Potentiality of Soybean Proteomics in Untying the Mechanism of Flood and Drought Stress Tolerance


Abstract
:1. Introduction
2. Protein Extraction

{kind=link}
| Stress | Cultivar/ Stress exposure | Organ/ Organelle | Protein extraction buffer | Protein solubilization /lysis buffer | Proteomic methodologies | Spot resolved Proteins | Differentially abundant protein classification | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| Function | Localization | ||||||||
| Flooding | Enrei (5 days) | Leaf Hypocotyl Root | 10% TCA, 0.07% 2-ME in acetone | 8 M urea, 2 M thiourea, 5% CHAPS, 2 mM tributyl-phosphine, 0.4% Ampholytes pH 3–10 | IEF, SDS-PAGE, nanoLC-MS/MS | 577 (L): 24↑26↓ 555 (H): 35↑31↓ 515 (R): 20↑27↓ | Met, Ene, ProtDesSt, DisDef, ProtSyn | Mito, Nucl, Cyto, Extr, ER, Cysk, PM | [9] |
| Enrei (2 days) | Hypocotyl Root mitochondria | - | 8 M urea, 2% NP-40, 5% 2-ME, 5% PVP 40, 0.4% Ampholytes pH 3–10 | IPG, SDS-PAGE, BN-PAGE, nanoLC-MS/MS | Matrix 327 29↑7↓ Membrane 72 5↑11↓ | Ene, DisDef | Mito, Chlo | [5] | |
| Enrei (2 days) | Hypocotyl Root cell wall | - | 8 M urea, 2% NP-40, 0.8% Ampholine pH 3.5–10, 5% 2-ME and 5% PVP 40 | IEF, SDS-PAGE, MALDI-TOF MS, nanoLC-MS/MS, protein sequencing | 204 4↑12↓ | Met, ProtDesSt, DisDef | Sec | [7] | |
| Enrei (1–4 days) | Hypocotyl Root | Phosphate saline buffer pH 7.6, 400 mM NaCl, 3 mM NaN3 followed by 10% TCA | 8 M urea, 2% NP-40, 0.8% Ampholine (pH 3.5–10), 5% 2-ME and 5% PVP 40 | IEF/IPG, SDS-PAGE, MALDI-TOF MS, protein sequencing | 803 21↑7↓ | ProtDesSt, DisDef, Ene, Pmet, CellSt, Trans | - | [8] | |
| Asoagari (3, 7 days) | Root | Cold acetone containing 10% TCA, 0.07% 2-ME | 8 M urea, 1% CHAPS, 0.5% IPG buffer pH 4–7, 20 mM DTT, BPB | IPG, SDS-PAGE, MALDI-TOF MS, ESI-MS/MS | ~900 14↑5↓5 Newly induced | Met, Ene, DisDef, ProtSyn | - | [19] | |
| Enrei (12–48 h) | Hypocotyl Root | - | 9.5M urea, 2% NP-40, 2% Ampholines pH 3–10, 5% 2-ME | IEF/IPG tube gel, 2-DE, MALDI-TOF MS, nanoLC-MS/MS, protein sequencing | 799 14↑20↓ | Ene, DisDef, Pmet, CellSt, Secmet, Sgnl | - | [20] | |
| Enrei (1 days) | Hypocotyl Root plasma membrane | - | 8 M urea, 2% NP-40, 0.8% Ampholine pH 3.5–10, 5% 2-ME and 5% PVP 40 | IEF tube gel, 2-DE, MALDI-TOF MS, nanoLC-MS/MS, protein sequencing | 150 12↑2↓ | ProtDesSt, ProtSyn, DisDef, CellDiv, Trans, Pmet, Ene, Secmet, Sgnl | - | [21] | |
