
Proteomics of Skeletal Muscle: Focus on Insulin Resistance and Exercise Biology


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Skeletal Muscle Proteomics—Technical Challenges
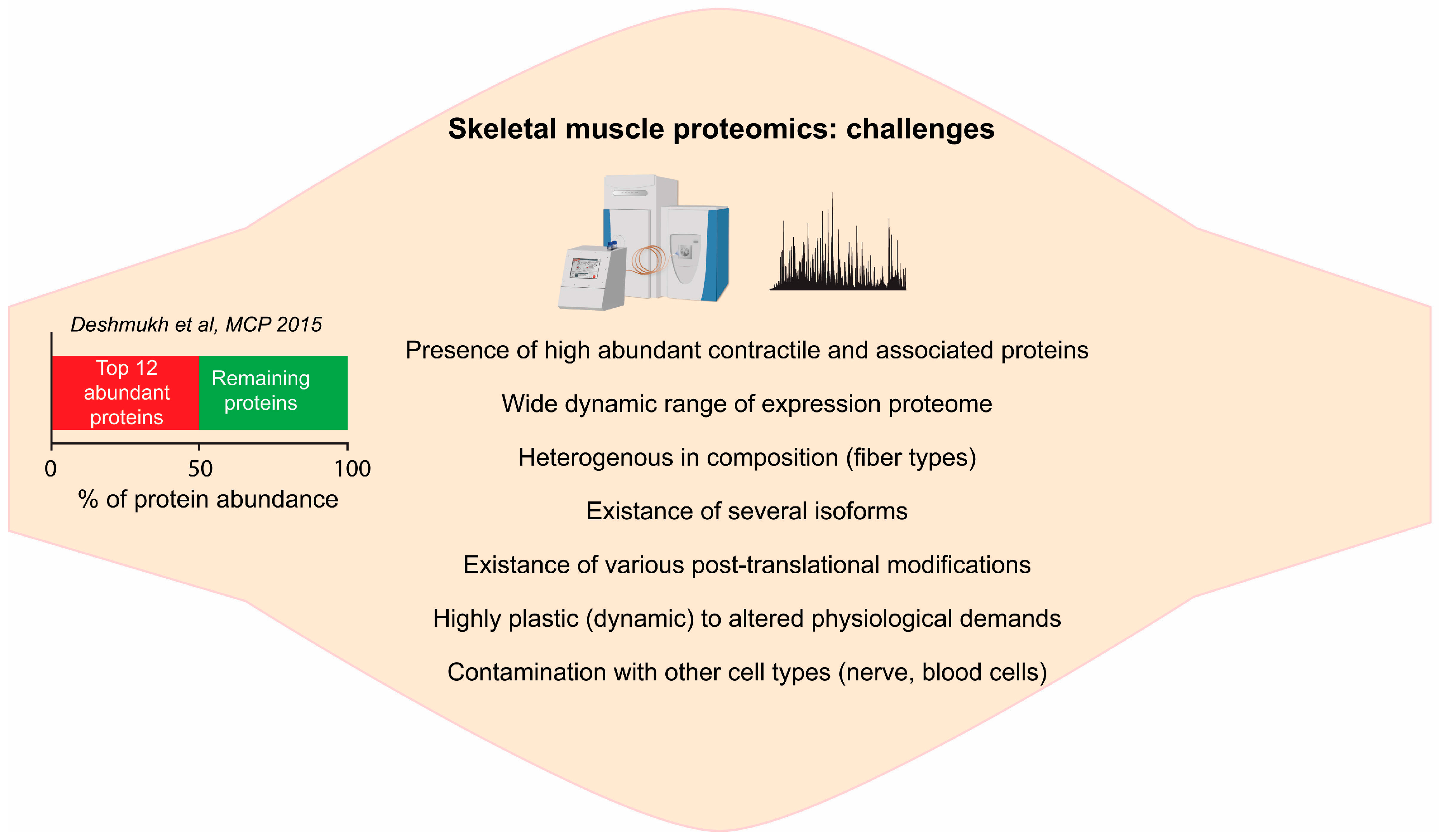
2.1. Complexity of Skeletal Muscle Tissue
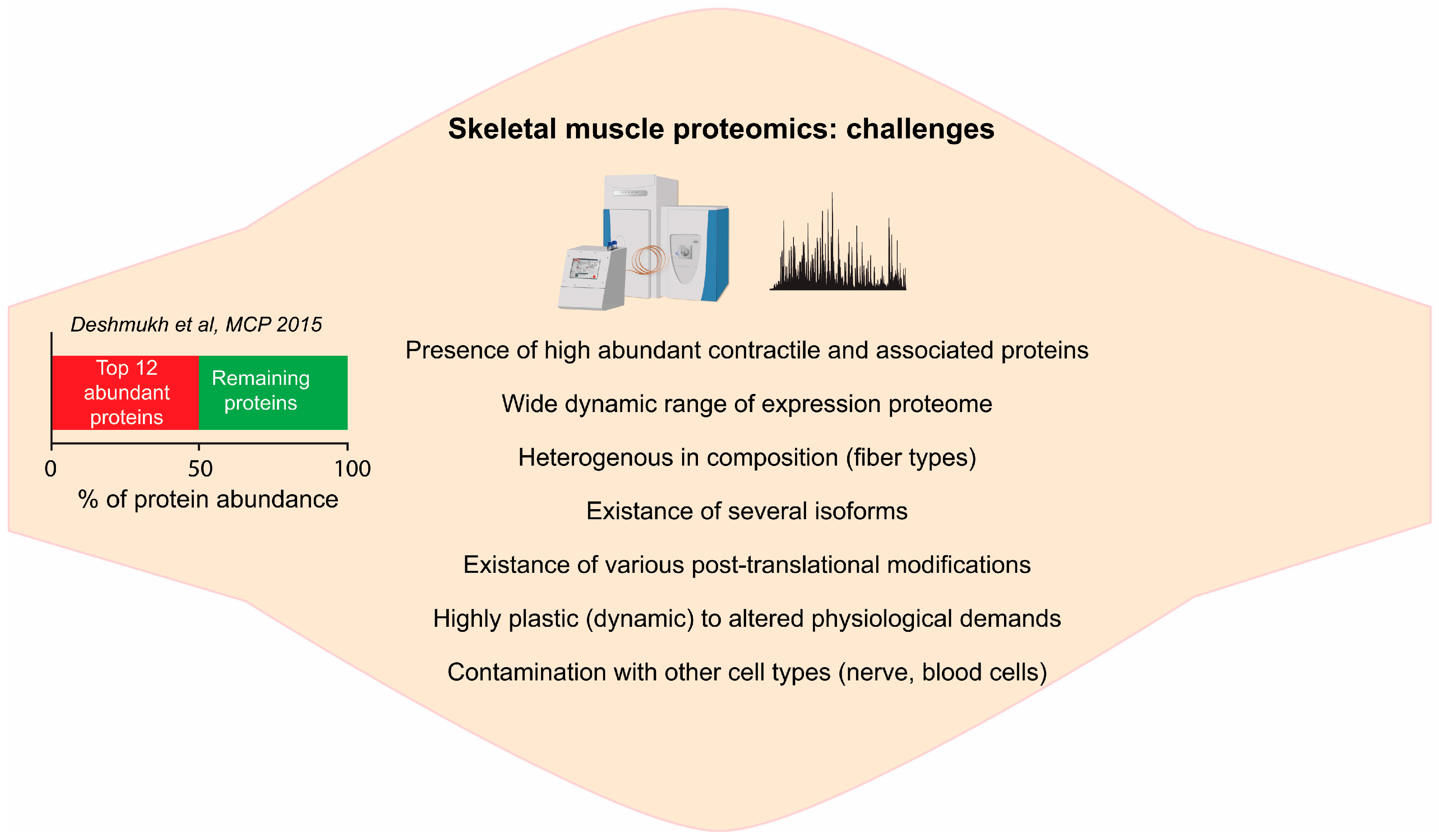
2.2. Deep Proteome of Skeletal Muscle Tissue
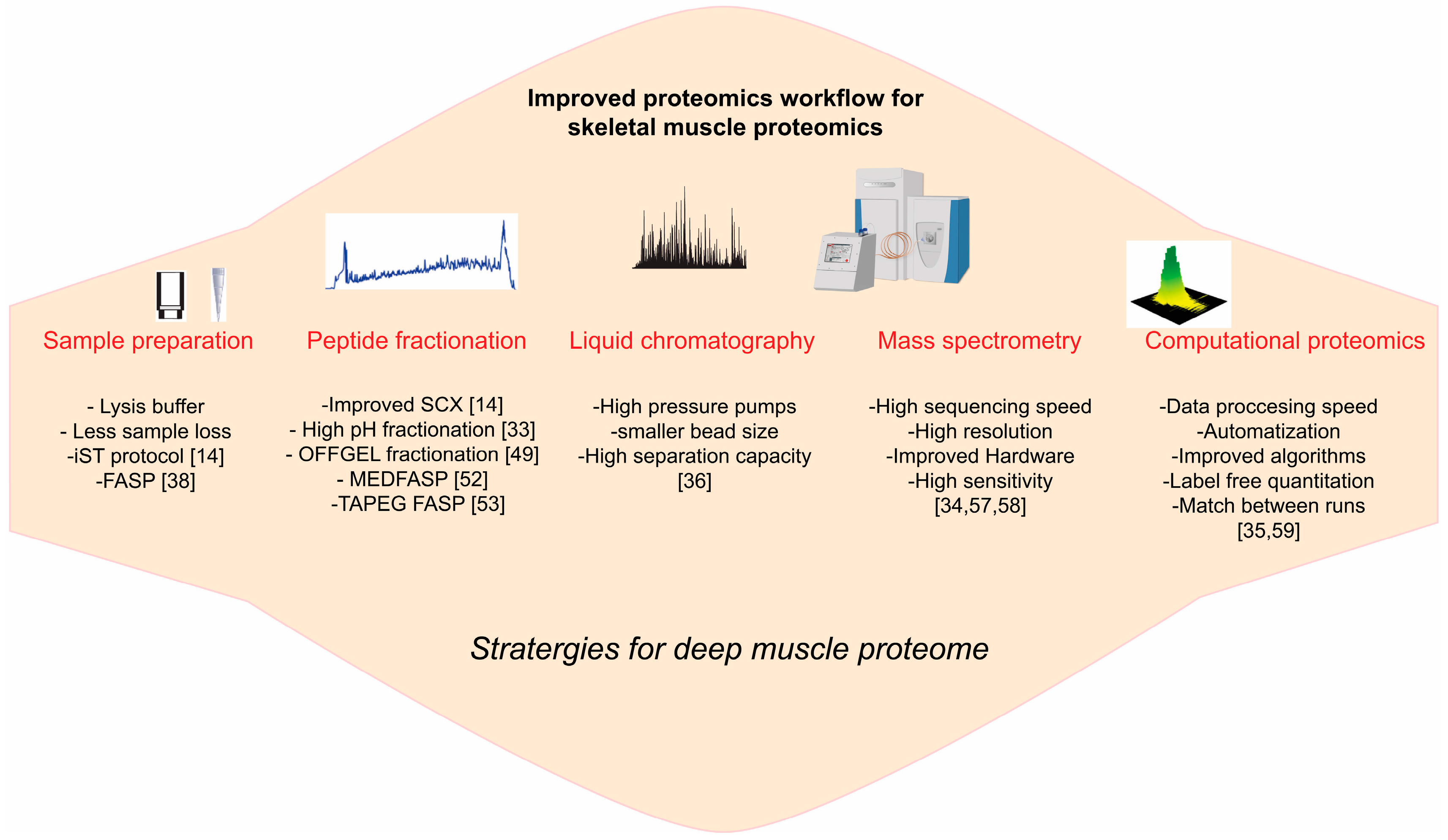
3. Emerging Technology for Skeletal Muscle Proteome
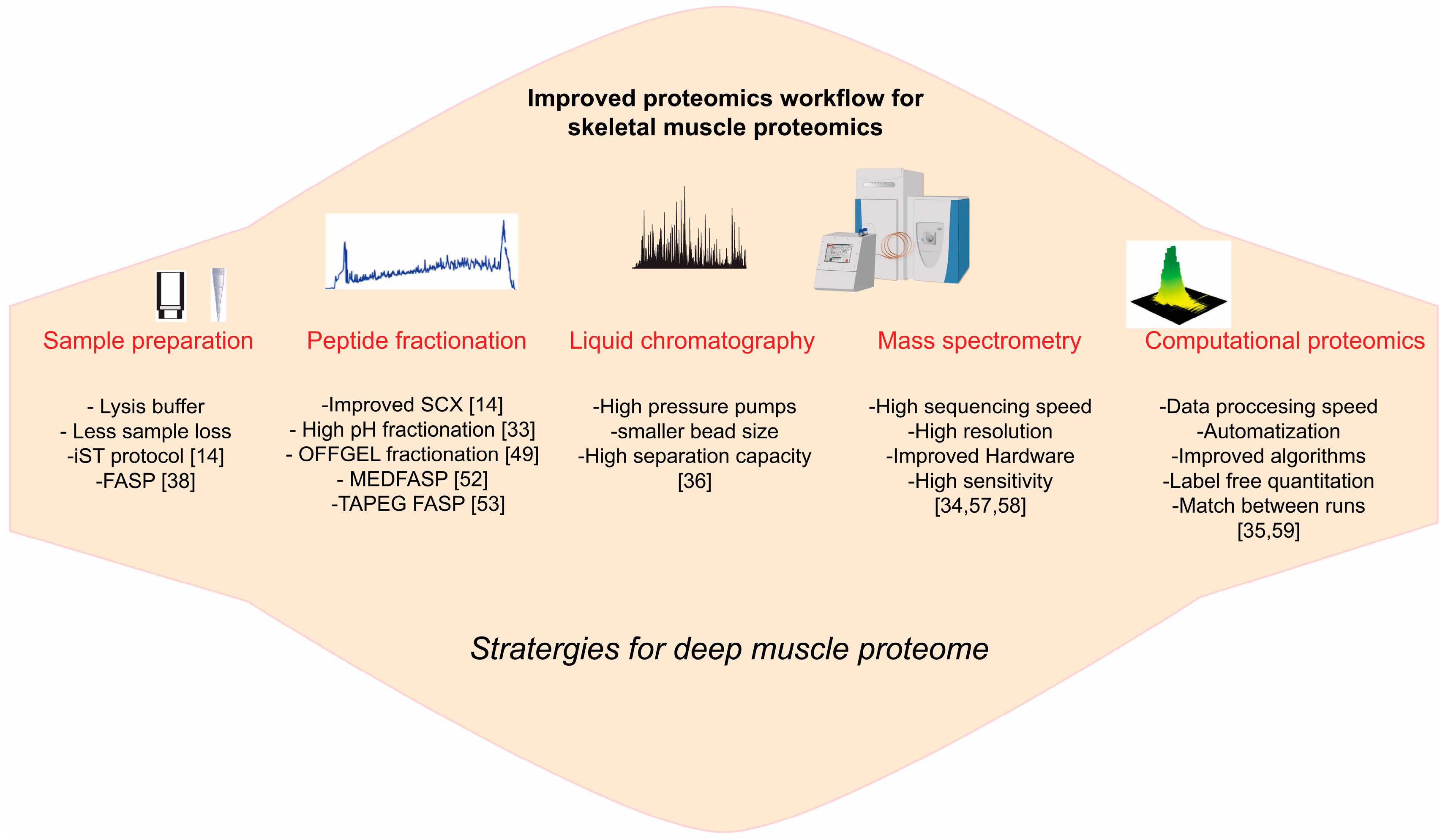
3.1. Sample Preparation
3.2. Protein, Peptide Fractionation
3.3. LCMS Instrumentations
3.4. Computational Proteomics
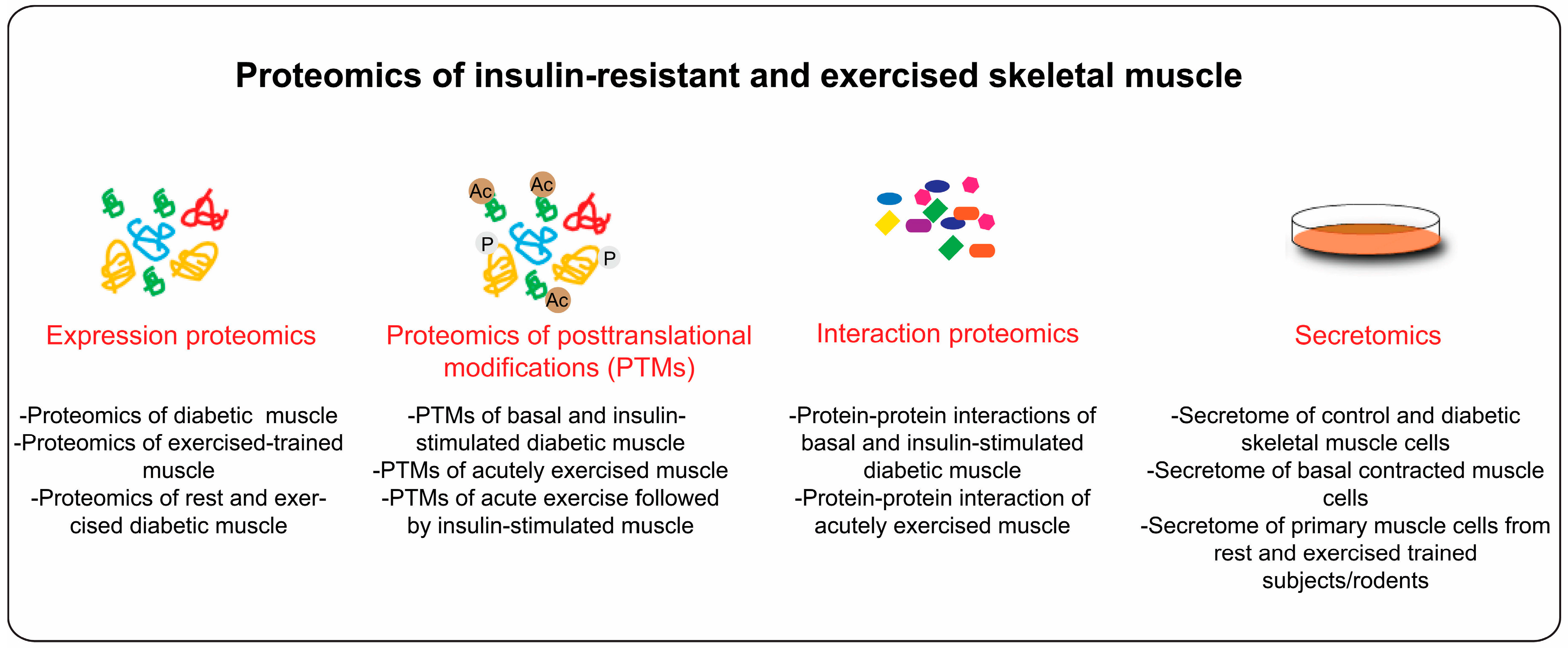
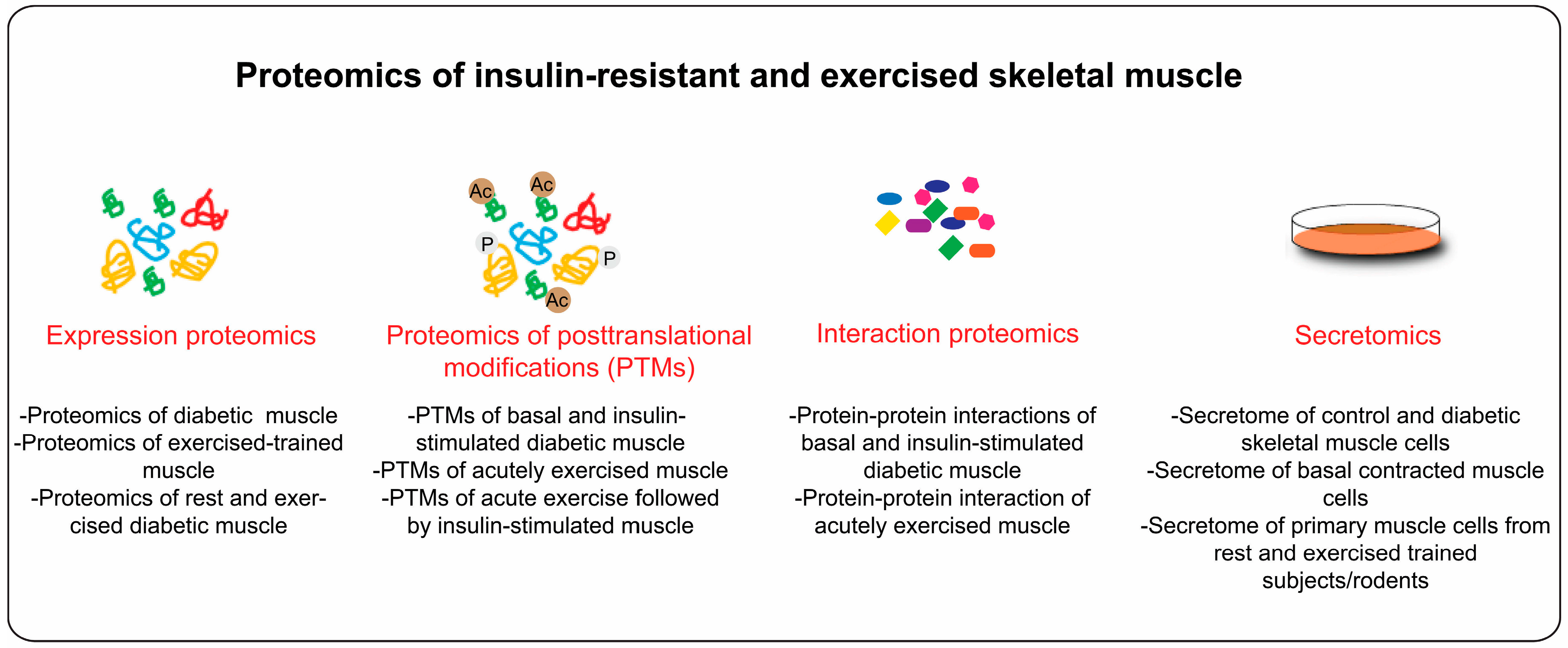
4. Proteome Profiling of Diabetic Skeletal Muscle
4.1. Diabetes and Skeletal Muscle PTMs
4.2. Diabetes, Skeletal Muscle Metabolism and Muscle Fiber Type
4.3. Diabetes and Muscle Strength
4.4. Skeletal Muscle Biomarkers for Diabetes
4.5. Interaction Proteomics
5. Proteomics Application to Study Exercise Biology
5.1. Acute Exercise (Muscle Contraction) and PTMs
5.2. Exercise Training and Skeletal Muscle Adaptations
6. Secretome of Insulin Resistant and Exercised Skeletal Muscle
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kahn, C.R. Banting Lecture. Insulin action, diabetogenes, and the cause of type II diabetes. Diabetes 1994, 43, 1066–1084. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.; Koranyi, L.; Bourey, R.; Schalin-Jantti, C.; Widen, E.; Mueckler, M.; Permutt, A.M.; Groop, L.C. Insulin resistance in Type 2 (non-insulin-dependent) diabetic patients and their relatives is not associated with a defect in the expression of the insulin-responsive glucose transporter (GLUT-4) gene in human skeletal muscle. Diabetologia 1992, 35, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, J.E.; Alford, F.; Handberg, A.; Vaag, A.; Ward, G.M.; Kalfas, A.; Beck-Nielsen, H. Increased glucose effectiveness in normoglycemic but insulin-resistant relatives of patients with non-insulin-dependent diabetes mellitus. A novel compensatory mechanism. J. Clin. Investig. 1994, 94, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Vaag, A.; Henriksen, J.E.; Beck-Nielsen, H. Decreased insulin activation of glycogen synthase in skeletal muscles in young nonobese Caucasian first-degree relatives of patients with non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1992, 89, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Bjorkman, O.; Reichard, G.A., Jr.; Pilo, A.; Olsson, M.; Wahren, J.; DeFronzo, R.A. The disposal of an oral glucose load in healthy subjects. A quantitative study. Diabetes 1985, 34, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, S.P.; Ragland, D.R.; Leung, R.W.; Paffenbarger, R.S., Jr. Physical activity and reduced occurrence of non-insulin-dependent diabetes mellitus. N. Engl. J. Med. 1991, 325, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Nathan, D.M.; Krolewski, A.S.; Stampfer, M.J.; Willett, W.C.; Hennekens, C.H. A prospective study of exercise and incidence of diabetes among US male physicians. JAMA 1992, 268, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Rimm, E.B.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C.; Krolewski, A.S.; Rosner, B.; Hennekens, C.H.; Speizer, F.E. Physical activity and incidence of non-insulin-dependent diabetes mellitus in women. Lancet 1991, 338, 774–778. [Google Scholar] [CrossRef]
