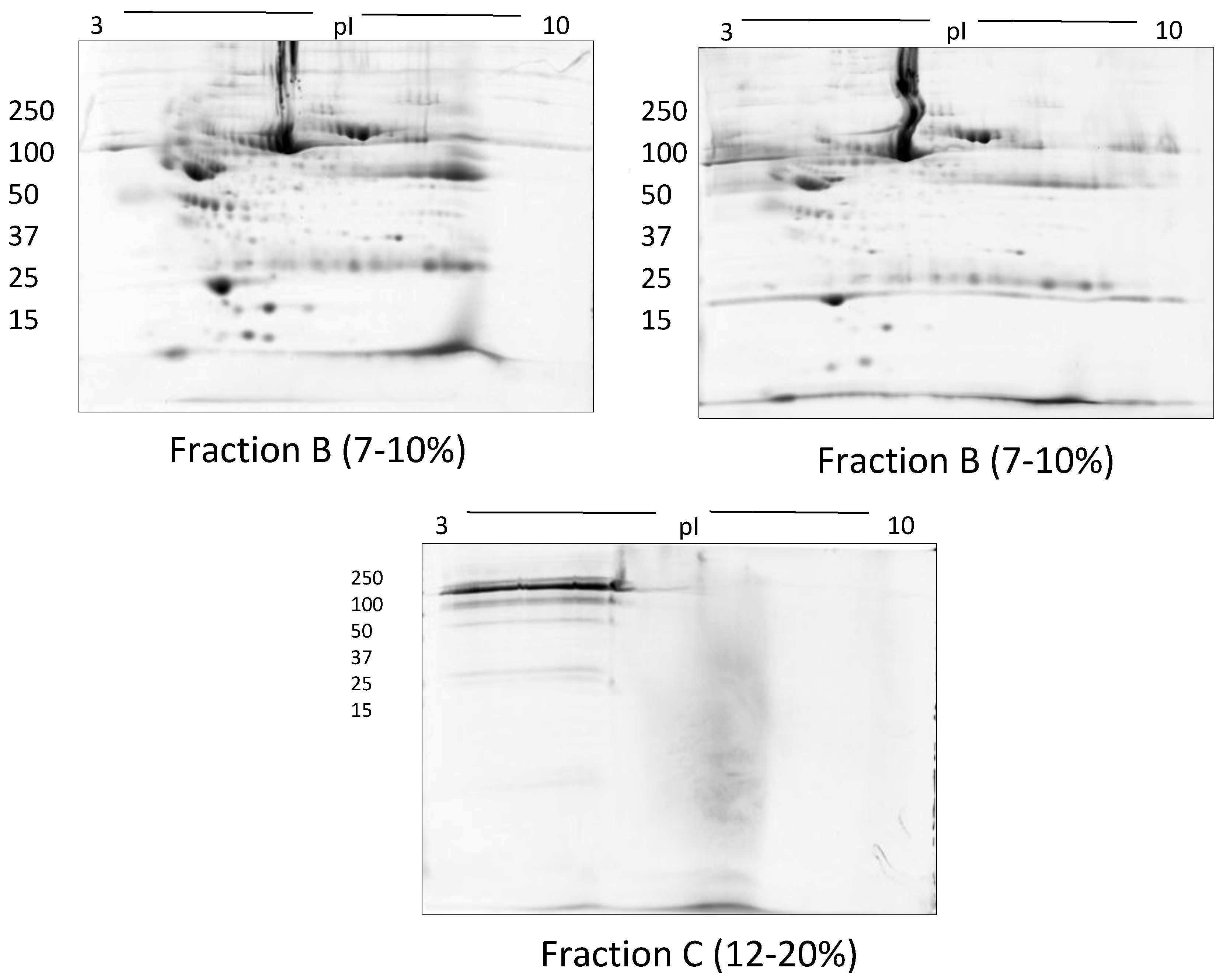
2.1.5. Size Exclusion Filters
2.1.6. Aurum Affi-Gel Blue Column
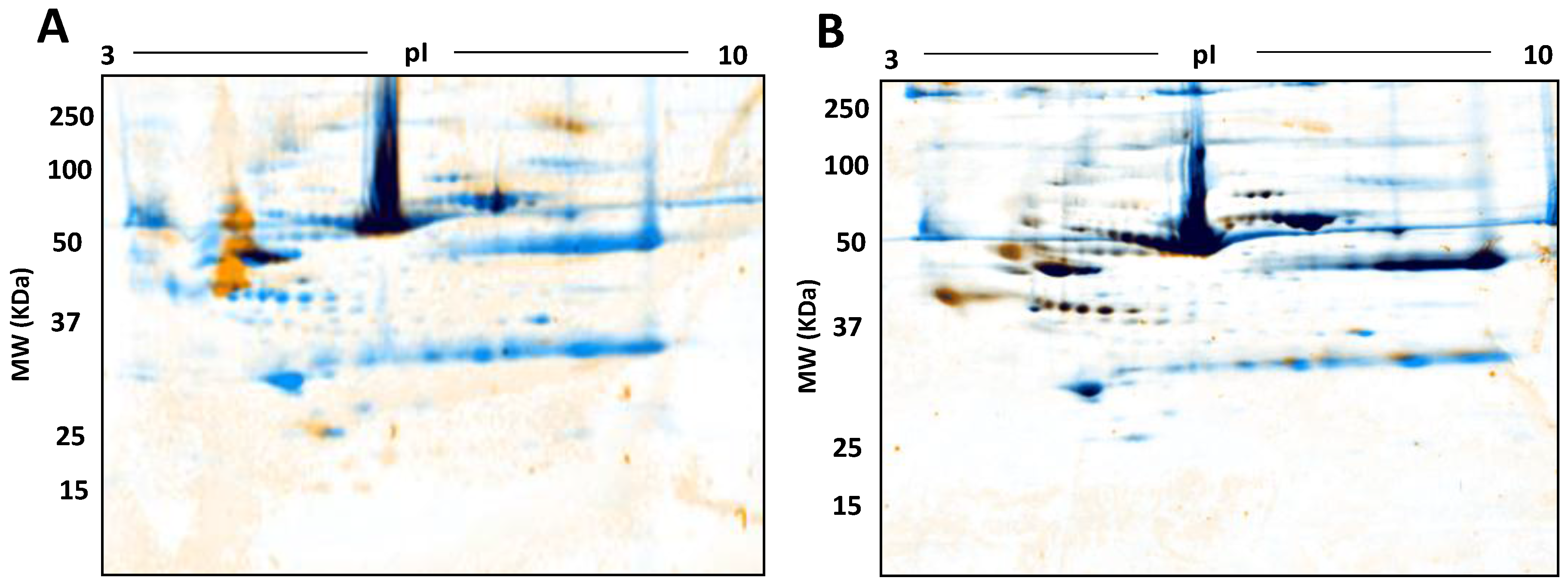
2.1.7. Lithium Dodecyl Sulfate (LDS) vs. Sodium Dodecyl Sulfate (SDS)
2.2. Phospho and Glyco Staining
2.3. Image and Statistical Analyses
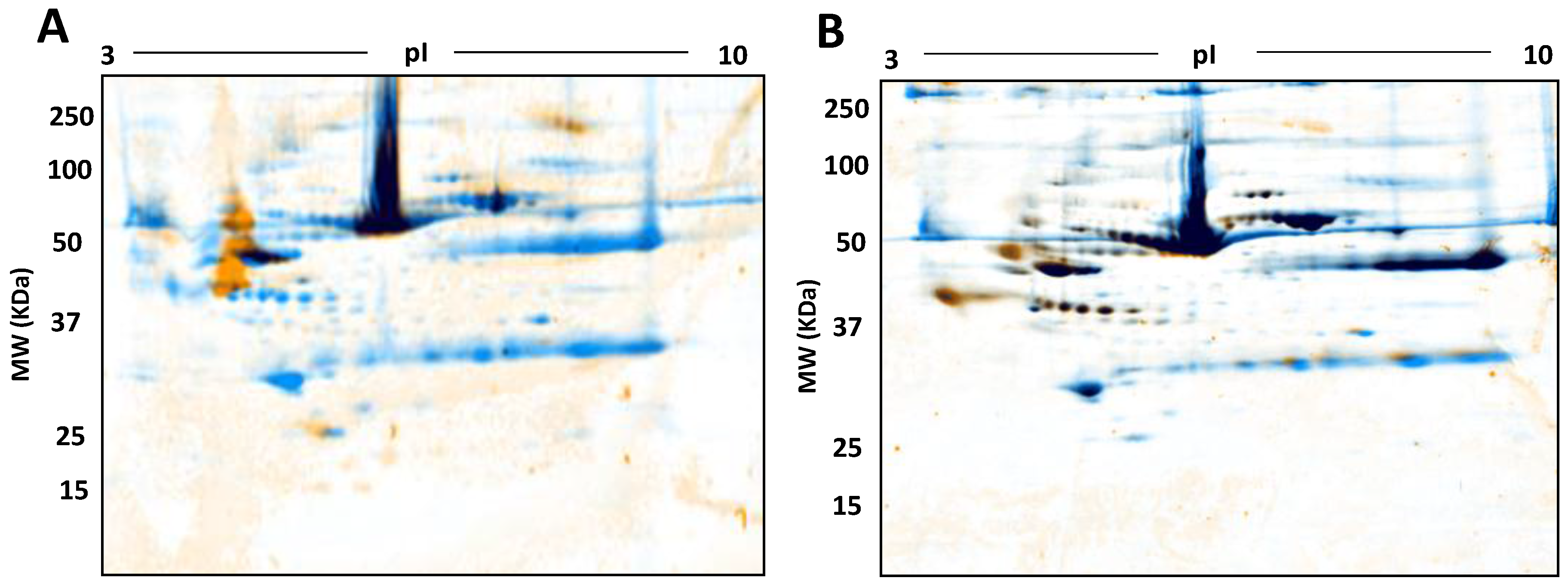
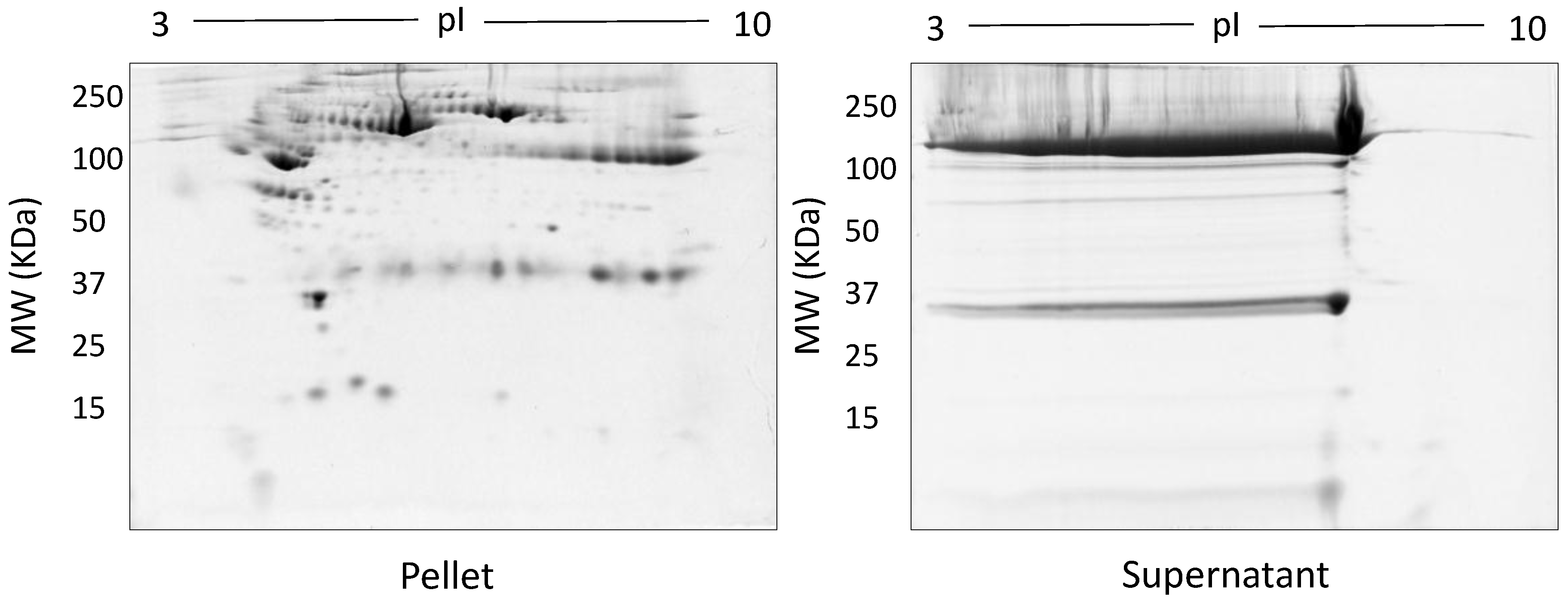
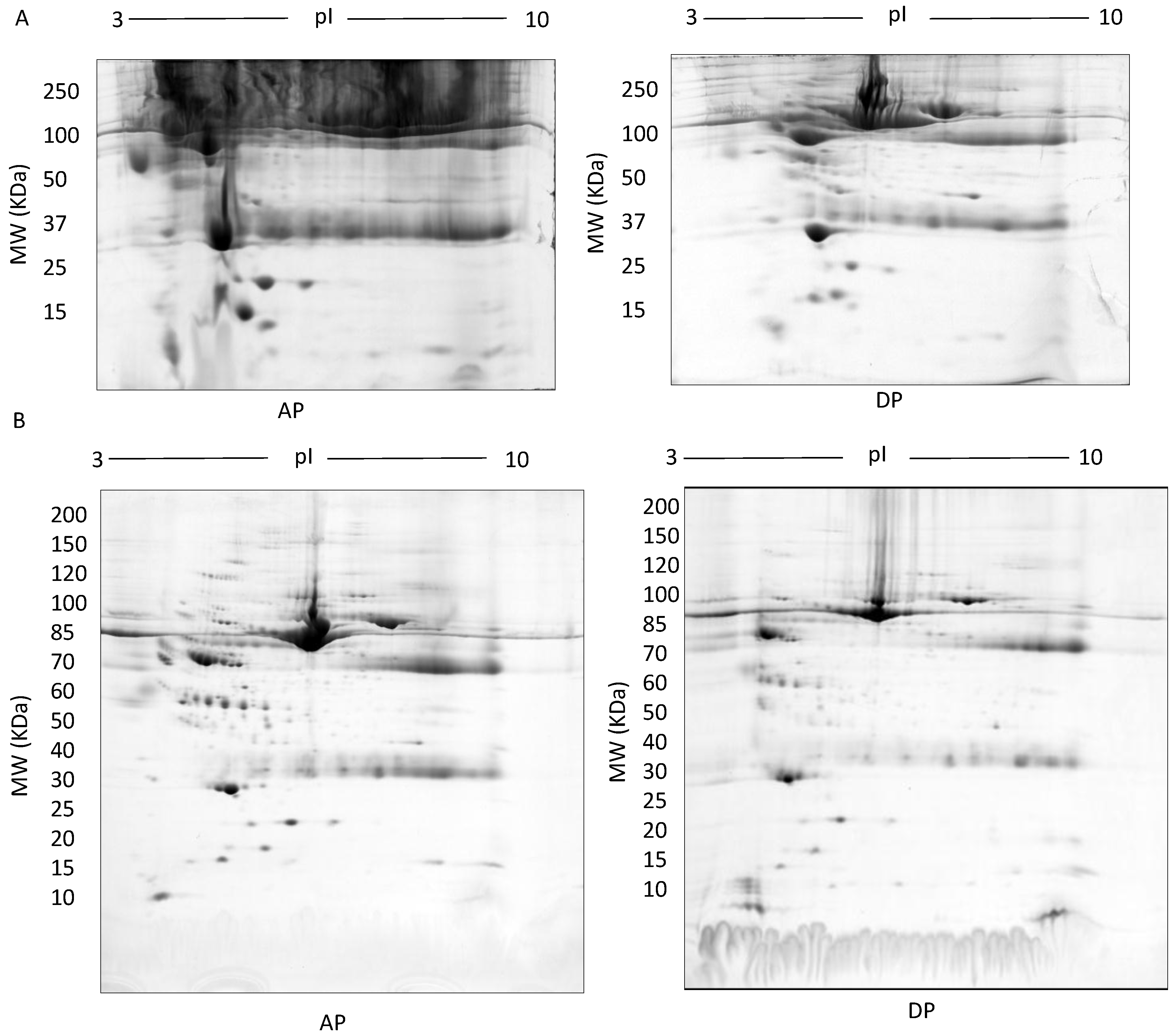
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Esplin, M.S.; Merrell, K.; Goldenberg, R.; Lai, Y.; Iams, J.D.; Mercer, B.; Spong, C.Y.; Miodovnik, M.; Simhan, H.N.; Van Dorsten, P.; et al. Proteomic identification of serum peptides predicting subsequent spontaneous preterm birth. Am. J. Obstet. Gynecol. 2011, 204, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.J.; Nelson, A.; Treumann, A.; Skeath, T.; Cummings, S.P.; Embleton, N.D.; Berrington, J.E. Metabolomic and proteomic analysis of serum from preterm infants with necrotising entercolitis and late-onset sepsis. Pediatr. Res. 2016, 79, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, M.d.S.; Torres-Romero, J.C.; Lobo, M.D.P.; Moreno, F.B.M.B.; Bezerra, L.P.; Lima, D.S.; Matos, J.C.; Moreira, R.d.A.; Monteiro-Moreira, A.C.d.O. A panel of glycoproteins as candidate biomarkers for early diagnosis and treatment evaluation of b-cell acute lymphoblastic leukemia. Biomark. Res. 2016, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Novelli, G.; Ciccacci, C.; Borgiani, P.; Papaluca Amati, M.; Abadie, E. Genetic tests and genomic biomarkers: Regulation, qualification and validation. Clin. Cases Miner. Bone Metab. 2008, 5, 149–154. [Google Scholar] [PubMed]