| Flooding Low oxygen | Enrei (3, 6 days Low oxygen) | Root | 10% TCA, 0.07% 2-ME in acetone | 8 M urea, 2 M thiourea, 5% CHAPS, 2 mM tributyl-phosphine, 0.4% Ampholytes pH 3–10 | IEF, SDS-PAGE , MALDI-TOF MS, nanoLC-MS/MS | 1,233 F: 4↑12↓ LO: 2↓ | Met, Ene, ProtDesSt, Sgnl, ProtSyn, DisDef | Cyto, Chlo, Nucl | [10] |
| Drought | Enrei (Stop watering 10% PEG 4 days) | Leaf Hypocotyl Root | 10% TCA, 0.07% 2-ME in acetone | 8 M urea, 2 M thiourea, 5% CHAPS, and 2 mM tributyl-phosphine, 0.4% Ampholytes pH 3–10 | IPG, SDS-PAGE, nanoLC-MS/MS | 549 (L): PEG: 20↑17↓ Drought: 20↑21↓ 451 (H): PEG: 20↑13↓ Drought: 18↑19↓ 632 (R): PEG: 20↑10↓ Drought: 33↑16↓ | Met, Ene, ProtSyn, DisDef | Chlo, Cyto, Nucl, Mito | [12] |
| Taegwang (withholding water - 5 days, rewatering - 4 days) | Root | Mg/NP-40 buffer [0.5 M Tris-HCl ( pH 8.3), 2% NP-40, 20 mM MgCl2, 1 mM PMSF , 2% 2-ME, 1% PVP], water-saturated phenol, followed by ammonium acetate in methanol | 8 M urea, 1% CHAPS, 0.5% IPG buffer (pH 4–7), 20 mM DTT, BPB | IPG, SDS-PAGE, MALDI-TOF MS | 1,350 6↑20↓2 New | Met, Ene, Sgnl, DisDef, CellSt, | - | [13] | |
| Osmotic stress | Enrei (10% PEG 1–4 days) | Hypocotyl Root plasma membrane | Plasma membrane proteins precipitated by TCA followed by cold acetone washing | 7 M urea, 0.2 M thiourea, 0.2mM tributylphosphine, 5% PVP-40, 0.4% CHAPS, 0.2% Ampholytes (pH 3.0–10.0) | IEF tube gel, SDS-PAGE, LC MS/MS, nanoLC-MS/MS | 202 11↑75↓ | Sgnl, Met, ProtSyn, DisDef, Trans | - | [22] |
| Osmotic stress | Enrei (0, 5, 10, 20% PEG 1–4 days) | Root | Phosphate saline buffer (pH 7.6): 65 mM K2HPO4, 2.6 mM KH2PO4, 400 mM NaCl and 3 mM NaN3 followed by 10% TCA | 8 M urea, 2% NP-40, 0.8% Ampholine (pH 3.5–10), 5% 2-ME and 5% PVP 40 | IEF tube gel, SDS-PAGE, MALDI-TOF MS, protein sequencing | 415 19↑18↓ | DisDef, Ene, ProtDesSt, Met, CellSt, Secmet. | - | [23] |
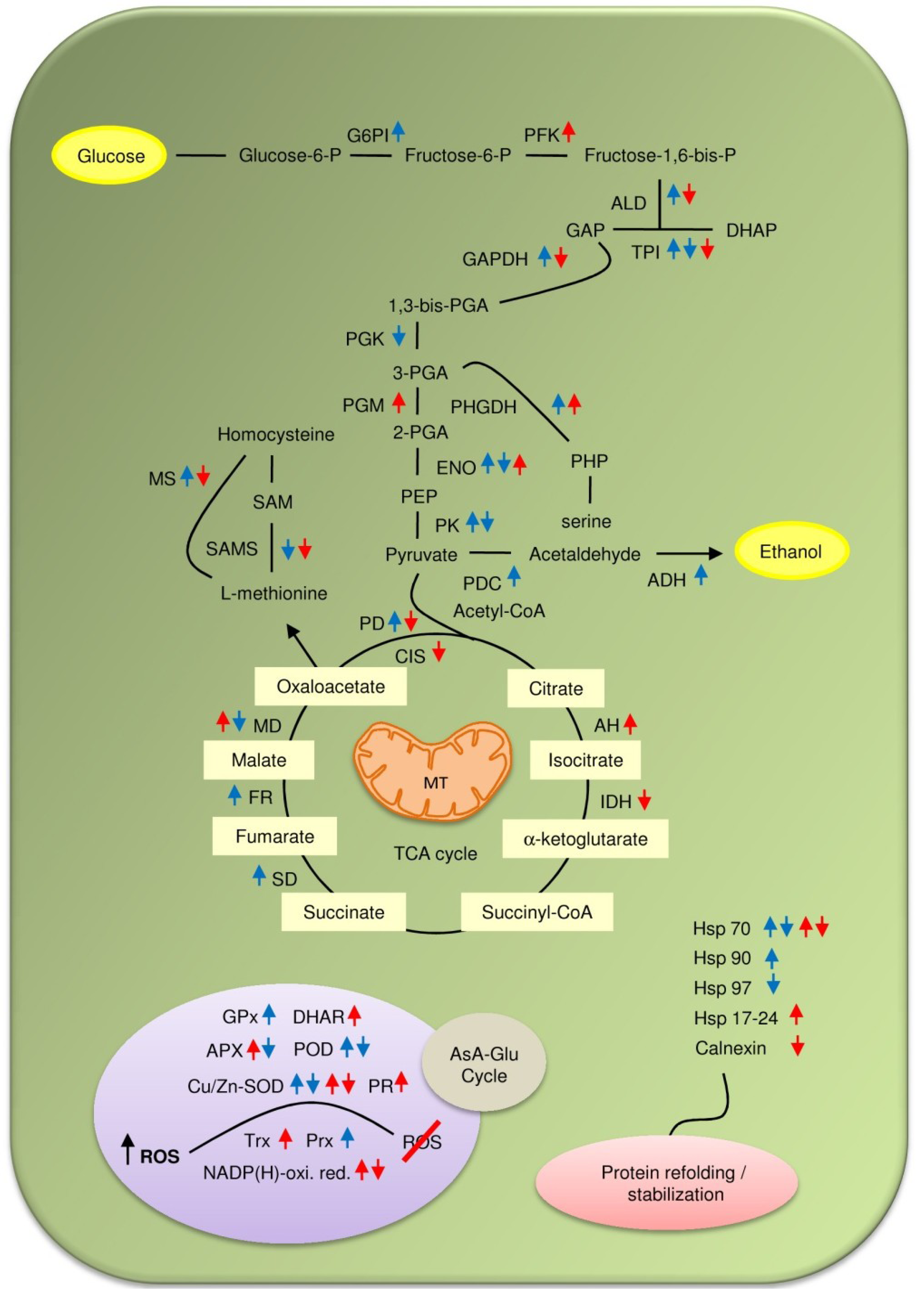
3. Changes in Soybean Proteome in Response to Flooding
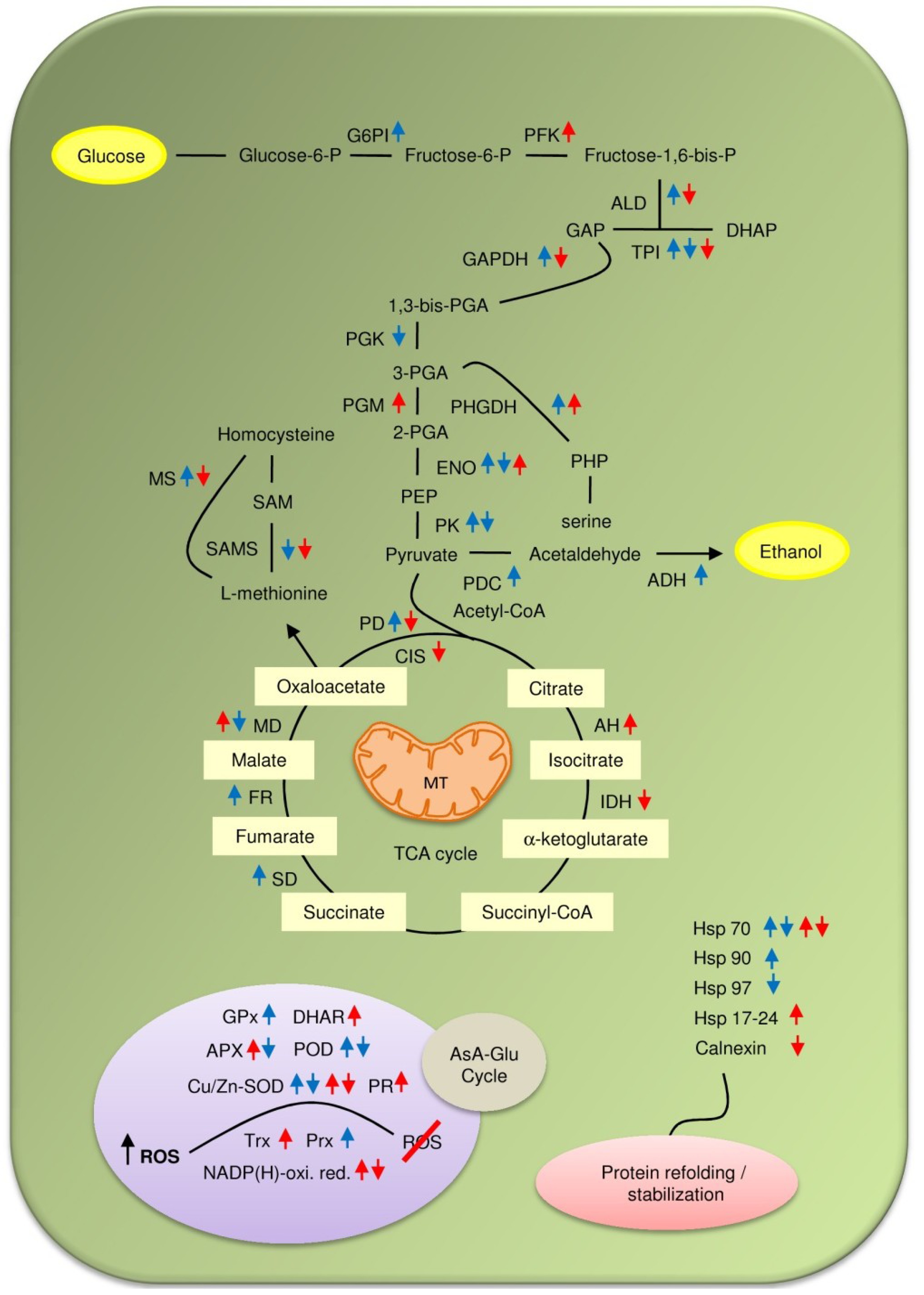
4. Drought Induced Modulation of Soybean Proteome Composition
5. Novel Methodological Approaches to Study Plant Proteomes
6. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Komatsu, S.; Nanjo, Y.; Nishimura, M. Proteomic analysis of the flooding tolerance mechanism in mutant soybean. J. Proteomics 2013, 79, 231–250. [Google Scholar] [CrossRef]