- Tuomilehto, J.; Lindstrom, J.; Eriksson, J.G.; Valle, T.T.; Hamalainen, H.; Ilanne-Parikka, P.; Keinanen-Kiukaanniemi, S.; Laakso, M.; Louheranta, A.; Rastas, M.; et al. Finnish Diabetes Prevention Study G. Prevention of Type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N. Engl. J. Med. 2001, 344, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, L.J.; Kahn, B.B. Exercise, glucose transport, and insulin sensitivity. Annu. Rev. Med. 1998, 49, 235–261. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kulak, N.A.; Nagaraj, N.; Cox, J. The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 2013, 49, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Doran, P.; Martin, G.; Dowling, P.; Jockusch, H.; Ohlendieck, K. Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics 2006, 6, 4610–4621. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomics of skeletal muscle differentiation, neuromuscular disorders and fiber aging. Expert Rev. Proteom. 2010, 7, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Schmidt, A.; Malmstroem, J.; Claassen, M.; Ori, A.; Szymborska, A.; Herzog, F.; Rinner, O.; Ellenberg, J.; Aebersold, R. The quantitative proteome of a human cell line. Mol. Syst. Biol. 2011, 7, 549. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Wisniewski, J.R.; Geiger, T.; Cox, J.; Kircher, M.; Kelso, J.; Paabo, S.; Mann, M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Syst. Biol. 2011, 7, 548. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Moghaddas Gholami, A.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [PubMed]
- Kim, M.S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Skeletal muscle proteomics: Current approaches, technical challenges and emerging techniques. Skelet. Muscle 2011, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.S.; Murgia, M.; Nagaraj, N.; Treebak, J.T.; Cox, J.; Mann, M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol. Cell. Proteom. 2015, 14, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Zierath, J.R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.S. Insulin-stimulated glucose uptake in healthy and insulin-resistant skeletal muscle. Horm. Mol. Biol. Clin. Investig. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Proteomic identification of biomarkers of skeletal muscle disorders. Biomark. Med. 2013, 7, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [PubMed]
- Kugelberg, E.; Edstrom, L. Differential histochemical effects of muscle contractions on phosphorylase and glycogen in various types of fibres: Relation to fatigue. J. Neurol. Neurosurg. Psychiatry 1968, 31, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Vetter, C.; Reichmann, H.; Pette, D. Microphotometric determination of enzyme activities in type-grouped fibres of reinnervated rat muscle. Histochemistry 1984, 80, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Clamp, M.; Fry, B.; Kamal, M.; Xie, X.; Cuff, J.; Lin, M.F.; Kellis, M.; Lindblad-Toh, K.; Lander, E.S. Distinguishing protein-coding and noncoding genes in the human genome. Proc. Natl. Acad. Sci. USA 2007, 104, 19428–19433. [Google Scholar] [CrossRef] [PubMed]
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Kalyanasundaram, A. SERCA pump isoforms: Their role in calcium transport and disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, M.E.; Huss, M.; Solnestam, B.W.; Kjellqvist, S.; Lundeberg, J.; Sundberg, C.J. The human skeletal muscle transcriptome: Sex differences, alternative splicing, and tissue homogeneity assessed with RNA sequencing. FASEB J. 2014, 28, 4571–4581. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Nagaraj, N.; Deshmukh, A.S.; Zeiler, M.; Cancellara, P.; Moretti, I.; Reggiani, C.; Schiaffino, S.; Mann, M. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Batth, T.S.; Francavilla, C.; Olsen, J.V. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J. Proteome Res. 2014, 13, 6176–6186. [Google Scholar] [CrossRef] [PubMed]
