
Comparative Skeletal Muscle Proteomics Using Two-Dimensional Gel Electrophoresis

Abstract
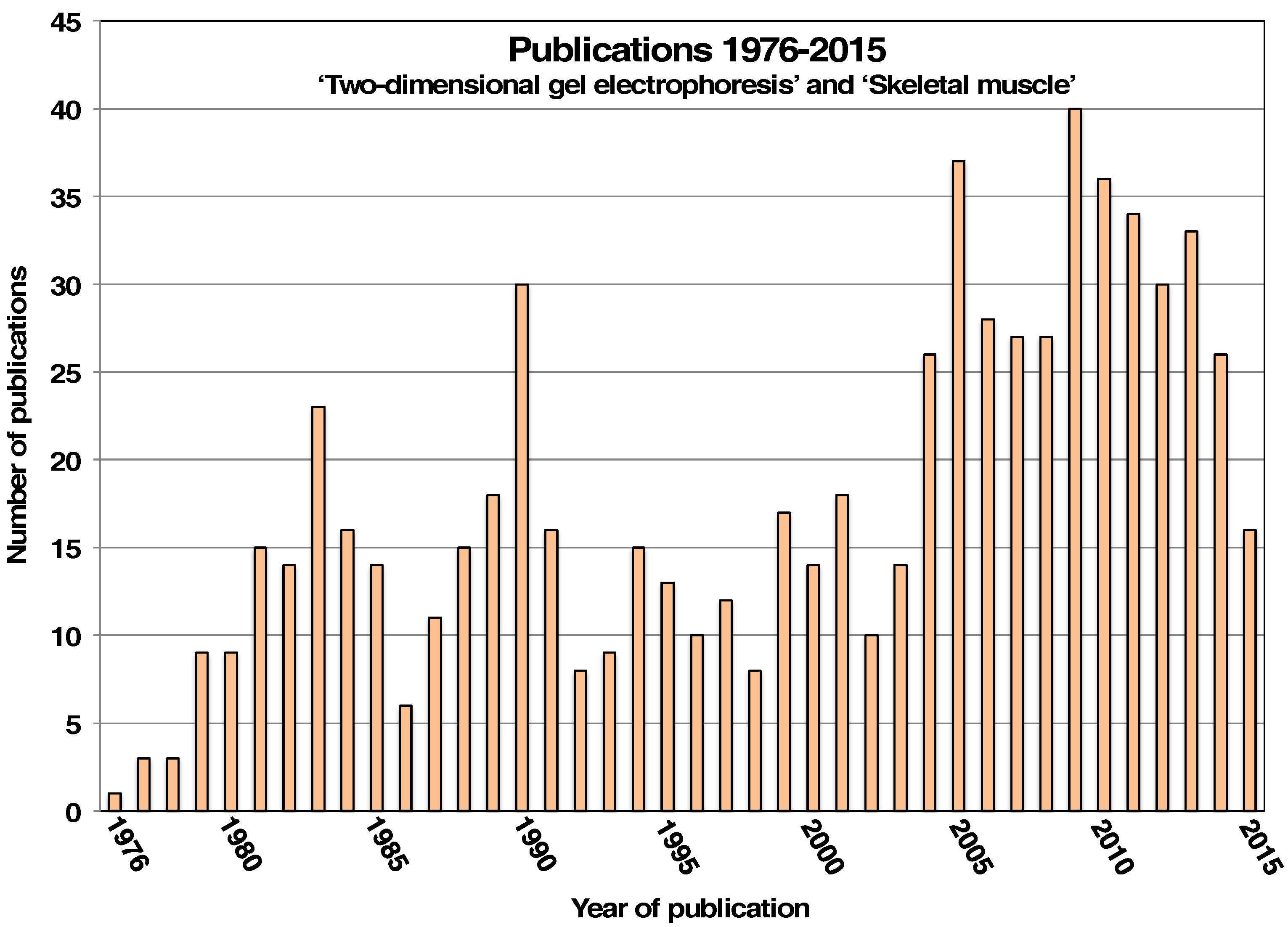
:1. Introduction
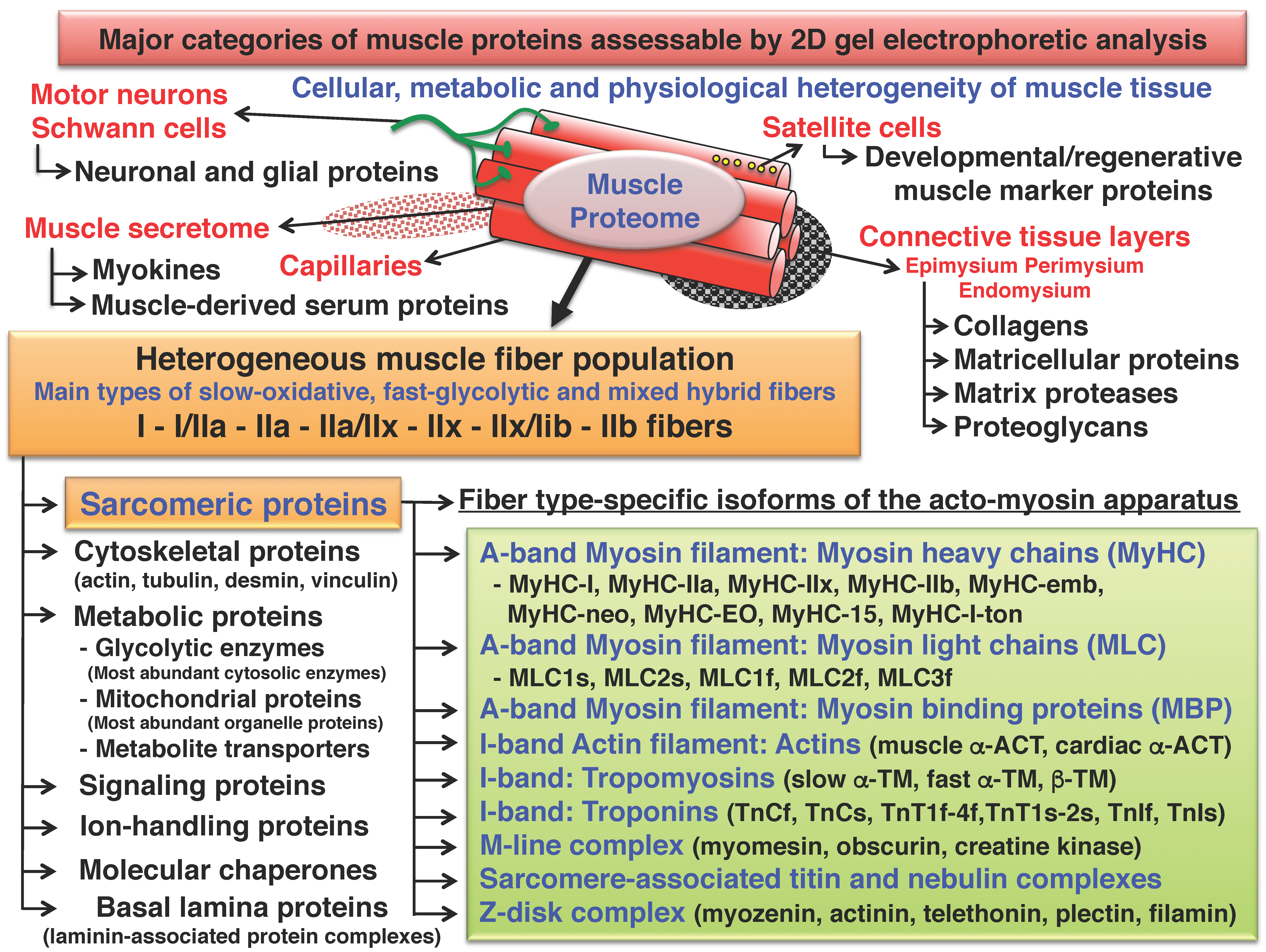
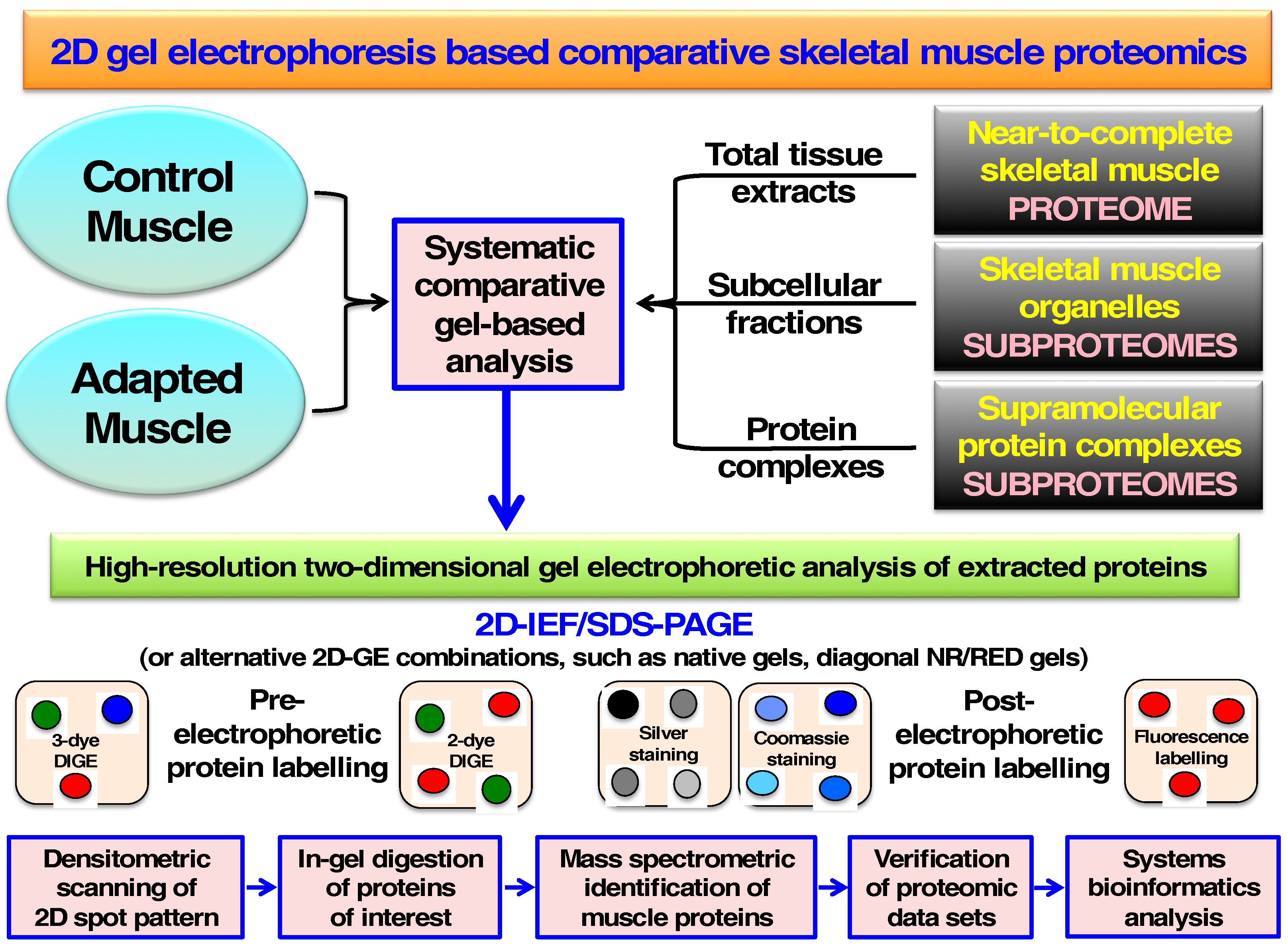
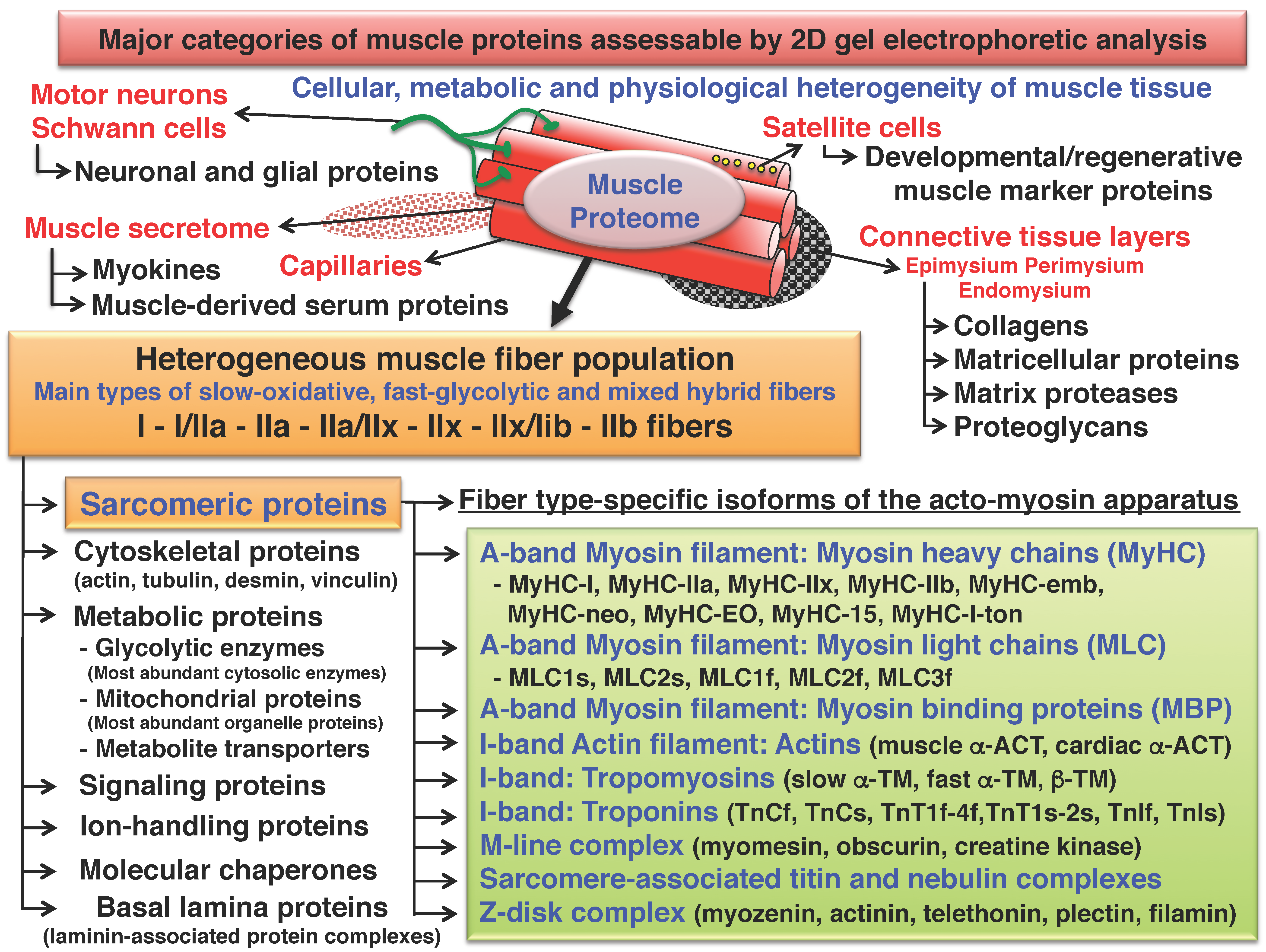
2. Two-Dimensional Gel Electrophoresis of Skeletal Muscle Proteins
3. Cataloguing of the Skeletal Muscle Proteome Using Two-Dimensional Gel Electrophoresis
- Possible under-estimation of particular types of proteins, including highly hydrophobic proteins, very high-molecular-mass proteins and low-copy-number proteins. Changing gel conditions, the introduction of suitable pre- and post-fractionation steps, as well as higher sensitivity detection protocols can often overcome some of these technical limitations [84,85,86,87].
- Hypothetical under-representation or 2D streaking of proteins with extreme pI-values, which however depends heavily on the particular IEF conditions employed in the first dimensional separation step. Often very acidic protein species form vertical streaking patterns at the pH 3 region and very basic proteins at the pH 11 region. To at least partially overcome this problem, the usage of narrow-range immobilized pH gradients can be applied for zooming in on protein species that do not fall into the commonly applied range of approximately pI 3 to 11 [88,89,90,91]. In addition, combining the findings from several different IEF gels in the first dimension with slightly overlapping pI-values can be advantageous for producing more comprehensive protein coverage [15,92,93].
- Potentially restricted separation of complex protein mixtures with greatly differing molecular masses using routine 2D-GE approaches. Often the usage of large-scale gels, optimized gradient SDS-PAGE slab gel systems in the second dimension and the reduction of sample complexity can overcome some of these technical problems and be used to cover protein species that do not fall into in the routinely analyzed range of approximately 10 to 250 kDa [67,94].
- Latent cross-contamination of individual 2D protein spots through highly abundant polypeptides that are dragged throughout the 2D gel system due to their exceedingly high density. These abnormal electrophoretic mobility patterns of particular proteins cause a certain degree of 2D streaking, which can be minimized by (i) decreasing the total amount of protein loading; (ii) using very large gel systems with a higher discriminatory capacity and/or (iii) applying optimized pre-fractionation techniques to decisively decrease sample complexity [95,96,97,98]. Artifacts can be kept to a minimum using 50 to 200 μg of total protein in first dimension gels. Lower protein concentrations usually result in weak staining patterns. Comparative studies with fluorescent dyes give optimum results with approximately 50 μg of protein per sample.