- Coorssen, J.R.; Yergey, A.L. Proteomics is analytical chemistry: Fitness-for-purpose in the application of top-down and bottom-up analyses. Proteomes 2015, 3, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Savaryn, J.P.; Catherman, A.D.; Thomas, P.M.; Abecassis, M.M.; Kelleher, N.L. The emergence of top-down proteomics in clinical research. Genome Med. 2013, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.L.; Anderson, N.G. The human plasma proteome: History, character, and diagnostic prospects. Mol. Cell. Proteom. 2002, 1, 845–867. [Google Scholar] [CrossRef]
- Bellei, E.; Bergamini, S.; Monari, E.; Fantoni, L.I.; Cuoghi, A.; Ozben, T.; Tomasi, A. High-abundance proteins depletion for serum proteomic analysis: Concomitant removal of non-targeted proteins. Amino Acids 2011, 40, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de-Souza, D. 2de: The phoenix of proteomics. J. Proteom. 2014, 104, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.N.; Pottala, J.V.; Nanjee, M.N. A comparative study of four independent methods to measure ldl particle concentration. Atherosclerosis 2015, 243, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Caradec, J.; Kharmate, G.; Hosseini-Beheshti, E.; Adomat, H.; Gleave, M.; Guns, E. Reproducibility and efficiency of serum-derived exosome extraction methods. Clin. Biochem. 2014, 47, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Lin, S.Y.; Yeh, Y.Y.; Hsiao, H.H.; Wu, C.Y.; Chen, S.T.; Wang, A.H. A modified protein precipitation procedure for efficient removal of albumin from serum. Electrophoresis 2005, 26, 2117–2127. [Google Scholar] [CrossRef] [PubMed]