- Scheltema, R.A.; Hauschild, J.P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteom. 2014, 13, 3698–3708. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. MaxLFQ allows accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.S.; Geiger, T.; Chatterjee, B.; Bandilla, P.; Frohlich, F.; Cox, J.; Mann, M. Deep and highly sensitive proteome coverage by LC-MS/MS without prefractionation. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Altelaar, A.F.; Heck, A.J. Trends in ultrasensitive proteomics. Curr. Opin. Chem. Biol. 2012, 16, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Huo, L.; Liu, Z.; Li, J.; Liu, Y.; He, Q.; Wang, X.; Liang, S. Sodium laurate, a novel protease- and mass spectrometry-compatible detergent for mass spectrometry-based membrane proteomics. PLoS ONE 2013, 8, e59779. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, J.W.; Madsen, C.T.; Young, C.; Poulsen, F.M.; Nielsen, M.L. Using guanidine-hydrochloride for fast and efficient protein digestion and single-step affinity-purification mass spectrometry. J. Proteome Res. 2013, 12, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Rakus, D.; Gizak, A.; Deshmukh, A.; Wisniewski, J.R. Absolute quantitative profiling of the key metabolic pathways in slow and fast skeletal muscle. J. Proteome Res. 2015, 14, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Shevchenko, A.; Wilm, M.; Vorm, O.; Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996, 68, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.I.; McClatchy, D.; Park, S.K.; Yates, J.R., 3rd. Comparisons of mass spectrometry compatible surfactants for global analysis of the mammalian brain proteome. Anal. Chem. 2008, 80, 8694–8701. [Google Scholar] [CrossRef] [PubMed]
- Leon, I.R.; Schwammle, V.; Jensen, O.N.; Sprenger, R.R. Quantitative assessment of in-solution digestion efficiency identifies optimal protocols for unbiased protein analysis. Mol. Cell. Proteom. 2013, 12, 2992–3005. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.; Kulak, N.A.; Cox, J.; Neuhauser, N.; Mayr, K.; Hoerning, O.; Vorm, O.; Mann, M. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell. Proteom. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, G.; Horvath, C. Ion-exchange chromatography. Methods Enzymol. 1996, 270, 47–82. [Google Scholar] [PubMed]
- Howard, G.A.; Martin, A.J. The separation of the C12-C18 fatty acids by reversed-phase partition chromatography. Biochem. J. 1950, 46, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Hjerten, S. Hydrophobic interaction chromatography of proteins, nucleic acids, viruses, and cells on noncharged amphiphilic gels. Methods Biochem. Anal. 1981, 27, 89–108. [Google Scholar] [PubMed]
- Hubner, N.C.; Ren, S.; Mann, M. Peptide separation with immobilized pI strips is an attractive alternative to in-gel protein digestion for proteome analysis. Proteomics 2008, 8, 4862–4872. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.P.; Wolters, D.; Yates, J.R., 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012, 84, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Prus, G. Homogenous Phase Enrichment of Cysteine-Containing Peptides for Improved Proteome Coverage. Anal. Chem. 2015, 87, 6861–6867. [Google Scholar] [CrossRef] [PubMed]