- Potential discrepancies between the findings from the densitometric scanning of gel images and the MS-based protein identification in case of a heterogeneous composition of a single 2D protein spot. For example, if a protein spot contains more than one protein species and the most abundant protein is not as susceptible to digestion as the low-copy-number proteins in its vicinity, then the concentration change of this 2D protein spot (as determined by densitometric scanning) may be misleading. However, this analytical complication is a relatively rare occurrence and the use of simple post-fractionation approaches and/or independent verification of gel-based proteomic data by immunoblotting surveys or immunofluorescence microscopical analysis can effectively assess the rate of these kinds of analytical discrepancies [21].
- Extremely reliable protein separation system that can be routinely used in large-scale and high-throughput proteomic surveys. Multi-gel systems using large buffer tanks can run a considerable number of 2D gels in parallel making this approach both cost-effective and highly reproducible for systematic biochemical studies [15,16,17].
- Technical provision of a bioanalytical platform that is ideally suited for the subsequent identification of specific protein isoforms and their PTMs [46,47,48]. Many in-gel staining or labeling methods can specifically highlight PTMs, such as enzyme-conjugated lectin labeling or Pro-Q Emerald staining for glycosylation or the fluorescent Pro-Q Diamond dye for phosphorylation [58,105,106,107].
- Direct visualization of proteins of interest as discrete 2D spots, enabling the exact evaluation of the characteristic combination of the pI-value and relative molecular mass of a particular protein subunit or isoform. This provides a unique analytical advantage over simpler 1D gel systems that display heterogeneous protein bands or LC methods. Often MS data from LC-based analyses do not given efficient information on sequence coverage to unequivocally determine whether a fully intact protein species or fragments have been detected. In contrast, proteomic data from the analysis of distinct 2D-GE spots can be directly correlated with the electrophoretic mobility and thereby the relative molecular mass of the protein of interest [21].
- Since potential discrepancies between the mass spectrometric identification of a protein and its position in a 2D gel in relation to its pI-value and/or molecular mass can be easily assessed, the rate of false positive protein hits can be conveniently measured and swiftly eliminated from the final list of altered protein species. Additional analyses can then determine whether an abnormal or unexpected electrophoretic mobility pattern is due to protein degradation, protein clustering or a technical artifact caused by 2D streaking and cross-contamination [18].
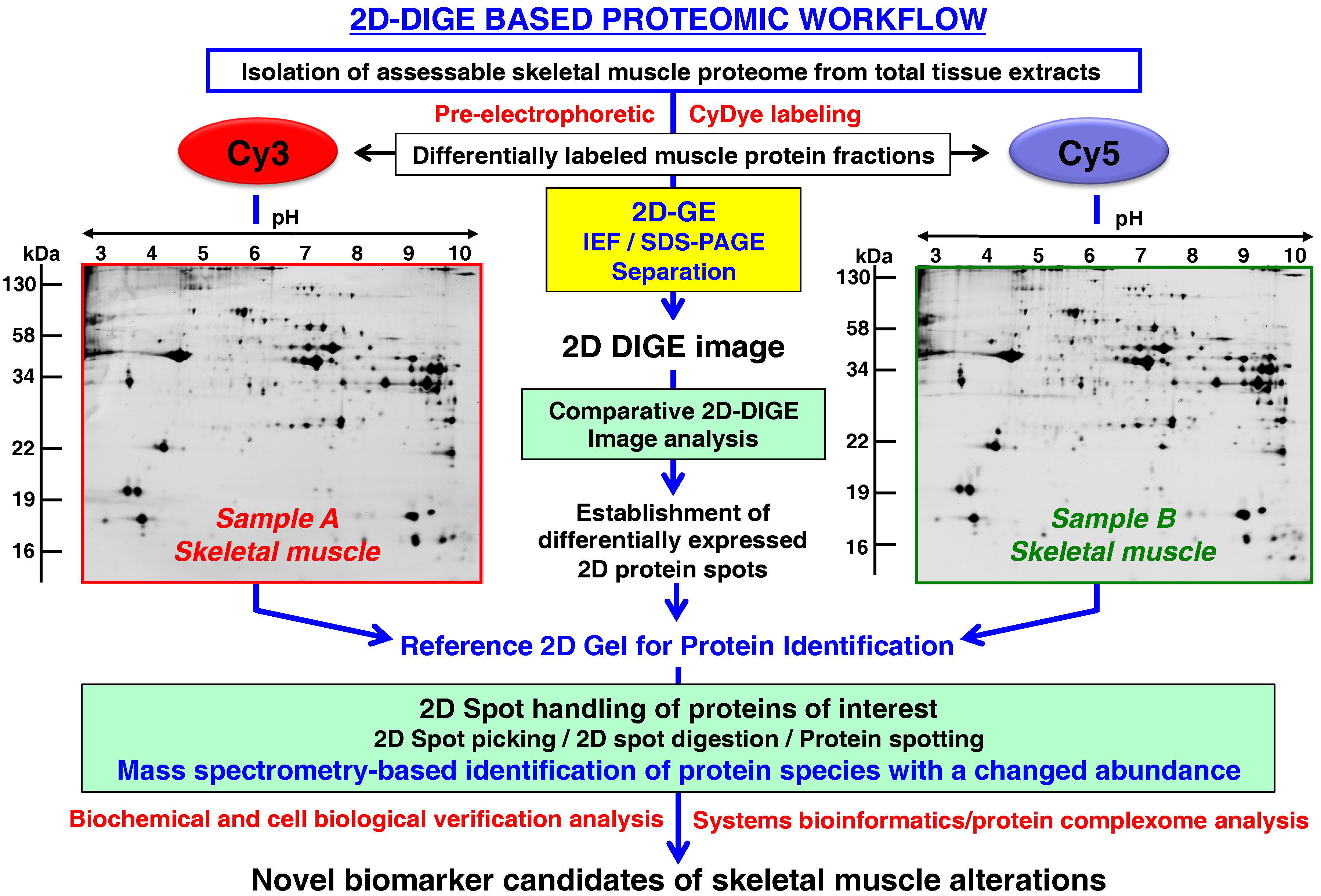
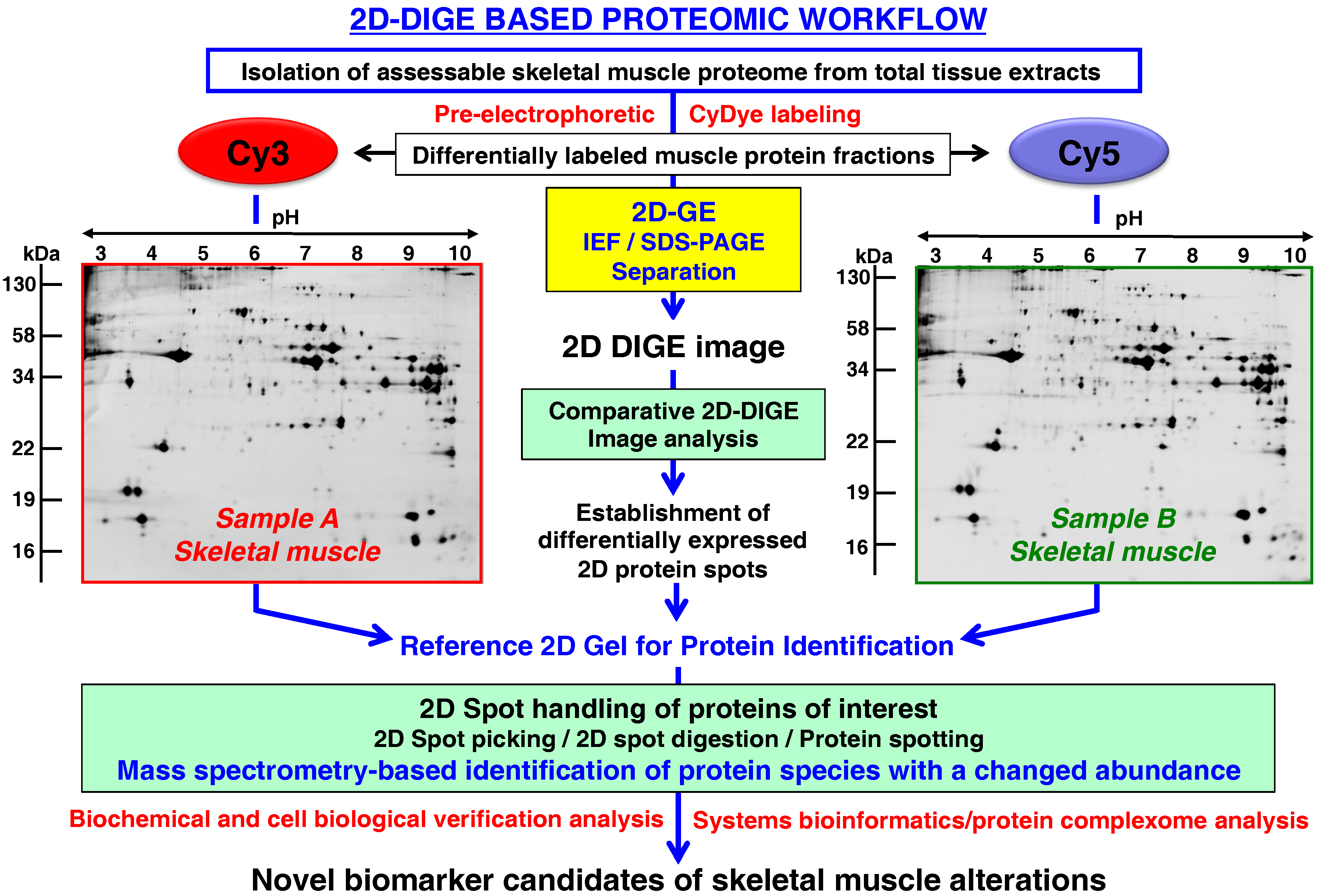
- Rapid and quantitative analyses of paired protein samples can be conducted. An example of an extremely powerful comparative 2D-GE method is the fluorescence 2D-DIGE technique [108] that eliminates gel-to-gel variations by the differential pre-electrophoretic labeling of protein fractions and the subsequent separation on the same 2D gel followed by image analysis [109]. See below section for details on the DIGE method and its application in skeletal muscle proteomics.