- Komatsu, S.; Han, C.; Nanjo, Y.; Altaf-Un-Nahar, M.; Wang, K.; He, D.; Yang, P. Label-free quantitative proteomic analysis of abscisic acid effect in early-stage soybean under flooding. J. Proteome Res. 2013, 12, 4769–4784. [Google Scholar] [CrossRef]
- Komatsu, S.; Hiraga, S.; Yanagawa, Y. Proteomics techniques for the development of flood tolerant crops. J. Proteome Res. 2012, 11, 68–78. [Google Scholar] [CrossRef]
- Komatsu, S.; Kuji, R.; Nanjo, Y.; Hiraga, S.; Furukawa, K. Comprehensive analysis of endoplasmic reticulum-enriched fraction in root tips of soybean under flooding stress using proteomics techniques. J. Proteomics 2012, 77, 531–560. [Google Scholar]
- Komatsu, S.; Yamamoto, A.; Nakamura, T.; Nouri, M.Z.; Nanjo, Y.; Nishizawa, K.; Furukawa, K. Comprehensive analysis of mitochondria in roots and hypocotyls of soybean under flooding stress using proteomics and metabolomics techniques. J. Proteome Res. 2011, 10, 3993–4004. [Google Scholar] [CrossRef]
- Komatsu, S.; Deschamps, T.; Hiraga, S.; Kato, M.; Chiba, M.; Hashiguchi, A.; Tougou, M.; Shimamura, S.; Yasue, H. Characterization of a novel flooding stress-responsive alcohol dehydrogenase expressed in soybean roots. Plant Mol. Biol. 2011, 77, 309–322. [Google Scholar] [CrossRef]
- Komatsu, S.; Kobayashi, Y.; Nishizawa, K.; Nanjo, Y.; Furukawa, K. Comparative proteomics analysis of differentially expressed proteins in soybean cell wall during flooding stress. Amino Acids 2010, 39, 1435–1449. [Google Scholar] [CrossRef]
- Komatsu, S.; Sugimoto, T.; Hoshino, T.; Nanjo, Y.; Furukawa, K. Identification of flooding stress responsible cascades in root and hypocotyl of soybean using proteome analysis. Amino Acids 2010, 38, 729–738. [Google Scholar] [CrossRef]
- Khatoon, A.; Rehman, S.; Hiraga, S.; Makino, T.; Komatsu, S. Organ-specific proteomics analysis for response mechanism in soybean seedlings under flooding stress. J. Proteomics 2012, 75, 5706–5723. [Google Scholar] [CrossRef]
- Khatoon, A.; Rehman, S.; Oh, M.W.; Woo, S.H.; Komatsu, S. Analysis of response mechanism in soybean under low oxygen and flooding stresses using gel-base proteomics technique. Mol. Biol. Rep. 2012, 39, 10581–10594. [Google Scholar] [CrossRef]