- Fonslow, B.R.; Stein, B.D.; Webb, K.J.; Xu, T.; Choi, J.; Park, S.K.; Yates, J.R., 3rd. Digestion and depletion of abundant proteins improves proteomic coverage. Nat. Methods 2013, 10, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Fonslow, B.R.; Stein, B.D.; Webb, K.J.; Xu, T.; Choi, J.; Park, S.K.; Yates, J.R., 3rd. Addendum: Digestion and depletion of abundant proteins improves proteomic coverage. Nat. Methods 2014, 11, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Kocher, T.; Swart, R.; Mechtler, K. Ultra-high-pressure RPLC hyphenated to an LTQ-Orbitrap Velos reveals a linear relation between peak capacity and number of identified peptides. Anal. Chem. 2011, 83, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.S.; Richards, A.L.; Bailey, D.J.; Ulbrich, A.; Coughlin, E.E.; Westphall, M.S.; Coon, J.J. The one hour yeast proteome. Mol. Cell. Proteom. 2014, 13, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Riley, N.M.; Mullen, C.; Weisbrod, C.R.; Sharma, S.; Senko, M.W.; Zabrouskov, V.; Westphall, M.S.; Syka, J.E.; Coon, J.J. Enhanced Dissociation of Intact Proteins with High Capacity Electron Transfer Dissociation. J. Am. Soc. Mass Spectrom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.E.; Mann, M. Stable isotope labeling by amino acids in cell culture for quantitative proteomics. Methods Mol. Biol. 2007, 359, 37–52. [Google Scholar] [PubMed]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Boersema, P.J.; Aye, T.T.; van Veen, T.A.; Heck, A.J.; Mohammed, S. Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 2008, 8, 4624–4632. [Google Scholar] [CrossRef] [PubMed]
- Eberl, H.C.; Spruijt, C.G.; Kelstrup, C.D.; Vermeulen, M.; Mann, M. A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol. Cell 2013, 49, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in Type 2 diabetes. Diabetes Care 2009, 32 (Suppl. 2), S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Warram, J.H.; Martin, B.C.; Krolewski, A.S.; Soeldner, J.S.; Kahn, C.R. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann. Intern. Med. 1990, 113, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Shulman, G.I. Pathogenesis of skeletal muscle insulin resistance in Type 2 diabetes mellitus. Am. J. Cardiol. 2002, 90, 11G–18G. [Google Scholar] [CrossRef]
- Giebelstein, J.; Poschmann, G.; Hojlund, K.; Schechinger, W.; Dietrich, J.W.; Levin, K.; Beck-Nielsen, H.; Podwojski, K.; Stuhler, K.; Meyer, H.E.; et al. The proteomic signature of insulin-resistant human skeletal muscle reveals increased glycolytic and decreased mitochondrial enzymes. Diabetologia 2012, 55, 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Zabielski, P.; Lanza, I.R.; Gopala, S.; Holtz Heppelmann, C.J.; Bergen, H.R., 3rd; Dasari, S.; Nair, K.S. Altered skeletal muscle mitochondrial proteome as the basis of disruption of mitochondrial function in diabetic mice. Diabetes 2015. [Google Scholar] [CrossRef] [PubMed]
- Stentz, F.B.; Kitabchi, A.E. Transcriptome and proteome expressions involved in insulin resistance in muscle and activated T-lymphocytes of patients with type 2 diabetes. Genom. Proteom. Bioinform. 2007, 5, 216–235. [Google Scholar] [CrossRef]
- Mullen, E.; O’Reilly, E.; Ohlendieck, K. Skeletal muscle tissue from the Goto-Kakizaki rat model of Type-2 diabetes exhibits increased levels of the small heat shock protein Hsp27. Mol. Med. Rep. 2011, 4, 229–236. [Google Scholar] [PubMed]