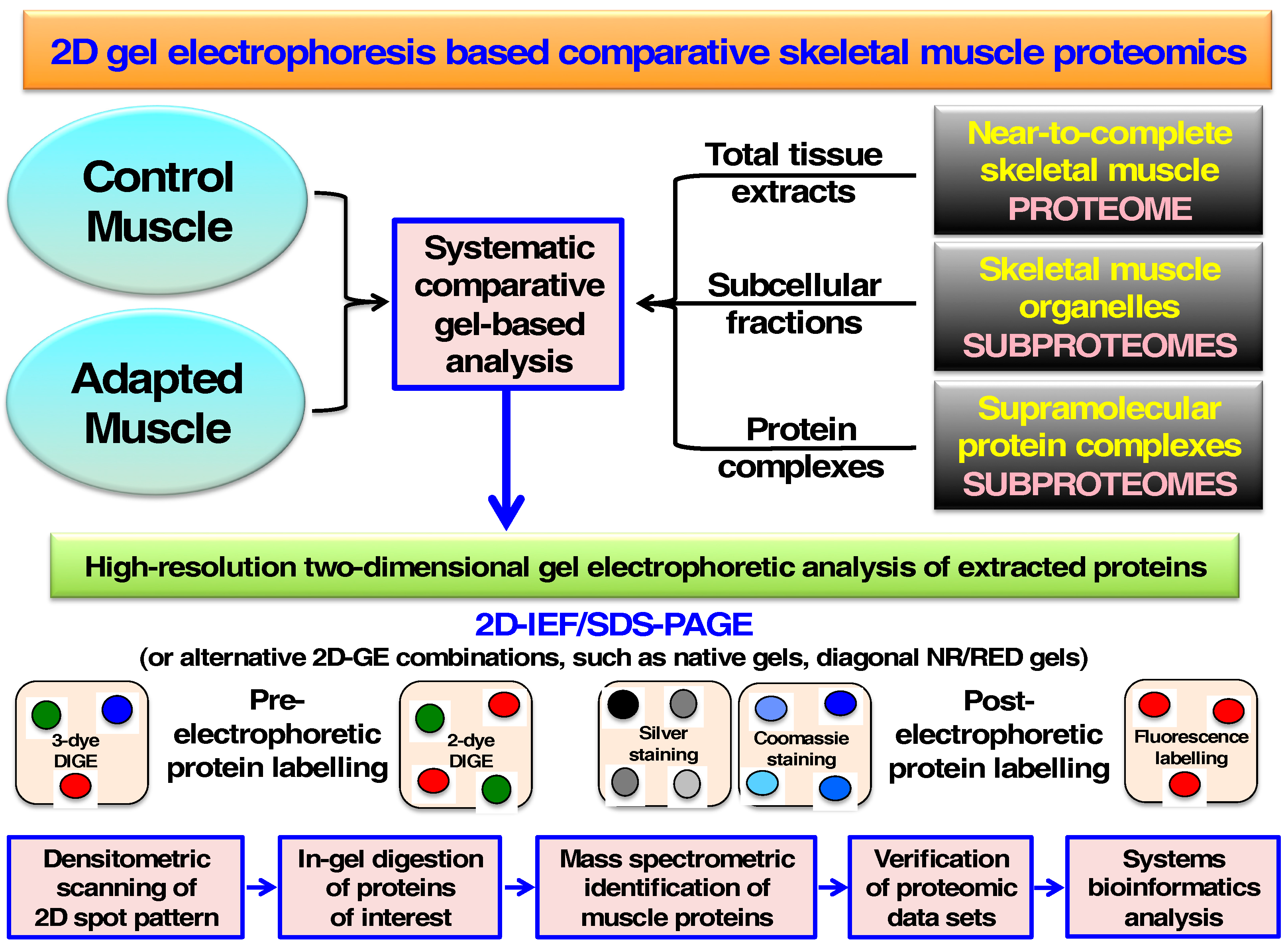
4. Comparative Skeletal Muscle Proteomics Using Two-Dimensional Gel Electrophoresis
4.1. Proteome Signature of Skeletal Muscle Development
4.2. Muscle Plasticity and Fiber Type Specification
4.3. Exercise-Induced Proteome Signature
4.4. Hypoxia-Related Muscle Adaptations
4.5. Proteome-Wide Changes during Disuse Atrophy
4.6. Sarcopenia of Old Age
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2D | two-dimensional |
| ACT | actin |
| DIGE | difference in-gel electrophoresis |
| GE | gel electrophoresis |
| Hsp | heat shock protein |
| IEF | isoelectric focusing |
| LC | liquid chromatography |
| MBP | myosin binding protein |
| MLC | myosin light chain |
| MS | mass spectrometry |
| MyHC | myosin heavy chain |
| PAGE | polyacrylamide gel electrophoresis |
| p I | isoelectric point |
| PTM | post-translational modification |
| SDS | sodium dodecyl sulfate |
| TM | tropomyosin |
| TN | troponin |
References
- Righetti, P.G. Bioanalysis: Its past, present, and some future. Electrophoresis 2004, 25, 2111–2127. [Google Scholar] [CrossRef] [PubMed]
- Rible, H. Historical and theoretical aspects of isoelectric focusing. Ann. N. Y. Acad. Sci. 1973, 209, 11–22. [Google Scholar] [PubMed]
- Weber, K.; Osborn, M. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J. Biol. Chem. 1969, 244, 4406–4412. [Google Scholar] [PubMed]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Chrambach, A.; Rodbard, D. Polyacrylamide gel electrophoresis. Science 1971, 172, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Kubota, K.; Kosaka, T.; Ichikawa, K. Combination of two-dimensional electrophoresis and shotgun peptide sequencing in comparative proteomics. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 815, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.B.; Hoving, S.; Westermeier, R. Isoelectric focusing and two-dimensional gel electrophoresis. Methods Enzymol. 2009, 463, 515–540. [Google Scholar] [PubMed]
- Klose, J. From 2-D electrophoresis to proteomics. Electrophoresis 2009, 30 (Suppl. 1), S142–S149. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar] [PubMed]
- Klose, J. Protein mapping by combined isoelectric focusing and electrophoresis of mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik 1975, 26, 231–243. [Google Scholar] [PubMed]
- Scheele, G.A. Two-dimensional gel analysis of soluble proteins. Charaterization of guinea pig exocrine pancreatic proteins. J. Biol. Chem. 1975, 250, 5375–5385. [Google Scholar] [PubMed]
- Iborra, F.; Buhler, J.M. Protein subunit mapping. A sensitive high resolution method. Anal. Biochem. 1976, 74, 503–511. [Google Scholar] [CrossRef]
- Anderson, L.; Anderson, N.G. High resolution two-dimensional electrophoresis of human plasma proteins. Proc. Natl. Acad. Sci. USA 1977, 74, 5421–5425. [Google Scholar] [CrossRef] [PubMed]
- Klose, J.; Kobalz, U. Two-dimensional electrophoresis of proteins: An updated protocol and implications for a functional analysis of the genome. Electrophoresis 1995, 16, 1034–1059. [Google Scholar] [CrossRef] [PubMed]
- Görg, A.; Weiss, W.; Dunn, M.J. Current two-dimensional electrophoresis technology for proteomics. Proteomics 2004, 4, 3665–3685. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Chevallet, M.; Luche, S.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: Past, present and future. J. Proteom. 2010, 73, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.M.; Coorssen, J.R.; Martins-de-Souza, D. 2DE: The phoenix of proteomics. J. Proteom. 2014, 104, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Donoghue, P.; O’Connell, K.; Gannon, J.; Ohlendieck, K. Proteomic profiling of pathological and aged skeletal muscle fibres by peptide mass fingerprinting (Review). Int. J. Mol. Med. 2007, 19, 547–564. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohlendieck, K. Proteomics of skeletal muscle differentiation, neuromuscular disorders and fiber aging. Expert Rev. Proteom. 2010, 7, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Gelfi, C.; Vasso, M.; Cerretelli, P. Diversity of human skeletal muscle in health and disease: Contribution of proteomics. J. Proteom. 2011, 74, 774–795. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Skeletal muscle proteomics: Current approaches, technical challenges and emerging techniques. Skelet. Muscle 2011, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.; Murphy, S.; Ohlendieck, K. Proteomic profiling of muscle fibre type shifting in neuromuscular diseases. Expert Rev. Proteom. 2016, 13, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomic identification of biomarkers of skeletal muscle disorders. Biomark. Med. 2013, 7, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Giometti, C.S. Muscle protein analysis by two-dimensional gel electrophoresis. Crit. Rev. Clin. Lab. Sci. 1982, 18, 79–109. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine, National Institutes of Health PubMed Site. Available online: http://www.ncbi.nlm.nih.gov/pubm (accessed on 15 August 2016).