- Nanjo, Y.; Nakamura, T.; Komatsu, S. Identification of indicator proteins associated with flooding injury in soybean seedlings using label-free quantitative proteomics. J. Proteome Res. 2013, 12, 4785–4798. [Google Scholar] [CrossRef]
- Mohammadi, P.P.; Moieni, A.; Hiraga, S.; Komatsu, S. Organ-specific proteomic analysis of drought-stressed soybean seedlings. J. Proteomics 2012, 75, 1906–1923. [Google Scholar] [CrossRef]
- Alam, I.; Sharmin, S.A.; Kim, K.H.; Yang, J.K.; Choi, M.S.; Lee, B.H. Proteome analysis of soybean roots subjected to short-term drought stress. Plant Soil 2010, 333, 491–505. [Google Scholar] [CrossRef]
- Hossain, Z.; Hajika, M.; Komatsu, S. Comparative proteome analysis of high and low cadmium accumulating soybeans under cadmium stress. Amino Acids 2012, 43, 2393–2416. [Google Scholar] [CrossRef]
- Swigonska, S.; Weidner, S. Proteomic analysis of response to long-term continuous stress in roots of germinating soybean seeds. J. Plant Physiol. 2013, 170, 470–479. [Google Scholar] [CrossRef]
- Nanjo, Y.; Skultety, L.; Ashraf, Y.; Komatsu, S. Comparative proteomic analysis of early-stage soybean seedlings responses to flooding by using gel and gel-free techniques. J. Proteome Res. 2010, 9, 3989–4002. [Google Scholar] [CrossRef]
- Nanjo, Y.; Skultety, L.; Uváčková, L.; Klubicová, K.; Hajduch, M.; Komatsu, S. Mass spectrometry-based analysis of proteomic changes in the root tips of flooded soybean seedlings. J. Proteome Res. 2012, 11, 372–385. [Google Scholar] [CrossRef]
- Komatsu, S.; Ahsan, N. Soybean proteomics and its application to functional analysis. J. Proteomics 2009, 72, 325–336. [Google Scholar] [CrossRef]
- Alam, I.; Lee, D.G.; Kim, K.H.; Park, C.H.; Sharmin, S.A.; Lee, H.; Oh, K.W.; Yun, B.W.; Lee, B.H. Proteome analysis of soybean roots under waterlogging stress at an early vegetative stage. J. Biosci. 2010, 35, 49–62. [Google Scholar] [CrossRef]
- Komatsu, S.; Yamamoto, R.; Nanjo, Y.; Mikami, Y.; Yunokawa, H.; Sakata, K. A comprehensive analysis of the soybean genes and proteins expressed under flooding stress using transcriptome and proteome techniques. J. Proteome Res. 2009, 8, 4766–4778. [Google Scholar] [CrossRef]
- Komatsu, S.; Wada, T.; Abaléa, Y.; Nouri, M. Z.; Nanjo, Y.; Nakayama, N.; Shimamura, S.; Yamamoto, R.; Nakamura, T.; Furukawa, K. Analysis of plasma membrane proteome in soybean and application to flooding stress response. J. Proteome Res. 2009, 8, 4487–4499. [Google Scholar] [CrossRef]