- Hwang, H.; Bowen, B.P.; Lefort, N.; Flynn, C.R.; De Filippis, E.A.; Roberts, C.; Smoke, C.C.; Meyer, C.; Hojlund, K.; Yi, Z.; Mandarino, L.J. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and Type 2 diabetes. Diabetes 2010, 59, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Choi, J.W.; Chaudhari, H.N.; Aseer, K.R.; Mukherjee, R.; Yun, J.W. Gender-dimorphic regulation of skeletal muscle proteins in streptozotocin-induced diabetic rats. Cell. Physiol. Biochem. 2013, 31, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalili, L.; de Castro Barbosa, T.; Ostling, J.; Massart, J.; Cuesta, P.G.; Osler, M.E.; Katayama, M.; Nystrom, A.C.; Oscarsson, J.; Zierath, J.R. Proteasome inhibition in skeletal muscle cells unmasks metabolic derangements in Type 2 diabetes. Am. J. Physiol. Cell Physiol. 2014, 307, C774–C787. [Google Scholar] [CrossRef] [PubMed]
- Hojlund, K.; Bowen, B.P.; Hwang, H.; Flynn, C.R.; Madireddy, L.; Geetha, T.; Langlais, P.; Meyer, C.; Mandarino, L.J.; Yi, Z. In vivo phosphoproteome of human skeletal muscle revealed by phosphopeptide enrichment and HPLC-ESI-MS/MS. J. Proteome Res. 2009, 8, 4954–4965. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Leon, I.R.; Bak, S.; Mogensen, M.; Wrzesinski, K.; Hojlund, K.; Jensen, O.N. Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, S.J.; Azimifar, S.B.; Mann, M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 2015, 33, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Lundby, A.; Secher, A.; Lage, K.; Nordsborg, N.B.; Dmytriyev, A.; Lundby, C.; Olsen, J.V. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 2012, 3, 876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, D.; Caruso, M.; Lewis, M.; Qi, Y.; Yi, Z. Quantitative phosphoproteomics reveals novel phosphorylation events in insulin signaling regulated by protein phosphatase 1 regulatory subunit 12A. J. Proteom. 2014, 109, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.E.; Mocanu, V.; Mocanu, M.; Dicheva, N.; Warren, M.R. Mass Spectrometry for Post-Translational Modifications. In Neuroproteomics; Alzate, O., Ed.; Frontiers in Neuroscience: Boca Raton, FL, USA, 2010. [Google Scholar]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.A. Exercise as a therapeutic intervention for the prevention and treatment of insulin resistance. Diabetes Metab. Res. Rev. 2004, 20, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Hesselink, M.K. Oxidative capacity, lipotoxicity, and mitochondrial damage in Type 2 diabetes. Diabetes 2004, 53, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Oberbach, A.; Bossenz, Y.; Lehmann, S.; Niebauer, J.; Adams, V.; Paschke, R.; Schon, M.R.; Bluher, M.; Punkt, K. Altered fiber distribution and fiber-specific glycolytic and oxidative enzyme activity in skeletal muscle of patients with Type 2 diabetes. Diabetes Care 2006, 29, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Freemont, A.J.; Hoyland, J.A. Morphology, mechanisms and pathology of musculoskeletal ageing. J. Pathol. 2007, 211, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Leenders, M.; Verdijk, L.B.; van der Hoeven, L.; Adam, J.J.; van Kranenburg, J.; Nilwik, R.; van Loon, L.J. Patients with Type 2 diabetes show a greater decline in muscle mass, muscle strength, and functional capacity with aging. J. Am. Med. Dir Assoc. 