- Pette, D. The adaptive potential of skeletal muscle fibers. Can. J. Appl. Physiol. 2002, 27, 423–448. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Pette, D.; Staron, R.S. Myosin isoforms, muscle fiber types, and transitions. Microsc. Res. Tech. 2000, 50, 500–509. [Google Scholar] [CrossRef]
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Weigert, C.; Lehmann, R.; Hartwig, S.; Lehr, S. The secretome of the working human skeletal muscle—A promising opportunity to combat the metabolic disaster? Proteom. Clin. Appl. 2014, 8, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Sherton, C.C.; Wool, I.G. A comparison of the proteins of rat skeletal muscle and liver ribosomes by two-dimensional polyacrylamide gel electrophoresis. Observations on the partition of proteins between ribosomal subunits and a description of two acidic proteins in the large subunit. J. Biol. Chem. 1974, 249, 2258–2267. [Google Scholar] [PubMed]
- Barrett, E.J.; Headon, D.R. Two-dimensional polyacrylamide gel electrophoresis of rabbit skeletal muscle microsomal proteins. FEBS Lett. 1975, 51, 12112–12115. [Google Scholar] [CrossRef] [Green Version]
- Izant, J.G.; Lazarides, E. Invariance and heterogeneity in the major structural and regulatory proteins of chick muscle cells revealed by two-dimensional gel electrophoresis. Proc. Natl. Acad. Sci. USA 1977, 74, 1450–1454. [Google Scholar] [CrossRef] [PubMed]
- Zechel, K. Localization of the charge differences in the actins of rabbit skeletal muscle and chicken gizzard by two-dimensional gel electrophoretic analysis of tryptic fragments. Hoppe Seylers Z. Physiol. Chem. 1979, 360, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Giometti, C.S.; Anderson, N.G.; Anderson, N.L. Muscle protein analysis. I. High-resolution two-dimensional electrophoresis of skeletal muscle proteins for analysis of small biopsy samples. Clin. Chem. 1979, 25, 1877–1884. [Google Scholar] [PubMed]
- Giometti, C.S.; Danon, M.J.; Anderson, N.G. Human muscle proteins: Analysis by two-dimensional electrophoresis. Neurology 1983, 33, 1152–1156. [Google Scholar] [CrossRef] [PubMed]
- Billeter, R.; Heizmann, C.W.; Howald, H.; Jenny, E. Analysis of myosin light and heavy chain types in single human skeletal muscle fibers. Eur. J. Biochem. 1981, 116, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Billeter, R.; Heizmann, C.W.; Reist, U.; Howald, H.; Jenny, E. Two-dimensional peptide analysis of myosin heavy chains and actin from single-typed human skeletal muscle fibers. FEBS Lett. 1982, 139, 45–48. [Google Scholar] [CrossRef]
- O’Farrell, P.H. The pre-omics era: The early days of two-dimensional gels. Proteomics 2008, 8, 4842–4852. [Google Scholar] [CrossRef] [PubMed]
- Bárány, K.; Bárány, M.; Giometti, C.S. Polyacrylamide gel electrophoretic methods in the separation of structural muscle proteins. J. Chromatogr. A 1995, 698, 301–332. [Google Scholar] [CrossRef]
- Dunn, M.J. Quantitative two-dimensional gel electrophoresis: From proteins to proteomes. Biochem. Soc. Trans. 1997, 25, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomic profiling of skeletal muscle plasticity. Muscles Ligaments Tendons J. 2012, 1, 119–126. [Google Scholar] [PubMed]
- Holland, A.; Ohlendieck, K. Proteomic profiling of the contractile apparatus from skeletal muscle. Expert Rev. Proteom. 2013, 10, 239–257. [Google Scholar] [CrossRef] [PubMed]
- López, J.L. Two-dimensional electrophoresis in proteome expression analysis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 849, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Rabilloud, T.; Lelong, C. Two-dimensional gel electrophoresis in proteomics: A tutorial. J. Proteom. 2011, 74, 1829–18241. [Google Scholar] [CrossRef] [PubMed]
- Rogowska-Wrzesinska, A.; Le Bihan, M.C.; Thaysen-Andersen, M.; Roepstorff, P. 2D gels still have a niche in proteomics. J. Proteom. 2013, 88, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Link, A.J. 2-D Proteome Analysis Protocols. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 1999; Volume 112, pp. 1–601. [Google Scholar]
- Reinders, J.; Sickmann, A. Proteomics Methods and Protocols. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; Volume 564, pp. 1–431. [Google Scholar]
- Cramer, R.; Westermeier, R. Difference Gel Electrophoresis (DIGE) Methods and Protocols. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 854, pp. 1–401. [Google Scholar]
- Kurien, B.T.; Scofield, R.H. Protein Electrophoresis Methods and Protocols. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 869, pp. 1–648. [Google Scholar]
- Marengo, E.; Robotti, E. 2D PAGE Map Analysis Methods and Protocols. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2016; Volume 1384, pp. 1–331. [Google Scholar]
- Patton, W.F. Detection technologies in proteome analysis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002, 771, 3–31. [Google Scholar] [CrossRef]
- Westermeier, R.; Marouga, R. Protein detection methods in proteomics research. Biosci. Rep. 2005, 25, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.; Crawford, J.; Gianazza, E. Protein stains for proteomic applications: Which, when, why? Proteomics 2006, 6, 5385–5408. [Google Scholar] [CrossRef] [PubMed]
- Riederer, B.M. Non-covalent and covalent protein labeling in two-dimensional gel electrophoresis. J. Proteom. 2008, 71, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.; Weiland, F.; Görg, A. Protein detection and quantitation technologies for gel-based proteome analysis. Methods Mol. Biol. 2009, 564, 59–82. [Google Scholar] [PubMed]
- Gauci, V.J.; Wright, E.P.; Coorssen, J.R. Quantitative proteomics: Assessing the spectrum of in-gel protein detection methods. J. Chem. Biol. 2011, 4, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Kurien, B.T.; Scofield, R.H. A brief review of other notable protein detection methods on acrylamide gels. Methods Mol. Biol. 2012, 869, 617–620. [Google Scholar] [PubMed]
- Panfoli, I.; Calzia, D.; Santucci, L.; Ravera, S.; Bruschi, M.; Candiano, G. A blue dive: From ‘blue fingers’ to ‘blue silver’. A comparative overview of staining methods for in-gel proteomics. Expert Rev. Proteom. 2012, 9, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Coomassie blue as a near-infrared fluorescent stain: A systematic comparison with Sypro Ruby for in-gel protein detection. Mol. Cell. Proteom. 2013, 12, 3834–3850. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Altelaar, A.F.; Heck, A.J. Trends in ultrasensitive proteomics. Curr. Opin. Chem. Biol. 2012, 16, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 2011, 80, 273–299. [Google Scholar] [CrossRef] [PubMed]
- Faini, M.; Stengel, F.; Aebersold, R. The Evolving Contribution of Mass Spectrometry to Integrative Structural Biology. J. Am. Soc. Mass Spectrom. 2016, 27, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Doran, P.; Ohlendieck, K. Proteomic analysis of dystrophic muscle. Methods Mol. Biol. 2012, 798, 357–369. [Google Scholar] [PubMed]
- Gelfi, C.; De Palma, S. 2D DIGE analysis of protein extracts from muscle tissue. Methods Mol. Biol. 2012, 854, 155–168. [Google Scholar] [PubMed]
- Reed, P.W.; Densmore, A.; Bloch, R.J. Optimization of large gel 2D electrophoresis for proteomic studies of skeletal muscle. Electrophoresis 2012, 33, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Ohlendieck, K. Gel electrophoresis-based proteomics of senescent tissues. Methods Mol. Biol. 2013, 1048, 229–246. [Google Scholar] [PubMed]
- Sanchez, J.C.; Chiappe, D.; Converset, V.; Hoogland, C.; Binz, P.A.; Paesano, S.; Appel, R.D.; Wang, S.; Sennitt, M.; Nolan, A.; et al. The mouse SWISS-2D PAGE database: A tool for proteomics study of diabetes and obesity. Proteomics 2001, 1, 136–163. [Google Scholar] [CrossRef]
- Yan, J.X.; Harry, R.A.; Wait, R.; Welson, S.Y.; Emery, P.W.; Preedy, V.R.; Dunn, M.J. Separation and identification of rat skeletal muscle proteins using two-dimensional gel electrophoresis and mass spectrometry. Proteomics 2001, 1, 424–434. [Google Scholar] [CrossRef]
- Appel, R.D.; Sanchez, J.C.; Bairoch, A.; Golaz, O.; Miu, M.; Vargas, J.R.; Hochstrasser, D.F. SWISS-2DPAGE: A database of two-dimensional gel electrophoresis images. Electrophoresis 1993, 14, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Hoogland, C.; Mostaguir, K.; Sanchez, J.C.; Hochstrasser, D.F.; Appel, R.D. SWISS-2DPAGE, ten years later. Proteomics 2004, 4, 2352–2356. [Google Scholar] [CrossRef] [PubMed]
- Lemkin, P.F. The 2DWG meta-database of two-dimensional electrophoretic gel images on the Internet. Electrophoresis 1997, 18, 2759–2773. [Google Scholar] [CrossRef] [PubMed]
- Lemkin, P.F.; Thornwall, G. Flicker image comparison of 2-D gel images for putative protein identification using the 2DWG meta-database. Mol. Biotechnol. 1999, 12, 159–172. [Google Scholar] [CrossRef]
- Babnigg, G.; Giometti, C.S. ProteomeWeb: A web-based interface for the display and interrogation of proteomes. Proteomics 2003, 3, 584–600. [Google Scholar] [CrossRef] [PubMed]
- Babnigg, G.; Giometti, C.S. GELBANK: A database of annotated two-dimensional gel electrophoresis patterns of biological systems with completed genomes. Nucleic Acids Res. 2004, 32, D582–D585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pernet, P.; Bruneel, A.; Baudin, B.; Vaubourdolle, M. PHProteomicDB: A module for two-dimensional gel electrophoresis database creation on personal web sites. Genom. Proteom. Bioinform. 2006, 4, 134–136. [Google Scholar] [CrossRef]
- Laukens, K.; Matthiesen, R.; Lemière, F.; Esmans, E.; Onckelen, H.V.; Jensen, O.N.; Witters, E. Integration of gel-based proteome data with pProRep. Bioinformatics 2006, 22, 2838–2840. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Piñeiro, C.; Vázquez, J.; Marina, A.I.; Barros-Velázquez, J.; Gallardo, J.M. Characterization and partial sequencing of species-specific sarcoplasmic polypeptides from commercial hake species by mass spectrometry following two-dimensional electrophoresis. Electrophoresis 2001, 22, 1545–1552. [Google Scholar] [CrossRef]
- Clarke, A.J.; Knight, C.; Bass, J.; Cooper, G.J. Identification and characterization of a bovine myosin light chain-1 fast polymorphism. Proteomics 2001, 1, 1495–1502. [Google Scholar] [CrossRef]
- Brewis, I.A.; Brennan, P. Proteomics technologies for the global identification and quantification of proteins. Adv. Protein Chem. Struct. Biol. 2010, 80, 1–44. [Google Scholar] [PubMed]
- Roepstorff, P. Mass spectrometry based proteomics, background, status and future needs. Protein Cell. 2012, 3, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomics of skeletal muscle glycolysis. Biochim. Biophys. Acta 2010, 1804, 2089–2101. [Google Scholar] [CrossRef] [PubMed]