- Qoronfleh, M.W.; Benton, B.; Ignacio, R.; Kaboord, B. Selective enrichment of membrane proteins by partition phase separation for proteomic studies. J. Biomed. Biotechnol. 2003, 2003, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Bjorhall, K.; Miliotis, T.; Davidsson, P. Comparison of different depletion strategies for improved resolution in proteomic analysis of human serum samples. Proteomics 2005, 5, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.R.; Churchward, M.A.; Butt, R.H.; Coorssen, J.R. Assessing detection methods for gel-based proteomic analyses. J. Proteome Res. 2007, 6, 1418–1425. [Google Scholar] [CrossRef] [PubMed]
- Gauci, V.J.; Wright, E.P.; Coorssen, J.R. Quantitative proteomics: Assessing the spectrum of in-gel protein detection methods. J. Chem. Biol. 2011, 4, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Gauci, V.J.; Padula, M.P.; Coorssen, J.R. Coomassie blue staining for high sensitivity gel-based proteomics. J. Proteom. 2013, 90, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Coomassie blue as a near-infrared fluorescent stain: A systematic comparison with sypro ruby for in-gel protein detection. Mol. Cell. Proteom. 2013, 12, 3834–3850. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xiong, E.; Wang, W.; Scali, M.; Cresti, M. Universal sample preparation method integrating trichloroacetic acid/acetone precipitation with phenol extraction for crop proteomic analysis. Nat. Protoc. 2014, 9, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Pfeifer, T.A.; Delaney, A.; Grigliatti, T.A.; Tetzlaff, W.G.; Coorssen, J.R. Enabling coupled quantitative genomics and proteomics analyses from rat spinal cord samples. Mol. Cell. Proteom. 2007, 6, 1574–1588. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Lee, M.W.; Pirshahid, S.A.; Backlund, P.S.; Wood, S.; Coorssen, J.R. An initial proteomic analysis of human preterm labor: Placental membranes. J. Proteome Res. 2006, 5, 3161–3172. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Postfractionation for enhanced proteomic analyses: Routine electrophoretic methods increase the resolution of standard 2d-page. J. Proteome Res. 2005, 4, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.P.; Prasad, K.A.G.; Padula, M.P.; Coorssen, J.R. Deep imaging: How much of the proteome does current top-down technology already resolve? PLoS ONE 2014, 9, e86058. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.P.; Partridge, M.A.; Padula, M.P.; Gauci, V.J.; Malladi, C.S.; Coorssen, J.R. Top-down proteomics: Enhancing 2d gel electrophoresis from tissue processing to high-sensitivity protein detection. Proteomics 2014, 14, 872–889. [Google Scholar] [CrossRef] [PubMed]
- Partridge, M.A.; Gopinath, S.; Myers, S.J.; Coorssen, J.R. An initial top-down proteomic analysis of the standard cuprizone mouse model of multiple sclerosis. J. Chem. Biol. 2016, 9, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Pre-extraction sample handling by automated frozen disruption significantly improves subsequent proteomic analyses. J. Proteome Res. 2006, 5, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Dung, N.T.; Van Chi, P. A survey of membrane proteins in human serum. Proteom. Insights 2012, 5, 1–19. [Google Scholar]
- Dan, P.D.; Thuong, T.T.; Minh, P.D.; Loi, D.D.; Nhi, N.B.; Chi, P.V. Analysis of the membrane proteins in human serum. J. Proteom. Bioinform. 2013, 6, 296–301. [Google Scholar]
- Churchward, M.A.; Butt, R.H.; Lang, J.C.; Hsu, K.K.; Coorssen, J.R. Enhanced detergent extraction for analysis of membrane proteomes by two-dimensional gel electrophoresis. Proteome Sci. 2005, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Bordier, C. Phase separation of integral membrane proteins in triton x-114 solution. J. Biol. Chem. 1981, 256, 1604–1607. [Google Scholar] [PubMed]