2013, 14, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.; Nielsen, S.; Mogensen, C.E.; Jakobsen, J. Muscle strength in Type 2 diabetes. Diabetes 2004, 53, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Goodpaster, B.H.; Strotmeyer, E.S.; de Rekeneire, N.; Harris, T.B.; Schwartz, A.V.; Tylavsky, F.A.; Newman, A.B. Decreased muscle strength and quality in older adults with type 2 diabetes: The health, aging, and body composition study. Diabetes 2006, 55, 1813–1818. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.I. Undiagnosed NIDDM: Clinical and public health issues. Diabetes Care 1993, 16, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Herman, W.H.; Ye, W.; Griffin, S.J.; Simmons, R.K.; Davies, M.J.; Khunti, K.; Rutten, G.E.; Sandbaek, A.; Lauritzen, T.; Borch-Johnsen, K.; et al. Early Detection and Treatment of Type 2 Diabetes Reduce Cardiovascular Morbidity and Mortality: A Simulation of the Results of the Anglo-Danish-Dutch Study of Intensive Treatment in People With Screen-Detected Diabetes in Primary Care (ADDITION-Europe). Diabetes Care 2015, 38, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Zendjabil, M. Biological diagnosis of diabetes mellitus. Pathol. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Stumpf, M.P.; Thorne, T.; de Silva, E.; Stewart, R.; An, H.J.; Lappe, M.; Wiuf, C. Estimating the size of the human interactome. Proc. Natl. Acad. Sci. USA 2008, 105, 6959–6964. [Google Scholar] [CrossRef] [PubMed]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Keilhauer, E.C.; Hein, M.Y.; Mann, M. Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol. Cell. Proteom. 2015, 14, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I. Computational and informatics strategies for identification of specific protein interaction partners in affinity purification mass spectrometry experiments. Proteomics 2012, 12, 1639–1655. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Ma, D.; Msallaty, Z.; Lewis, M.; Seyoum, B.; Al-janabi, W.; Diamond, M.; Abou-Samra, A.B.; Hojlund, K.; Tagett, R.; et al. Increased interaction with insulin receptor substrate 1, a novel abnormality in insulin resistance and Type 2 diabetes. Diabetes 2014, 63, 1933–1947. [Google Scholar] [CrossRef] [PubMed]
- Krogh-Madsen, R.; Thyfault, J.P.; Broholm, C.; Mortensen, O.H.; Olsen, R.H.; Mounier, R.; Plomgaard, P.; van Hall, G.; Booth, F.W.; Pedersen, B.K. A 2-wk reduction of ambulatory activity attenuates peripheral insulin sensitivity. J. Appl. Physiol. (1985) 2010, 108, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.H.; Krogh-Madsen, R.; Thomsen, C.; Booth, F.W.; Pedersen, B.K. Metabolic responses to reduced daily steps in healthy nonexercising men. JAMA 2008, 299, 1261–1263. [Google Scholar] [PubMed]
- Garetto, L.P.; Richter, E.A.; Goodman, M.N.; Ruderman, N.B. Enhanced muscle glucose metabolism after exercise in the rat: The two phases. Am. J. Physiol. 1984, 246, E471–E475. [Google Scholar] [PubMed]
- Richter, E.A.; Garetto, L.P.; Goodman, M.N.; Ruderman, N.B. Enhanced muscle glucose metabolism after exercise: Modulation by local factors. Am. J. Physiol. 1984, 246, E476–E482. [Google Scholar] [PubMed]
- Holloszy, J.O. Exercise-induced increase in muscle insulin sensitivity. J. Appl. Physiol. (1985) 2005, 99, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Christ-Roberts, C.Y.; Pratipanawatr, T.; Pratipanawatr, W.; Berria, R.; Belfort, R.; Mandarino, L.J. Increased insulin receptor signaling and glycogen synthase activity contribute to the synergistic effect of exercise on insulin action. J. Appl. Physiol. (1985) 2003, 95, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Wallberg-Henriksson, H.; Holloszy, J.O. Contractile activity increases glucose uptake by muscle in severely diabetic rats. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 57, 1045–1049. [Google Scholar] [PubMed]