- Kashino, Y.; Harayama, T.; Pakrasi, H.B.; Satoh, K. Preparation of membrane proteins for analysis by two-dimensional gel electrophoresis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 849, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Tan, H.T.; Chung, M.C. Membrane proteins and membrane proteomics. Proteomics 2008, 8, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, P.G.; Groen, A.J.; Dupree, P.; Lilley, K.S. Subcellular localization of membrane proteins. Proteomics 2008, 8, 3991–4011. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.Z.; Foster, L.J. Biochemical and proteomic approaches for the study of membrane microdomains. J. Proteom. 2009, 72, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Manadas, B.; English, J.A.; Wynne, K.J.; Cotter, D.R.; Dunn, M.J. Comparative analysis of OFFGel, strong cation exchange with pH gradient, and RP at high pH for first-dimensional separation of peptides from a membrane-enriched protein fraction. Proteomics 2009, 9, 5194–5198. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.M.; Dunn, M.J.; Posch, A.; Görg, A. Positional reproducibility of protein spots in two-dimensional polyacrylamide gel electrophoresis using immobilised pH gradient isoelectric focusing in the first dimension: An interlaboratory comparison. Electrophoresis 1994, 15, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, J.A.; Yan, J.X.; Wait, R.; Welson, S.Y.; Dunn, M.J. Zooming-in on the proteome: Very narrow-range immobilised pH gradients reveal more protein species and isoforms. Electrophoresis 2001, 22, 2865–2871. [Google Scholar] [CrossRef]
- Zuo, X.; Speicher, D.W. Comprehensive analysis of complex proteomes using microscale solution isoelectrofocusing prior to narrow pH range two-dimensional electrophoresis. Proteomics 2002, 2, 58–68. [Google Scholar] [CrossRef]
- Görg, A.; Drews, O.; Lück, C.; Weiland, F.; Weiss, W. 2-DE with IPGs. Electrophoresis 2009, 30 (Suppl. 1), S122–S132. [Google Scholar] [CrossRef] [PubMed]
- Staunton, L.; Jockusch, H.; Wiegand, C.; Albrecht, T.; Ohlendieck, K. Identification of secondary effects of hyperexcitability by proteomic profiling of myotonic mouse muscle. Mol. Biosyst. 2011, 7, 2480–2489. [Google Scholar] [CrossRef] [PubMed]
- Jarrold, B.; DeMuth, J.; Greis, K.; Burt, T.; Wang, F. An effective skeletal muscle prefractionation method to remove abundant structural proteins for optimized two-dimensional gel electrophoresis. Electrophoresis 2005, 26, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- Nordhoff, E.; Egelhofer, V.; Giavalisco, P.; Eickhoff, H.; Horn, M.; Przewieslik, T.; Theiss, D.; Schneider, U.; Lehrach, H.; Gobom, J. Large-gel two-dimensional electrophoresis-matrix assisted laser desorption/ionization-time of flight-mass spectrometry: An analytical challenge for studying complex protein mixtures. Electrophoresis 2001, 22, 2844–2855. [Google Scholar] [CrossRef]
- Righetti, P.G.; Castagna, A.; Herbert, B.; Candiano, G. How to bring the “unseen” proteome to the limelight via electrophoretic pre-fractionation techniques. Biosci. Rep. 2005, 25, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, F.G.; Arckens, L.; Quirion, R. Applications and current challenges of proteomic approaches, focusing on two-dimensional electrophoresis. Amino Acids 2007, 33, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Groen, A.J.; Lilley, K.S. Proteomics of total membranes and subcellular membranes. Expert Rev. Proteom. 2010, 7, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.; Ohlendieck, K. Mass spectrometric identification of dystrophin isoform Dp427 by on-membrane digestion of sarcolemma from skeletal muscle. Anal. Biochem. 2010, 404, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, P.; Staunton, L.; Mullen, E.; Manning, G.; Ohlendieck, K. DIGE analysis of rat skeletal muscle proteins using nonionic detergent phase extraction of young adult versus aged gastrocnemius tissue. J. Proteom. 2010, 73, 1441–1453. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Staunton, L.; Ohlendieck, K. Mass spectrometric characterization of the sarcoplasmic reticulum from rabbit skeletal muscle by on-membrane digestion. Protein Pept. Lett. 2012, 19, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. On-Membrane Digestion Technology for Muscle Proteomics. J. Membr. Sep. Technol. 2013, 2, 1–12. [Google Scholar] [CrossRef]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Comparative proteomic analysis of the contractile-protein-depleted fraction from normal versus dystrophic skeletal muscle. Anal. Biochem. 2014, 446, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Gauci, V.J.; Padula, M.P.; Coorssen, J.R. Coomassie blue staining for high sensitivity gel-based proteomics. J. Proteom. 2013, 90, 96–106. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.; Doran, P.; Gannon, J.; Ohlendieck, K. Lectin-based proteomic profiling of aged skeletal muscle: Decreased pyruvate kinase isozyme M1 exhibits drastically increased levels of N-glycosylation. Eur. J. Cell Biol. 2008, 87, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Gannon, J.; Staunton, L.; O’Connell, K.; Doran, P.; Ohlendieck, K. Phosphoproteomic analysis of aged skeletal muscle. Int. J. Mol. Med. 2008, 22, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, G.K.; Thelen, J.J. A high-resolution two dimensional Gel- and Pro-Q DPS-based proteomics workflow for phosphoprotein identification and quantitative profiling. Methods Mol. Biol. 2009, 527, 3–19. [Google Scholar] [PubMed]
- Minden, J.S.; Dowd, S.R.; Meyer, H.E.; Stühler, K. Difference gel electrophoresis. Electrophoresis 2009, 30 (Suppl. 1), S156–S161. [Google Scholar] [CrossRef] [PubMed]
- Arentz, G.; Weiland, F.; Oehler, M.K.; Hoffmann, P. State of the art of 2D DIGE. Proteom. Clin. Appl. 2015, 9, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Isfort, R.J. Proteomic analysis of striated muscle. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002, 771, 155–165. [Google Scholar] [CrossRef]
- Gelfi, C.; De Palma, S.; Cerretelli, P.; Begum, S.; Wait, R. Two-dimensional protein map of human vastus lateralis muscle. Electrophoresis 2003, 24, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.B.; Lehar, M.; Braga, N.; Westra, W.; Liu, L.H.; Flint, P.W. Study of human laryngeal muscle protein using two-dimensional electrophoresis and mass spectrometry. Proteomics 2003, 3, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Bouley, J.; Chambon, C.; Picard, B. Mapping of bovine skeletal muscle proteins using two-dimensional gel electrophoresis and mass spectrometry. Proteomics 2004, 4, 1811–1824. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Joh, J.H.; Park, H.R.; Kim, O.H.; Park, B.Y.; Lee, C.S. Differential expression profiling of the proteomes and their mRNAs in porcine white and red skeletal muscles. Proteomics 2004, 4, 3422–3428. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Viganò, A.; Ricci, E.; Cerretelli, P.; Wait, R.; Gelfi, C. Comparison of protein expression in human deltoideus and vastus lateralis muscles using two-dimensional gel electrophoresis. Proteomics 2005, 5, 2577–2586. [Google Scholar] [CrossRef] [PubMed]
- Okumura, N.; Hashida-Okumura, A.; Kita, K.; Matsubae, M.; Matsubara, T.; Takao, T.; Nagai, K. Proteomic analysis of slow- and fast-twitch skeletal muscles. Proteomics 2005, 5, 2896–2906. [Google Scholar] [CrossRef] [PubMed]
- Le Bihan, M.C.; Hou, Y.; Harris, N.; Tarelli, E.; Coulton, G.R. Proteomic analysis of fast and slow muscles from normal and kyphoscoliotic mice using protein arrays, 2-DE and MS. Proteomics 2006, 6, 4646–4661. [Google Scholar] [CrossRef] [PubMed]
- Gelfi, C.; Viganò, A.; De Palma, S.; Ripamonti, M.; Begum, S.; Cerretelli, P.; Wait, R. 2-D protein maps of rat gastrocnemius and soleus muscles: A tool for muscle plasticity assessment. Proteomics 2006, 6, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Chaze, T.; Bouley, J.; Chambon, C.; Barboiron, C.; Picard, B. Mapping of alkaline proteins in bovine skeletal muscle. Proteomics 2006, 6, 2571–2575. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.H.; Huang, H.W.; Mok, H.K. Differential proteome analysis of hagfish dental and somatic skeletal muscles. Mar. Biotechnol (N.Y.) 2007, 9, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, K.; Albrecht, D.; Hochgräfe, F.; Hecker, M.; Gotthardt, M. A proteome map of murine heart and skeletal muscle. Proteomics 2008, 8, 1885–1897. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Campos, A.; van Harten, S.; Cardoso, L.A.; Coelho, A.V. Establishment of a proteomic reference map for the gastrocnemius muscle in the rabbit (Oryctolagus cuniculus). Res. Vet. Sci. 2009, 87, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Addis, M.F.; Tanca, A.; Pagnozzi, D.; Rocca, S.; Uzzau, S. 2-D PAGE and MS analysis of proteins from formalin-fixed, paraffin-embedded tissues. Proteomics 2009, 9, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Kovalyova, M.A.; Kovalyov, L.I.; Toropygin, I.Y.; Shigeev, S.V.; Ivanov, A.V.; Shishkin, S.S. Proteomic analysis of human skeletal muscle (m. vastus lateralis) proteins: Identification of 89 gene expression products. Biochemistry 2009, 74, 1239–1252. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zheng, J.; Liu, H.; Li, J.; Chen, H.; Chen, K. Protein profiling analysis of skeletal muscle of a pufferfish, Takifugu rubripes. Mol. Biol. Rep. 2010, 37, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Abbaraju, N.V.; Boutaghou, M.N.; Townley, I.K.; Zhang, Q.; Wang, G.; Cole, R.B.; Rees, B.B. Analysis of tissue proteomes of the Gulf killifish, Fundulus grandis, by 2D electrophoresis and MALDI-TOF/TOF mass spectrometry. Integr. Comp. Biol. 2012, 52, 626–635. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohlendieck, K. Organelle proteomics in skeletal muscle biology. J. Integr. OMICS 2012, 2, 27–38. [Google Scholar] [CrossRef]
- Vitorino, R.; Ferreira, R.; Neuparth, M.; Guedes, S.; Williams, J.; Tomer, K.B.; Domingues, P.M.; Appell, H.J.; Duarte, J.A.; Amado, F.M. Subcellular proteomics of mice gastrocnemius and soleus muscles. Anal. Biochem. 2007, 366, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Reifschneider, N.H.; Goto, S.; Nakamoto, H.; Takahashi, R.; Sugawa, M.; Dencher, N.A.; Krause, F. Defining the mitochondrial proteomes from five rat organs in a physiologically significant context using 2D blue-native/SDS-PAGE. J. Proteome Res. 2006, 5, 1117–1132. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.; Ohlendieck, K. Proteomic DIGE analysis of the mitochondria-enriched fraction from aged rat skeletal muscle. Proteomics 2009, 9, 5509–5524. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Silvestri, E.; Cioffi, F.; Senese, R.; Lanni, A.; Goglia, F.; de Lange, P.; Moreno, M. Defining the transcriptomic and proteomic profiles of rat ageing skeletal muscle by the use of a cDNA array, 2D- and Blue native-PAGE approach. J. Proteom. 2009, 72, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Vitorino, R.; Alves, R.M.; Appell, H.J.; Powers, S.K.; Duarte, J.A.; Amado, F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 2010, 10, 3142–3154. [Google Scholar] [CrossRef] [PubMed]
- Gannon, J.; Doran, P.; Kirwan, A.; Ohlendieck, K. Drastic increase of myosin light chain MLC-2 in senescent skeletal muscle indicates fast-to-slow fibre transition in sarcopenia of old age. Eur. J. Cell Biol. 2009, 88, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Gajendran, N.; Frey, J.R.; Lefkovits, I.; Kuhn, L.; Fountoulakis, M.; Krapfenbauer, K.; Brenner, H.R. Proteomic analysis of secreted muscle components: Search for factors involved in neuromuscular synapse formation. Proteomics 2002, 2, 1601–1615. [Google Scholar] [CrossRef]