- Shaw, M.M.; Riederer, B.M. Sample preparation for two-dimensional gel electrophoresis. Proteomics 2003, 3, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, T.; Saito, Y.; Nakamura, Y.; Tomonaga, T.; Murakami, Y.; Kondo, T. Combined use of a solid-phase hexapeptide ligand library with liquid chromatography and two-dimensional difference gel electrophoresis for intact plasma proteomics. Int. J. Proteom. 2011, 2011, 739615. [Google Scholar] [CrossRef] [PubMed]
- Millioni, R.; Tolin, S.; Puricelli, L.; Sbrignadello, S.; Fadini, G.P.; Tessari, P.; Arrigoni, G. High abundance proteins depletion vs low abundance proteins enrichment: Comparison of methods to reduce the plasma proteome complexity. PLoS ONE 2011, 6, e19603. [Google Scholar] [CrossRef] [PubMed]
- Gundry, R.L.; Fu, Q.; Jelinek, C.A.; Van Eyk, J.E.; Cotter, R.J. Investigation of an albumin-enriched fraction of human serum and its albuminome. Proteom. Clin. Appl. 2007, 1, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Garnham, C.P.; Elliott, S.T.; Bovenkamp, D.E.; Van Eyk, J.E. A robust, streamlined, and reproducible method for proteomic analysis of serum by delipidation, albumin and igg depletion, and two-dimensional gel electrophoresis. Proteomics 2005, 5, 2656–2664. [Google Scholar] [CrossRef] [PubMed]