- Nouri, M.Z.; Komatsu, S. Comparative analysis of soybean plasma membrane proteins under osmotic stress using gel-based and LC MS/MS-based proteomics approaches. Proteomics 2010, 10, 1930–1945. [Google Scholar] [CrossRef]
- Toorchi, M.; Yukawa, K.; Nouri, M.Z.; Komatsu, S. Proteomics approach for identifying osmotic-stress-related proteins in soybean roots. Peptides 2009, 30, 2108–2117. [Google Scholar] [CrossRef]
- Buttery, B.R.; Buzzell, R.I. Soybean flavonol glycosides: Identification and biochemical genetics. Can. J. Bot. 1973, 53, 309–313. [Google Scholar]
- Saravanan, R.S.; Rose, J.K. A critical evaluation of sample extraction techniques for enhanced proteomic analysis of recalcitrant plant tissues. Proteomics 2004, 4, 2522–2532. [Google Scholar] [CrossRef]
- Krishnan, H.B.; Oehrle, N.W.; Natarajan, S.S. A rapid and simple procedure for the depletion of abundant storage proteins from legume seeds to advance proteome analysis: A case study using Glycine max. Proteomics 2009, 9, 3174–3188. [Google Scholar] [CrossRef]
- Mooney, B.P.; Thelen, J.J. High-throughput peptide mass fingerprinting of soybean seed proteins: Automated workflow and utility of UniGene expressed sequence tag databases for protein identification. Phytochemistry 2004, 65, 1733–1744. [Google Scholar] [CrossRef]
- Hajduch, M.; Ganapathy, A.; Stein, J.W.; Thelen, J.J. A systematic proteomic study of seed filling in soybean. Establishment of high-resolution two-dimensional reference maps, expression profiles, and an interactive proteome database. Plant Physiol. 2005, 137, 214–220. [Google Scholar]
- Natarajan, S.; Xu, C.; Caperna, T.J.; Garrett, W.M. Comparison of protein solubilization methods suitable for proteomic analysis of soybean seed proteins. Anal. Biochem. 2005, 342, 214–220. [Google Scholar] [CrossRef]
- Barbosa, H.S.; Arruda, S.C.; Azevedo, R.A.; Arruda, M.A. New insights on proteomics of transgenic soybean seeds: Evaluation of differential expressions of enzymes and proteins. Anal. Bioanal. Chem. 2012, 402, 299–314. [Google Scholar] [CrossRef]
- Ahsan, N.; Komatsu, S. Comparative analyses of the proteomes of leaves and flowers at various stages of development reveal organ-specific functional differentiation of proteins in soybean. Proteomics 2009, 9, 4889–4907. [Google Scholar] [CrossRef]