- Hartwig, S.; Raschke, S.; Knebel, B.; Scheler, M.; Irmler, M.; Passlack, W.; Muller, S.; Hanisch, F.G.; Franz, T.; Li, X.; et al. Secretome profiling of primary human skeletal muscle cells. Biochim. Biophys. Acta 2014, 1844, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Kanski, J.; Alterman, M.A.; Schöneich, C. Proteomic identification of age-dependent protein nitration in rat skeletal muscle. Free Radic. Biol. Med. 2003, 35, 1229–1239. [Google Scholar] [CrossRef]
- Cieniewski-Bernard, C.; Bastide, B.; Lefebvre, T.; Lemoine, J.; Mounier, Y.; Michalski, J.C. Identification of O-linked N-acetylglucosamine proteins in rat skeletal muscle using two-dimensional gel electrophoresis and mass spectrometry. Mol. Cell. Proteom. 2004, 3, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Maughan, D.W.; Henkin, J.A.; Vigoreaux, J.O. Concentrations of glycolytic enzymes and other cytosolic proteins in the diffusible fraction of a vertebrate muscle proteome. Mol. Cell. Proteom. 2005, 4, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Picariello, G.; De Martino, A.; Mamone, G.; Ferranti, P.; Addeo, F.; Faccia, M.; Spagnamusso, S.; Di Luccia, A. Proteomic study of muscle sarcoplasmic proteins using AUT-PAGE/SDS-PAGE as two-dimensional gel electrophoresis. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006, 833, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Forner, F.; Foster, L.J.; Campanaro, S.; Valle, G.; Mann, M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol. Cell. Proteom. 2006, 5, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Højlund, K.; Yi, Z.; Hwang, H.; Bowen, B.; Lefort, N.; Flynn, C.R.; Langlais, P.; Weintraub, S.T.; Mandarino, L.J. Characterization of the human skeletal muscle proteome by one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. Mol. Cell. Proteom. 2008, 7, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.C.; Walsh, R.J.; Salajegheh, M.; Amato, A.A.; Krastins, B.; Sarracino, D.A.; Greenberg, S.A. Characterization of human skeletal muscle biopsy samples using shotgun proteomics. J. Proteome Res. 2009, 8, 3265–3277. [Google Scholar] [CrossRef] [PubMed]
- Lefort, N.; Yi, Z.; Bowen, B.; Glancy, B.; De Filippis, E.A.; Mapes, R.; Hwang, H.; Flynn, C.R.; Willis, W.T.; Civitarese, A.; et al. Proteome profile of functional mitochondria from human skeletal muscle using one-dimensional gel electrophoresis and HPLC-ESI-MS/MS. J. Proteom. 2009, 72, 1046–1060. [Google Scholar] [CrossRef] [PubMed]
- Højlund, K.; Bowen, B.P.; Hwang, H.; Flynn, C.R.; Madireddy, L.; Geetha, T.; Langlais, P.; Meyer, C.; Mandarino, L.J.; Yi, Z. In vivo phosphoproteome of human skeletal muscle revealed by phosphopeptide enrichment and HPLC-ESI-MS/MS. J. Proteome Res. 2009, 8, 4954–4965. [Google Scholar] [CrossRef] [PubMed]
- Malik, Z.A.; Cobley, J.N.; Morton, J.P.; Close, G.L.; Edwards, B.J.; Koch, L.G.; Britton, S.L.; Burniston, J.G. Label-Free LC-MS Profiling of Skeletal Muscle Reveals Heart-Type Fatty Acid Binding Protein as a Candidate Biomarker of Aerobic Capacity. Proteomes 2013, 1, 290–308. [Google Scholar] [CrossRef] [PubMed]
- Burniston, J.G.; Connolly, J.; Kainulainen, H.; Britton, S.L.; Koch, L.G. Label-free profiling of skeletal muscle using high-definition mass spectrometry. Proteomics 2014, 14, 2339–2344. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Du, X.; Deng, J.; Gu, M.; Hu, H.; Gui, M.; Yin, C.C.; Chang, Z. The interactions between mitochondria and sarcoplasmic reticulum and the proteome characterization of mitochondrion-associated membrane from rabbit skeletal muscle. Proteomics 2015, 15, 2701–2704. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Nagaraj, N.; Deshmukh, A.S.; Zeiler, M.; Cancellara, P.; Moretti, I.; Reggiani, C.; Schiaffino, S.; Mann, M. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Rakus, D.; Gizak, A.; Deshmukh, A.; Wiśniewski, J.R. Absolute quantitative profiling of the key metabolic pathways in slow and fast skeletal muscle. J. Proteome Res. 2015, 14, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Deshmukhm, A.S.; Murgia, M.; Nagaraj, N.; Treebak, J.T.; Cox, J.; Mann, M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol. Cell. Proteom. 2015, 14, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Le Bihan, M.C.; Barrio-Hernandez, I.; Mortensen, T.P.; Henningsen, J.; Jensen, S.S.; Bigot, A.; Blagoev, B.; Butler-Browne, G.; Kratchmarova, I. Cellular Proteome Dynamics during Differentiation of Human Primary Myoblasts. J. Proteome Res. 2015, 14, 3348–3361. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Rigby, P.W. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Dev. Cell. 2014, 28, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Dayanidhi, S.; Lieber, R.L. Skeletal muscle satellite cells: Mediators of muscle growth during development and implications for developmental disorders. Muscle Nerve 2014, 50, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Hoppeler, H. Molecular networks in skeletal muscle plasticity. J. Exp. Biol. 2016, 219, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Tannu, N.S.; Rao, V.K.; Chaudhary, R.M.; Giorgianni, F.; Saeed, A.E.; Gao, Y.; Raghow, R. Comparative proteomes of the proliferating C(2)C(12) myoblasts and fully differentiated myotubes reveal the complexity of the skeletal muscle differentiation program. Mol. Cell. Proteom. 2004, 3, 1065–1082. [Google Scholar] [CrossRef] [PubMed]
- Casadei, L.; Vallorani, L.; Gioacchini, A.M.; Guescini, M.; Burattini, S.; D’Emilio, A.; Biagiotti, L.; Falcieri, E.; Stocchi, V. Proteomics-based investigation in C2C12 myoblast differentiation. Eur. J. Histochem. 2009, 53, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, T.; Ding, F.; Hu, N.; Gu, X. Proteomic studies of rat tibialis anterior muscle during postnatal growth and development. Mol. Cell. Biochem. 2009, 332, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qian, H.; Feng, X.; Xiong, Y.; Lei, M.; Ren, Z.; Zuo, B.; Xu, D.; Ma, Y.; Yuan, H. Differential proteome and transcriptome analysis of porcine skeletal muscle during development. J. Proteom. 2012, 75, 2093–2108. [Google Scholar] [CrossRef] [PubMed]
- Chan, X.C.; McDermott, J.C.; Siu, K.W. Identification of secreted proteins during skeletal muscle development. J. Proteome Res. 2007, 6, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Henningsen, J.; Rigbolt, K.T.; Blagoev, B.; Pedersen, B.K.; Kratchmarova, I. Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol. Cell. Proteom. 2010, 9, 2482–2496. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Masui, O.; Krakovska, O.; Belozerov, V.E.; Voisin, S.; Ghanny, S.; Chen, J.; Moyez, D.; Zhu, P.; Evans, K.R.; et al. Identification of differentially regulated secretome components during skeletal myogenesis. Mol. Cell. Proteom. 2011, 10, M110.004804. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; McFarlane, C.; Kambadur, R.; Kukreti, H.; Bonala, S.; Srinivasan, S. Myostatin: Expanding horizons. IUBMB Life 2015, 67, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Bouley, J.; Meunier, B.; Chambon, C.; De Smet, S.; Hocquette, J.F.; Picard, B. Proteomic analysis of bovine skeletal muscle hypertrophy. Proteomics 2005, 5, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Keady, S.M.; Kenny, D.A.; Ohlendieck, K.; Doyle, S.; Keane, M.G.; Waters, S.M. Proteomic profiling of bovine M. longissimus lumborum from Crossbred Aberdeen Angus and Belgian Blue sired steers varying in genetic merit for carcass weight. J. Anim. Sci. 2013, 91, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Gehlert, S.; Bloch, W.; Suhr, F. Ca2+-dependent regulations and signaling in skeletal muscle: From electro-mechanical coupling to adaptation. Int. J. Mol. Sci. 2015, 16, 1066–1095. [Google Scholar] [CrossRef] [PubMed]
- Blaauw, B.; Schiaffino, S.; Reggiani, C. Mechanisms modulating skeletal muscle phenotype. Compr. Physiol. 2013, 3, 1645–1687. [Google Scholar] [PubMed]
- Burniston, J.G.; Hoffman, E.P. Proteomic responses of skeletal and cardiac muscle to exercise. Expert Rev. Proteom. 2011, 8, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Petriz, B.A.; Gomes, C.P.; Rocha, L.A.; Rezende, T.M.; Franco, O.L. Proteomics applied to exercise physiology: A cutting-edge technology. J. Cell. Physiol. 2012, 227, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomics of exercise-induced skeletal muscle adaptations. OA Sports Med. 2013, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Padrão, A.I.; Ferreira, R.; Amado, F.; Vitorino, R.; Duarte, J.A. Uncovering the exercise-related proteome signature in skeletal muscle. Proteomics 2016, 16, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Holloway, K.V.; O’Gorman, M.; Woods, P.; Morton, J.P.; Evans, L.; Cable, N.T.; Goldspink, D.F.; Burniston, J.G. Proteomic investigation of changes in human vastus lateralis muscle in response to interval-exercise training. Proteomics 2009, 9, 5155–5174. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Dowling, P.; O’Connor, P.L.; Henry, M.; Meleady, P.; Zierath, J.R.; O’Gorman, D.J. 2-D DIGE analysis of the mitochondrial proteome from human skeletal muscle reveals time course-dependent remodelling in response to 14 consecutive days of endurance exercise training. Proteomics 2011, 11, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Moriggi, M.; Vasso, M.; Fania, C.; Capitanio, D.; Bonifacio, G.; Salanova, M.; Blottner, D.; Rittweger, J.; Felsenberg, D.; Cerretelli, P.; et al. Long term bed rest with and without vibration exercise countermeasures: Effects on human muscle protein dysregulation. Proteomics 2010, 10, 3756–3774. [Google Scholar] [CrossRef] [PubMed]
- Salanova, M.; Gelfi, C.; Moriggi, M.; Vasso, M.; Viganò, A.; Minafra, L.; Bonifacio, G.; Schiffl, G.; Gutsmann, M.; Felsenberg, D.; et al. Disuse deterioration of human skeletal muscle challenged by resistive exercise superimposed with vibration: Evidence from structural and proteomic analysis. FASEB J. 2014, 28, 4748–4763. [Google Scholar] [CrossRef] [PubMed]
- Hody, S.; Leprince, P.; Sergeant, K.; Renaut, J.; Croisier, J.L.; Wang, F.; Rogister, B. Human muscle proteome modifications after acute or repeated eccentric exercises. Med. Sci. Sports Exerc. 2011, 43, 2281–2296. [Google Scholar] [CrossRef] [PubMed]
- Malm, C.; Yu, J.G. Exercise-induced muscle damage and inflammation: Re-evaluation by proteomics. Histochem. Cell Biol. 2012, 138, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Burniston, J.G. Changes in the rat skeletal muscle proteome induced by moderate-intensity endurance exercise. Biochim. Biophys. Acta 2008, 1784, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, P.; Doran, P.; Dowling, P.; Ohlendieck, K. Differential expression of the fast skeletal muscle proteome following chronic low-frequency stimulation. Biochim. Biophys. Acta 2005, 1752, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, P.; Doran, P.; Wynne, K.; Pedersen, K.; Dunn, M.J.; Ohlendieck, K. Proteomic profiling of chronic low-frequency stimulated fast muscle. Proteomics 2007, 7, 3417–3430. [Google Scholar] [CrossRef] [PubMed]
- Guelfi, K.J.; Casey, T.M.; Giles, J.J.; Fournier, P.A.; Arthur, P.G. A proteomic analysis of the acute effects of high-intensity exercise on skeletal muscle proteins in fasted rats. Clin. Exp. Pharmacol. Physiol. 2006, 33, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, W.; Fujimoto, E.; Higuchi, M.; Tabata, I. A DIGE proteomic analysis for high-intensity exercise-trained rat skeletal muscle. J. Biochem. 2010, 148, 327–333. [Google Scholar] [PubMed]