- Deshmukh, A.S.; Hawley, J.A.; Zierath, J.R. Exercise-induced phospho-proteins in skeletal muscle. Int. J. Obes. 2008, 32 (Suppl. 4), S18–S23. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [PubMed]
- McGee, S.L.; Fairlie, E.; Garnham, A.P.; Hargreaves, M. Exercise-induced histone modifications in human skeletal muscle. J. Physiol. 2009, 587, 5951–5958. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Diabetes Prevention Program Research G. Reduction in the incidence of Type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [PubMed]
- Coffey, V.G.; Hawley, J.A. The molecular bases of training adaptation. Sports Med. 2007, 37, 737–763. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.R.; Hather, B.M.; Baldwin, K.M.; Dudley, G.A. Skeletal muscle myosin heavy chain composition and resistance training. J. Appl. Physiol. 1993, 74, 911–915. [Google Scholar] [PubMed]
- Widrick, J.J.; Stelzer, J.E.; Shoepe, T.C.; Garner, D.P. Functional properties of human muscle fibers after short-term resistance exercise training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R408–R416. [Google Scholar] [CrossRef] [PubMed]
- Spina, R.J.; Chi, M.M.; Hopkins, M.G.; Nemeth, P.M.; Lowry, O.H.; Holloszy, J.O. Mitochondrial enzymes increase in muscle in response to 7–10 days of cycle exercise. J. Appl. Physiol. (1985) 1996, 80, 2250–2254. [Google Scholar]
- Green, H.J.; Helyar, R.; Ball-Burnett, M.; Kowalchuk, N.; Symon, S.; Farrance, B. Metabolic adaptations to training precede changes in muscle mitochondrial capacity. J. Appl. Physiol. (1985) 1992, 72, 484–491. [Google Scholar]
- Benziane, B.; Burton, T.J.; Scanlan, B.; Galuska, D.; Canny, B.J.; Chibalin, A.V.; Zierath, J.R.; Stepto, N.K. Divergent cell signaling after short-term intensified endurance training in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1427–E1438. [Google Scholar] [CrossRef] [PubMed]
- Pilegaard, H.; Saltin, B.; Neufer, P.D. Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J. Physiol. 2003, 546, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Holloway, K.V.; O’Gorman, M.; Woods, P.; Morton, J.P.; Evans, L.; Cable, N.T.; Goldspink, D.F.; Burniston, J.G. Proteomic investigation of changes in human vastus lateralis muscle in response to interval-exercise training. Proteomics 2009, 9, 5155–5174. [Google Scholar] [CrossRef] [PubMed]
- Egan, B.; Dowling, P.; O’Connor, P.L.; Henry, M.; Meleady, P.; Zierath, J.R.; O’Gorman, D.J. 2-D DIGE analysis of the mitochondrial proteome from human skeletal muscle reveals time course-dependent remodelling in response to 14 consecutive days of endurance exercise training. Proteomics 2011, 11, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Schild, M.; Ruhs, A.; Beiter, T.; Zugel, M.; Hudemann, J.; Reimer, A.; Krumholz-Wagner, I.; Wagner, C.; Keller, J.; Eder, K.; et al. Basal and exercise induced label-free quantitative protein profiling of m. vastus lateralis in trained and untrained individuals. J. Proteom. 2015, 122, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.M.; Vitorino, R.; Figueiredo, P.; Duarte, J.A.; Ferreira, R.; Amado, F. Lifelong physical activity modulation of the skeletal muscle mitochondrial proteome in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Moriggi, M.; Vasso, M.; Fania, C.; Capitanio, D.; Bonifacio, G.; Salanova, M.; Blottner, D.; Rittweger, J.; Felsenberg, D.; Cerretelli, P.; et al. Long term bed rest with and without vibration exercise countermeasures: Effects on human muscle protein dysregulation. Proteomics 2010, 10, 3756–3774. [Google Scholar] [CrossRef] [PubMed]