- Fountoulakis, M.; Juranville, J.F.; Jiang, L.; Avila, D.; Roder, D.; Jakob, P.; Berndt, P.; Evers, S.; Langen, H. Depletion of the high-abundance plasma proteins. Amino Acids 2004, 27, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Govorukhina, N.I.; Reijmers, T.H.; Nyangoma, S.O.; van der Zee, A.G.; Jansen, R.C.; Bischoff, R. Analysis of human serum by liquid chromatography-mass spectrometry: Improved sample preparation and data analysis. J. Chromatogr. A 2006, 1120, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Martosella, J.; Zolotarjova, N.; Liu, H.; Nicol, G.; Boyes, B.E. Reversed-phase high-performance liquid chromatographic prefractionation of immunodepleted human serum proteins to enhance mass spectrometry identification of lower-abundant proteins. J. Proteome Res. 2005, 4, 1522–1537. [Google Scholar] [CrossRef] [PubMed]
- Guerrier, L.; Righetti, P.G.; Boschetti, E. Reduction of dynamic protein concentration range of biological extracts for the discovery of low-abundance proteins by means of hexapeptide ligand library. Nat. Protoc. 2008, 3, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Boschetti, E.; Giorgio Righetti, P. Hexapeptide combinatorial ligand libraries: The march for the detection of the low-abundance proteome continues. BioTechniques 2008, 44, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Sennels, L.; Salek, M.; Lomas, L.; Boschetti, E.; Righetti, P.G.; Rappsilber, J. Proteomic analysis of human blood serum using peptide library beads. J. Proteome Res. 2007, 6, 4055–4062. [Google Scholar] [CrossRef] [PubMed]
- Sussulini, A.; Dihazi, H.; Banzato, C.E.; Arruda, M.A.; Stuhmer, W.; Ehrenreich, H.; Jahn, O.; Kratzin, H.D. Apolipoprotein a-i as a candidate serum marker for the response to lithium treatment in bipolar disorder. Proteomics 2011, 11, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Zolotarjova, N.; Martosella, J.; Nicol, G.; Bailey, J.; Boyes, B.E.; Barrett, W.C. Differences among techniques for high-abundant protein depletion. Proteomics 2005, 5, 3304–3313. [Google Scholar] [CrossRef] [PubMed]