- Gandra, P.G.; Valente, R.H.; Perales, J.; Pacheco, A.G.; Macedo, D.V. Proteomic profiling of skeletal muscle in an animal model of overtraining. Proteomics 2012, 12, 2663–2667. [Google Scholar] [CrossRef] [PubMed]
- Gandra, P.G.; Valente, R.H.; Perales, J.; Pacheco, A.G.; Macedo, D.V. Proteomic analysis of rat skeletal muscle submitted to one bout of incremental exercise. Scand. J. Med. Sci. Sports 2012, 22, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Magherini, F.; Abruzzo, P.M.; Puglia, M.; Bini, L.; Gamberi, T.; Esposito, F.; Veicsteinas, A.; Marini, M.; Fiorillo, C.; Gulisano, M.; et al. Proteomic analysis and protein carbonylation profile in trained and untrained rat muscles. J. Proteom. 2012, 75, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, F.G.; van Ginneken, M.M.; Noben, J.P.; Royackers, E.; de Graaf-Roelfsema, E.; Wijnberg, I.D.; van der Kolk, J.H.; Mariman, E.C.; van Breda, E. Differential expression of equine muscle biopsy proteins during normal training and intensified training in young standardbred horses using proteomics technology. Comp. Biochem. Physiol. Part D Genom. Proteom. 2010, 5, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Macedo, A.; Moriggi, M.; Vasso, M.; De Palma, S.; Sturnega, M.; Friso, G.; Gelfi, C.; Giacca, M.; Zacchigna, S. Enhanced athletic performance on multisite AAV-IGF1 gene transfer coincides with massive modification of the muscle proteome. Hum. Gene Ther. 2012, 23, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Burniston, J.G.; Kenyani, J.; Gray, D.; Guadagnin, E.; Jarman, I.H.; Cobley, J.N.; Cuthbertson, D.J.; Chen, Y.W.; Wastling, J.M.; Lisboa, P.J.; et al. Conditional independence mapping of DIGE data reveals PDIA3 protein species as key nodes associated with muscle aerobic capacity. J. Proteom. 2014, 106, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Flueck, M. Plasticity of the muscle proteome to exercise at altitude. High Alt. Med. Biol. 2009, 10, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Favier, F.B.; Britto, F.A.; Freyssenet, D.G.; Bigard, X.A.; Benoit, H. HIF-1-driven skeletal muscle adaptations to chronic hypoxia: Molecular insights into muscle physiology. Cell. Mol. Life Sci. 2015, 72, 4681–4696. [Google Scholar] [CrossRef] [PubMed]
- Bosworth, C.A.; Chou, C.W.; Cole, R.B.; Rees, B.B. Protein expression patterns in zebrafish skeletal muscle: Initial characterization and the effects of hypoxic exposure. Proteomics 2005, 5, 1362–1371. [Google Scholar] [CrossRef] [PubMed]
- De Palma, S.; Ripamonti, M.; Vigano, A.; Moriggi, M.; Capitanio, D.; Samaja, M.; Milano, G.; Cerretelli, P.; Wait, R.; Gelfi, C. Metabolic modulation induced by chronic hypoxia in rats using a comparative proteomic analysis of skeletal muscle tissue. J. Proteome Res. 2007, 6, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Vigano, A.; Ripamonti, M.; De Palma, S.; Capitanio, D.; Vasso, M.; Wait, R.; Lundby, C.; Cerretelli, P.; Gelfi, C. Proteins modulation in human skeletal muscle in the early phase of adaptation to hypobaric hypoxia. Proteomics 2008, 8, 4668–4679. [Google Scholar] [CrossRef] [PubMed]
- Levett, D.Z.; Viganò, A.; Capitanio, D.; Vasso, M.; De Palma, S.; Moriggi, M.; Martin, D.S.; Murray, A.J.; Cerretelli, P.; Grocott, M.P.; et al. Changes in muscle proteomics in the course of the Caudwell Research Expedition to Mt. Everest. Proteomics 2015, 15, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Carmeli, E.; Aizenbud, D.; Rom, O. How Do Skeletal Muscles Die? An Overview. Adv. Exp. Med. Biol. 2015, 861, 99–111. [Google Scholar] [PubMed]
- Isfort, R.J.; Hinkle, R.T.; Jones, M.B.; Wang, F.; Greis, K.D.; Sun, Y.; Keough, T.W.; Anderson, N.L.; Sheldon, R.J. Proteomic analysis of the atrophying rat soleus muscle following denervation. Electrophoresis 2000, 21, 2228–2234. [Google Scholar] [CrossRef]
- Isfort, R.J.; Wang, F.; Greis, K.D.; Sun, Y.; Keough, T.W.; Farrar, R.P.; Bodine, S.C.; Anderson, N.L. Proteomic analysis of rat soleus muscle undergoing hindlimb suspension-induced atrophy and reweighting hypertrophy. Proteomics 2002, 2, 543–550. [Google Scholar] [CrossRef]
- Isfort, R.J.; Wang, F.; Greis, K.D.; Sun, Y.; Keough, T.W.; Bodine, S.C.; Anderson, N.L. Proteomic analysis of rat soleus and tibialis anterior muscle following immobilization. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002, 769, 323–332. [Google Scholar] [CrossRef]
- Seo, Y.; Lee, K.; Park, K.; Bae, K.; Choi, I. A proteomic assessment of muscle contractile alterations during unloading and reloading. J. Biochem. 2006, 139, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Moriggi, M.; Cassano, P.; Vasso, M.; Capitanio, D.; Fania, C.; Musicco, C.; Pesce, V.; Gadaleta, M.N.; Gelfi, C. A DIGE approach for the assessment of rat soleus muscle changes during unloading: Effect of acetyl-l-carnitine supplementation. Proteomics 2008, 8, 3588–3604. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Vitorino, R.; Neuparth, M.J.; Appell, H.J.; Duarte, J.A.; Amado, F. Proteolysis activation and proteome alterations in murine skeletal muscle submitted to 1 week of hindlimb suspension. Eur. J. Appl. Physiol. 2009, 107, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Basco, D.; Nicchia, G.P.; Desaphy, J.F.; Camerino, D.C.; Frigeri, A.; Svelto, M. Analysis by two-dimensional Blue Native/SDS-PAGE of membrane protein alterations in rat soleus muscle after hindlimb unloading. Eur. J. Appl. Physiol. 2010, 110, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, P.; Liu, H.; Fan, M.; Chen, X. Proteomic analysis of mouse soleus muscles affected by hindlimb unloading and reloading. Muscle Nerve 2015, 52, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.B.; Lehar, M.; Samlan, R.; Flint, P.W. Proteomic analysis of rat laryngeal muscle following denervation. Proteomics 2005, 5, 4764–4776. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Shimizu, M.; Mizunoya, W.; Wariishi, H.; Tatsumi, R.; Buchman, V.L.; Ikeuchi, Y. Differential expression of sarcoplasmic and myofibrillar proteins of rat soleus muscle during denervation atrophy. Biosci. Biotechnol. Biochem. 2009, 73, 1748–1756. [Google Scholar] [CrossRef] [PubMed]
- Budui, S.L.; Rossi, A.P.; Zamboni, M. The pathogenetic bases of sarcopenia. Clin. Cases Miner. Bone Metab. 2015, 12, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. A Biol. Sci. Med. Sci. 1995, 50, 11–16. [Google Scholar] [PubMed]
- Landi, F.; Calvani, R.; Cesari, M.; Tosato, M.; Martone, A.M.; Bernabei, R.; Onder, G.; Marzetti, E. Sarcopenia as the Biological Substrate of Physical Frailty. Clin. Geriatr. Med. 2015, 31, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.J.; Braunschweig, C.A. Prevalence of Sarcopenia and Associated Outcomes in the Clinical Setting. Nutr. Clin. Pract. 2016, 31, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Chaves, D.F.; Carvalho, P.C.; Lima, D.B.; Nicastro, H.; Lorenzeti, F.M.; Siqueira-Filhom, M.; Hirabara, S.M.; Alves, P.H.; Moresco, J.J.; Yates, J.R., III; et al. Comparative proteomic analysis of the aging soleus and extensor digitorum longus rat muscles using TMT labeling and mass spectrometry. J. Proteome Res. 2013, 12, 4532–4546. [Google Scholar] [CrossRef] [PubMed]
- Gueugneau, M.; Coudy-Gandilhon, C.; Gourbeyre, O.; Chambon, C.; Combaret, L.; Polge, C.; Taillandier, D.; Attaix, D.; Friguet, B.; et al. Proteomics of muscle chronological ageing in post-menopausal women. BMC Genom. 2014, 15, 1165. [Google Scholar] [CrossRef] [PubMed]
- Théron, L.; Gueugneau, M.; Coudy, C.; Viala, D.; Bijlsma, A.; Butler-Browne, G.; Maier, A.; Béchet, D.; Chambon, C. Label-free quantitative protein profiling of vastus lateralis muscle during human aging. Mol. Cell. Proteom. 2014, 13, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Donoghue, P.; O’Connell, K.; Gannon, J.; Ohlendieck, K. Proteomics of skeletal muscle aging. Proteomics 2009, 9, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomic Profiling of Fast-To-Slow Muscle Transitions during Aging. Front Physiol. 2011, 2, 105. [Google Scholar] [CrossRef] [PubMed]
- Baraibar, M.A.; Gueugneau, M.; Duguez, S.; Butler-Browne, G.; Bechet, D.; Friguet, B. Expression and modification proteomics during skeletal muscle ageing. Biogerontology 2013, 14, 339–352. [Google Scholar] [CrossRef] [PubMed]
- O'Connell, K.; Gannon, J.; Doran, P.; Ohlendieck, K. Proteomic profiling reveals a severely perturbed protein expression pattern in aged skeletal muscle. Int. J. Mol. Med. 2007, 20, 145–153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Doran, P.; Gannon, J.; O’Connell, K.; Ohlendieck, K. Aging skeletal muscle shows a drastic increase in the small heat shock proteins alphaB-crystallin/HspB5 and cvHsp/HspB7. Eur. J. Cell Biol. 2007, 86, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Timms, J.F.; Cramer, R. Difference gel electrophoresis. Proteomics 2008, 8, 4886–4897. [Google Scholar] [CrossRef] [PubMed]
- Gelfi, C.; Vigano, A.; Ripamonti, M.; Pontoglio, A.; Begum, S.; Pellegrino, M.A.; Grassi, B.; Bottinelli, R.; Wait, R.; Cerretelli, P. The human muscle proteome in aging. J. Proteome Res. 2006, 5, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; O’Connell, K.; Gannon, J.; Kavanagh, M.; Ohlendieck, K. Opposite pathobiochemical fate of pyruvate kinase and adenylate kinase in aged rat skeletal muscle as revealed by proteomic DIGE analysis. Proteomics 2008, 8, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Vasso, M.; Fania, C.; Moriggi, M.; Viganò, A.; Procacci, P.; Magnaghi, V.; Gelfi, C. Comparative proteomic profile of rat sciatic nerve and gastrocnemius muscle tissues in ageing by 2-D DIGE. Proteomics 2009, 9, 2004–2020. [Google Scholar] [CrossRef] [PubMed]
- Staunton, L.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Mass spectrometry-based proteomic analysis of middle-aged vs. aged vastus lateralis reveals increased levels of carbonic anhydrase isoform 3 in senescent human skeletal muscle. Int. J. Mol. Med. 2012, 30, 723–733. [Google Scholar] [PubMed]
- Capitanio, D.; Vasso, M.; De Palma, S.; Fania, C.; Torretta, E.; Cammarata, F.P.; Magnaghi, V.; Procacci, P.; Gelfi, C. Specific protein changes contribute to the differential muscle mass loss during ageing. Proteomics 2016, 16, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Unlü, M.; Morgan, M.E.; Minden, J.S. Difference gel electrophoresis: A single gel method for detecting changes in protein extracts. Electrophoresis 1997, 18, 2071–2077. [Google Scholar] [CrossRef] [PubMed]
- Alban, A.; David, S.O.; Bjorkesten, L.; Andersson, C.; Sloge, E.; Lewis, S.; Currie, I. A novel experimental design for comparative two-dimensional gel analysis: Two-dimensional difference gel electrophoresis incorporating a pooled internal standard. Proteomics 2003, 3, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.; Unlü, M.; Minden, J.S. Two-dimensional difference gel electrophoresis. Nat. Protoc. 2006, 1, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Minden, J.S. Two-dimensional difference gel electrophoresis. Methods Mol. Biol. 2012, 869, 287–304. [Google Scholar] [PubMed]