- Toorchi, M.; Nouri, M.Z.; Tsumura, M.; Komatsu, S. Acoustic technology for high-performance disruption and extraction of plant proteins. J. Proteome Res. 2008, 7, 3035–3041. [Google Scholar] [CrossRef]
- Hurkman, W.J.; Tanaka, C.K. Solubilization of plant membrane proteins for analysis by two-dimensional gel electrophoresis. Plant Physiol. 1986, 81, 802–806. [Google Scholar] [CrossRef]
- Salavati, A.; Khatoon, A.; Nanjo, Y.; Komatsu, S. Analysis of proteomic changes in roots of soybean seedlings during recovery after flooding. J. Proteomics 2012, 75, 878–893. [Google Scholar] [CrossRef]
- Zhen, Y.; Qi, J.L.; Wang, S.S.; Su, J.; Xu, G.H.; Zhang, M.S.; Miao, L.; Peng, X.X.; Tian, D.; Yang, Y.H. Comparative proteome analysis of differentially expressed proteins induced by Al toxicity in soybean. Physiol. Plant 2007, 131, 542–554. [Google Scholar] [CrossRef]
- Mathesius, U.; Djordjevic, M.A.; Oakes, M.; Goffard, N.; Haerizadeh, F.; Weiller, G.F.; Singh, M.B.; Bhalla, P.L. Comparative proteomic profiles of the soybean (Glycine max) root apex and differentiated root zone. Proteomics 2011, 11, 1707–1719. [Google Scholar] [CrossRef]
- Rose, J.C.; Bashir, S.; Giovannoni, J.J.; Jahn, M.M.; Saravanan, R.S. Tackling the plant proteome: Practical approaches, hurdles and experimental tools. Plant J. 2004, 39, 715–733. [Google Scholar] [CrossRef]
- Espagne, C.; Martinez, A.; Valot, B.; Meinnel, T.; Giglione, C. Alternative and effective proteomic analysis in Arabidopsis. Proteomics 2007, 7, 3788–3799. [Google Scholar] [CrossRef]
- Hossain, Z.; López-Climent, M.F.; Arbona, V.; Pérez-Clemente, R.M.; Gómez-Cadenas, A. Modulation of the antioxidant system in Citrus under waterlogging and subsequent drainage. J. Plant Physiol. 2009, 166, 1391–404. [Google Scholar] [CrossRef]
- Hole, D.J.; Greg Cobb, B; Hole, P.S.; Drew, M.C. Enhancement of anaerobic respiration in root tips of Zea mays following low oxygen (hypoxic) acclimation. Plant Physiol. 1992, 99, 213–218. [Google Scholar] [CrossRef]
- Bailey-Serres, J.; Voesenek, L.A.C.J. Flooding stress: Acclimations and genetic diversity. Annu. Rev. Plant Biol. 2008, 59, 313–339. [Google Scholar] [CrossRef]