- Yocum, A.K.; Yu, K.; Oe, T.; Blair, I.A. Effect of immunoaffinity depletion of human serum during proteomic investigations. J. Proteome Res. 2005, 4, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Granger, J.; Siddiqui, J.; Copeland, S.; Remick, D. Albumin depletion of human plasma also removes low abundance proteins including the cytokines. Proteomics 2005, 5, 4713–4718. [Google Scholar] [CrossRef] [PubMed]
- Stempfer, R.; Kubicek, M.; Lang, I.M.; Christa, N.; Gerner, C. Quantitative assessment of human serum high-abundance protein depletion. Electrophoresis 2008, 29, 4316–4323. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.; Shapiguzov, A.; Fucile, G.; Rochaix, J.D.; Goldschmidt-Clermont, M.; Eichacker, L.A. Separation of membrane protein complexes by native lds-page. Methods Mol. Biol. 2014, 1072, 667–676. [Google Scholar] [PubMed]
- Delepelaire, P.; Chua, N.-H. Lithium dodecyl sulfate/polyacrylamide gel electrophoresis of thylakoid membranes at 4 °C: Characterizations of two additional chlorophyll a-protein complexes. Proc. Natl. Acad. Sci. USA 1979, 76, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Fountoulakis, M.; Gasser, R. Proteomic analysis of the cell envelope fraction of escherichia coli. Amino Acids 2003, 24, 19–41. [Google Scholar] [PubMed]
- Fountoulakis, M.; Juranville, J.F. Enrichment of low-abundance brain proteins by preparative electrophoresis. Anal. Biochem. 2003, 313, 267–282. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Type of Gel | Gel % | Protein Conc. | Protein Species Detected |
|---|---|---|---|---|
| Native serum (Baseline for statistical comparison) | Mini | 12.5% | 100 µg | † 367 ± 2 |