- Malm, C.; Hadrevi, J.; Bergström, S.A.; Pedrosa-Domellöf, F.; Antti, H.; Svensson, M.; Frängsmyr, L. Evaluation of 2-D DIGE for skeletal muscle: Protocol and repeatability. Scand. J. Clin. Lab. Investig. 2008, 68, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Carberry, S.; Zweyer, M.; Swandulla, D.; Ohlendieck, K. Application of fluorescence two-dimensional difference in-gel electrophoresis as a proteomic biomarker discovery tool in muscular dystrophy research. Biology 2013, 2, 1438–1464. [Google Scholar] [CrossRef] [PubMed]
- Tonge, R.; Shaw, J.; Middleton, B.; Rowlinson, R.; Rayner, S.; Young, J.; Pognan, F.; Hawkins, E.; Currie, I.; Davison, M. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics 2001, 1, 377–396. [Google Scholar] [CrossRef]
- Marouga, R.; David, S.; Hawkins, E. The development of the DIGE system: 2D fluorescence difference gel analysis technology. Anal. Bioanal. Chem. 2005, 382, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Karp, N.A.; Kreil, D.P.; Lilley, K.S. Determining a significant change in protein expression with DeCyder during a pair-wise comparison using two-dimensional difference gel electrophoresis. Proteomics 2004, 4, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Karp, N.A.; Lilley, K.S. Maximising sensitivity for detecting changes in protein expression: Experimental design using minimal CyDyes. Proteomics 2005, 5, 3105–3115. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef] [PubMed]
- Staunton, L.; O’Connell, K.; Ohlendieck, K. Proteomic Profiling of Mitochondrial Enzymes during Skeletal Muscle Aging. J. Aging Res. 2011, 2011, 908035. [Google Scholar] [CrossRef] [PubMed]
- Ibebunjo, C.; Chick, J.M.; Kendall, T.; Eash, J.K.; Li, C.; Zhang, Y.; Vickers, C.; Wu, Z.; Clarke, B.A.; Shi, J.; et al. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013, 33, 194–212. [Google Scholar] [CrossRef] [PubMed]
- Lourenço Dos Santos, S.; Baraibar, M.A.; Lundberg, S.; Eeg-Olofsson, O.; Larsson, L.; Friguet, B. Oxidative proteome alterations during skeletal muscle ageing. Redox Biol. 2015, 5, 267–274. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, B.; Sakellariou, G.K.; Smith, N.T.; Brownridge, P.; Jackson, M.J. Redox proteomic analysis of the gastrocnemius muscle from adult and old mice. Data Brief 2015, 4, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Xie, H.; Meany, D.L.; Thompson, L.V.; Arriaga, E.A.; Griffin, T.J. Quantitative proteomic profiling of muscle type-dependent and age-dependent protein carbonylation in rat skeletal muscle mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 1137–1152. [Google Scholar] [CrossRef] [PubMed]
- Piec, I.; Listrat, A.; Alliot, J.; Chambon, C.; Taylor, R.G.; Bechet, D. Differential proteome analysis of aging in rat skeletal muscle. FASEB J. 2005, 19, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Teltathum, T.; Mekchay, S. Proteome changes in Thai indigenous chicken muscle during growth period. Int. J. Biol. Sci. 2009, 5, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.K.; Lee, S.H.; Cho, Y.M.; Son, E.S.; Kim, K.Y.; Lee, C.S.; Yoon, D.; Im, S.K.; Oh, S.J.; Park, E.W. Proteome analysis of the m. longissimus dorsi between fattening stages in Hanwoo steer. BMB Rep. 2009, 42, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Hildrum, K.I.; Westad, F.; Kummen, E.; Aass, L.; Hollung, K. Changes in enzymes associated with energy metabolism during the early post mortem period in longissimus thoracis bovine muscle analyzed by proteomics. J. Proteome Res. 2006, 5, 1763–1769. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Ekman, M.; Grove, H.; Faergestad, E.M.; Aass, L.; Hildrum, K.I.; Hollung, K. Proteome changes in bovine longissimus thoracis muscle during the early postmortem storage period. J. Proteome Res. 2007, 6, 2720–2731. [Google Scholar] [CrossRef] [PubMed]
- Di Luca, A.; Elia, G.; Hamill, R.; Mullen, A.M. 2D DIGE proteomic analysis of early post mortem muscle exudate highlights the importance of the stress response for improved water-holding capacity of fresh pork meat. Proteomics 2013, 13, 1528–1544. [Google Scholar] [CrossRef] [PubMed]
- Canto, A.C.; Suman, S.P.; Nair, M.N.; Li, S.; Rentfrow, G.; Beach, C.M.; Silva, T.J.; Wheeler, T.L.; Shackelford, S.D.; Grayson, A.; et al. Differential abundance of sarcoplasmic proteome explains animal effect on beef Longissimus lumborum color stability. Meat Sci. 2015, 102, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Mato, A.; Salgado, F.J.; López-Pedrouso, M.; Carrera, M.; Bravo, S.; Parrado, M.; Gallardo, J.M.; Zapata, C. Tackling proteome changes in the longissimus thoracis bovine muscle in response to pre-slaughter stress. J. Proteom. 2015, 122, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xu, X.L.; Ma, H.M.; Jiang, J. Integrative analysis of transcriptomics and proteomics of skeletal muscles of the Chinese indigenous Shaziling pig compared with the Yorkshire breed. BMC Genet. 2016, 17, 80. [Google Scholar] [CrossRef] [PubMed]
- Paredi, G.; Raboni, S.; Bendixen, E.; de Almeida, A.M.; Mozzarelli, A. “Muscle to meat” molecular events and technological transformations: The proteomics insight. J. Proteom. 2012, 75, 4275–4289. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, A.; Zolla, L. Meat science: From proteomics to integrated omics towards system biology. J. Proteom. 2013, 78, 558–577. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.; Lana, A.; Bottero, M.T.; Zolla, L. Apoptosis in muscle-to-meat aging process: The omic witness. J. Proteom. 2015, 125, 29–40. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteomic Analysis | Tissue and Species | References |
|---|---|---|
| Human 2D gel reference maps | Human vastus lateralis and laryngeal muscle | Gelfi et al. [111]; Li et al. [112]; Kovalyova et al. [124] |
| Human fast versus slow muscle fibre type specification | Normal human deltoideus and vastus lateralis muscles | Capitanio et al. [115] |
| Mouse 2D gel reference maps | Normal mouse gastrocnemius and quadriceps muscle | Sanchez et al. [69]; Raddatz et al. [121] |
| Mouse fast versus slow muscle fibre type specification | Normal and kyphoscoliotic mouse soleus and vastus lateralis muscles | Le Bihan et al. [117] |
| Rat 2D gel reference map | Normal rat skeletal muscle from the abdominal wall | Yan et al. [70] |
| Rat fast versus slow muscle fibre type specification | Normal rat soleus, gastrocnemius and extensor digitorum longus muscles | Okumura et al. [116]; Gelfi et al. [118] |
| Rabbit 2D gel reference map | Rabbit gastrocnemius muscle | Almeida et al. [122] |
| Bovine 2D gel reference maps | Bovine semitendinosus muscle | Bouley et al. [113]; Chaze et al. [119] |
| Pig fast versus slow muscle fibre type specification | Normal pig longissimus dorsi and soleus muscles | Kim et al. [114] |
| Pufferfish and killifish 2D gel reference maps | Skeletal muscles from Takifugu rubripes and Fundulus grandis | Lu et al. [125]; Abbaraju et al. [126] |
| Mitochondrial 2D gel maps | Subsarcolemmal and intermyofibrillar mitochondria from various rat muscles | Reifschneider et al. [129]; O’Connell et al. [130]; Lombardi et al. [131]; Ferreira et al. [132] |
| Contractile apparatus 2D gel map | Enriched acto-myosin apparatus from rat gastrocnemius muscle | Gannon et al. [133] |
| Cytosol and nucleus 2D gel map | Nucleus and cytosolic fraction from mouse gastrocnemius and soleus muscles | Vitorino et al. [128] |
| Muscle secretome 2D gel maps | Seretome from cultured muscle cells | Gajendran et al. [134]; Hartwig et al. [135] |
| 2D PTM gel maps of protein glycosylation | Rat leg skeletal muscles | O’Connell et al. [105]; Cieniewski-Bernard et al. [137] |
| 2D PTM gel map of protein phosphorylation | Rat gastrocnemius muscle | Gannon et al. [106,133] |
| 2D PTM gel map of protein nitration | Rat leg skeletal muscles | Kanski et al. [136] |
| Proteomic Analysis | Skeletal Muscle Tissue | References |
|---|---|---|
| Postnatal development | Rat tibialis anterior and porcine longissimus dorsi muscle | Sun et al. [158]; Xu et al. [159] |
| Myoblast differentiation and myotube formation | C2C12 cell culture model | Tannu et al. [156]; Casadei et al. [157] |
| Interval training | Human vastus lateralis muscle | Holoway et al. [172] |
| Endurance training | Human vastus lateralis muscle | Egan et al. [173] |
| Vibration exercise during long-term bed rest | Human soleus and vastus lateralis | Moriggi et al. [174]; Salanova et al. [175] |
| Repeated eccentric exercises | Human rectus femoris muscle | Hody et al. [176] |
| Downhill running-induced muscle damage | Human vastus lateralis muscle | Malm and Yu [177] |
| Various types of animal endurance training | Rat plantaris, gastrocnemius, tibialis anterior, soleus and epitrochlearis muscles; and horse vastus lateralis muscle | Burniston [178]; Guelfi et al. [181]; Yamaguchi et al. [182]; Gandra et al. [183]; Magherini et al. [185]; Bouwman et al. [186] |
| One bout of an exhaustive exercise | Rat gastrocnemius muscle | Gandra et al. [184] |
| Endurance training following gene doping | Various mouse leg muscles | Macedo et al. [187] |
| Chronic low-frequency electro-stimulation | Rabbit tibialis anterior muscle | Donoghue et al. [179,180] |
| High-capacity versus low-capacity runners | Rat soleus muscles | Burniston et al. [188] |
| Myostatin-related muscle hypertrophy | Belgium Blue bulls semitendious muscle lacking myostatin | Bouley et al. [164]; Keady et al. [165] |
| Hypoxia-induced muscle adaptations | Zebrafish, rat and human vastus lateralis muscle | Bosworth et al. [191]; De Palma et al. [192]; Vigano et al. [193]; Levett et al. [194] |
| Disuse atrophy due to neuromuscular unloading, immobilization or denervation | Rat soleus, tibialis anterior, laryngeal and gastrocnemius muscles | Isfort et al. [196,197,198]; Seo et al. [199]; Moriggi et al. [200]; Ferreira et al. [201]; Basco et al. [202]; Wang et al. [203]; Li et al. [204]; Sato et al. [205] |
| Skeletal muscle aging | Various aged rat skeletal muscles, including the gastrocnemius muscle | O’Connell et al. [105,216]; Gannon et al. [106,133] Kanski et al. [136]; Feng et al. [239]; Doran et al. [217,220]; Piec et al. [240]; Capitanio et al. [221,223] |
| Sarcopenia of old age | Various aged human skeletal muscles, including the vastus lateralis muscle | Gelfi et al. [219]; Staunton et al. [222] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, S.; Dowling, P.; Ohlendieck, K. Comparative Skeletal Muscle Proteomics Using Two-Dimensional Gel Electrophoresis. Proteomes 2016, 4, 27. https://doi.org/10.3390/proteomes4030027
Murphy S, Dowling P, Ohlendieck K. Comparative Skeletal Muscle Proteomics Using Two-Dimensional Gel Electrophoresis. Proteomes. 2016; 4(3):27. https://doi.org/10.3390/proteomes4030027
Chicago/Turabian StyleMurphy, Sandra, Paul Dowling, and Kay Ohlendieck. 2016. "Comparative Skeletal Muscle Proteomics Using Two-Dimensional Gel Electrophoresis" Proteomes 4, no. 3: 27. https://doi.org/10.3390/proteomes4030027
APA StyleMurphy, S., Dowling, P., & Ohlendieck, K. (2016). Comparative Skeletal Muscle Proteomics Using Two-Dimensional Gel Electrophoresis. Proteomes, 4(3), 27. https://doi.org/10.3390/proteomes4030027