- Hussey, S.E.; Sharoff, C.G.; Garnham, A.; Yi, Z.; Bowen, B.P.; Mandarino, L.J.; Hargreaves, M. Effect of exercise on the skeletal muscle proteome in patients with Type 2 diabetes. Med. Sci. Sports Exerc. 2013, 45, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Niu, Y.; Liu, X.; Yang, F.; Niu, W.; Fu, L. Proteomic analysis of skeletal muscle in insulin-resistant mice: Response to 6-week aerobic exercise. PLoS ONE 2013, 8, e53887. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.K.; Febbraio, M.A. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Scheltema, R.A.; Mollenkopf, H.J.; Mann, M. Direct proteomic quantification of the secretome of activated immune cells. Science 2013, 340, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.S.; Cox, J.; Jensen, L.J.; Meissner, F.; Mann, M. Secretome Analysis of Lipid-Induced Insulin Resistance in Skeletal Muscle Cells by a Combined Experimental and Bioinformatics Workflow. J. Proteome Res. 2015, 14, 4885–4895. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Salehzadeh, F.; Metayer-Coustard, S.; Fahlman, R.; Nair, K.S.; Al-Khalili, L. Post-transcriptional gene silencing of ribosomal protein S6 kinase 1 restores insulin action in leucine-treated skeletal muscle. Cell. Mol. Life Sci. 2009, 66, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, N.; Watson, M.; Sakamoto, K.; Hundal, H.S. Differential effects of palmitate and palmitoleate on insulin action and glucose utilization in rat L6 skeletal muscle cells. Biochem. J. 2006, 399, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Somwar, R.; Patel, N.; Niu, W.; Torok, D.; Klip, A. Sustained exposure of L6 myotubes to high glucose and insulin decreases insulin-stimulated GLUT4 translocation but upregulates GLUT4 activity. Diabetes 2002, 51, 2090–2098. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Michell, B.J.; van Denderen, B.J.; Watt, M.J.; Carey, A.L.; Fam, B.C.; Andrikopoulos, S.; Proietto, J.; Gorgun, C.Z.; Carling, D.; et al. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006, 4, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Nedachi, T.; Fujita, H.; Kanzaki, M. Contractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1191–E1204. [Google Scholar] [CrossRef] [PubMed]
- Raschke, S.; Eckardt, K.; Bjorklund Holven, K.; Jensen, J.; Eckel, J. Identification and validation of novel contraction-regulated myokines released from primary human skeletal muscle cells. PLoS ONE 2013, 8, e62008. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshmukh, A.S. Proteomics of Skeletal Muscle: Focus on Insulin Resistance and Exercise Biology. Proteomes 2016, 4, 6. https://doi.org/10.3390/proteomes4010006
Deshmukh AS. Proteomics of Skeletal Muscle: Focus on Insulin Resistance and Exercise Biology. Proteomes. 2016; 4(1):6. https://doi.org/10.3390/proteomes4010006
Chicago/Turabian StyleDeshmukh, Atul S. 2016. "Proteomics of Skeletal Muscle: Focus on Insulin Resistance and Exercise Biology" Proteomes 4, no. 1: 6. https://doi.org/10.3390/proteomes4010006
APA StyleDeshmukh, A. S. (2016). Proteomics of Skeletal Muscle: Focus on Insulin Resistance and Exercise Biology. Proteomes, 4(1), 6. https://doi.org/10.3390/proteomes4010006