| Ultra 3 h | 12.5% | 100 µg | 424 ± 21 | |
| Trichloroacetic acid (TCA) | 12.5% | 100 µg | 358 ± 6 | |
| Tx-114 | 12.5% | 100 µg | 415 ± 3 | |
| Size exclusion filters | Frac A and Frac B (7–10%) | 100 µg | † 392 ± 24 | |
| Frac C (12–20%) | ||||
| Aurum Affi-Gel Blue column | 12.5% | 100 µg | 285 ± 7 | |
| Tx-114 | Large | 7–20% | 500 µg | † 779 ± 51 * |
| Method | Type of Gel | Gel % | Protein Concentration | Number of Protein Species Identified |
|---|---|---|---|---|
| Sodium dodecyl sulfate (SDS) (0.1%) | Large gel | 12.5% | 500 µg | 709 ± 15 |
| No gradient (Baseline for statistical comparison) | ||||
| SDS (0.1%) | 7–20% | 500 µg | 864 ± 11 * | |
| Lithium dodecyl sulfate (LDS) (0.1%) | 7–20% | 500 µg | 870 ± 12 * | |
| SDS (0.1%) + LDS (0.1%) | 7–20% | 500 µg | 919 ± 15 * | |
| Deep imaging | 7–20% | 500 µg | 942 ± 7 * | |
| SDS (0.1%) + LDS (0.1%) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Silva, A.M.; Hyett, J.A.; Coorssen, J.R. A Routine ‘Top-Down’ Approach to Analysis of the Human Serum Proteome. Proteomes 2017, 5, 13. https://doi.org/10.3390/proteomes5020013
D’Silva AM, Hyett JA, Coorssen JR. A Routine ‘Top-Down’ Approach to Analysis of the Human Serum Proteome. Proteomes. 2017; 5(2):13. https://doi.org/10.3390/proteomes5020013
Chicago/Turabian StyleD’Silva, Arlene M., Jon A. Hyett, and Jens R. Coorssen. 2017. "A Routine ‘Top-Down’ Approach to Analysis of the Human Serum Proteome" Proteomes 5, no. 2: 13. https://doi.org/10.3390/proteomes5020013
APA StyleD’Silva, A. M., Hyett, J. A., & Coorssen, J. R. (2017). A Routine ‘Top-Down’ Approach to Analysis of the Human Serum Proteome. Proteomes, 5(2), 13. https://doi.org/10.3390/proteomes5020013