- Hashiguchi, A.; Sakata, K.; Komatsu, S. Proteome analysis of early-stage soybean seedlings under flooding stress. J. Proteome Res. 2009, 8, 2058–2069. [Google Scholar] [CrossRef]
- Yanagawa, Y.; Komatsu, S. Ubiquitin/proteasome-mediated proteolysis is involved in the response to flooding stress in soybean roots, independent of oxygen limitation. Plant Sci. 2012, 185–186, 250–258. [Google Scholar] [CrossRef]
- Shi, F.; Yamamoto, R.; Shimamura, S.; Hiraga, S.; Nakayama, N.; Nakamura, T.; Yukawa, K.; Hachinohe, M.; Matsumoto, H.; Komatsu, S. Cytosolic ascorbate peroxidase 2 (cAPX 2) is involved in the soybean response to flooding. Phytochemistry 2008, 69, 1295–1303. [Google Scholar] [CrossRef]
- Mielke, M.S.; de Almeida, A.A.F.; Gomes, F.P.; Aguilarb, M.A.G.; Mangabeiraa, P.A.O. Leaf gas exchange, chlorophyll fluorescence and growth responses of Genipa americana seedlings to soil flooding. Envinron. Exp. Bot. 2003, 50, 221–231. [Google Scholar] [CrossRef]
- Maayan, I.; Shaya, F.; Ratner, K.; Mani, Y.; Lavee, S.; Avidan, B.; Shahak, Y.; Ostersetzer-Biran, O. Photosynthetic activity during olive (Olea europaea) leaf development correlates with plastid biogenesis and RuBisCO levels. Physiol. Plant. 2008, 134, 547–558. [Google Scholar] [CrossRef]
- Donnelly, B.E.; Madden, R.D.; Ayoubi, P.; Porter, D.R.; Dillwith, J.W. The wheat (Triticum aestivum L.) leaf proteome. Proteomics 2005, 5, 1624–1633. [Google Scholar] [CrossRef]
- Oh, M.; Nanjo, Y.; Komatsu, S. Identification of nuclear proteins in soybean under flooding stress using proteomic technique. Protein Pept. Lett. 2014, 21, 458–467. [Google Scholar]
- Bota, J.; Flexas, J.; Medrano, H. Is photosynthesis limited by decreased Rubisco activity and RuBP content under progressive water stress? New Phytol. 2004, 162, 671–681. [Google Scholar] [CrossRef]
- Loreto, F.; Tricoli, D.; di Marco, G. On the relationship between electron transport rate and photosynthesis in leaves of the C4 plant Sorghum bicolor exposed to water stress, temperature changes and carbon metabolism inhibition. Aust. J. Plant Physiol. 1995, 22, 885–892. [Google Scholar] [CrossRef]
- Ribas-Carbo, M.; Taylor, N.L.; Giles, L.V.; Busquets, S.; Finnegan, P.M.; Day, D.A. Effects of water stress on respiration in soybean leaves. Plant Physiol. 2005, 139, 466–473. [Google Scholar] [CrossRef]
- Stacey, G.; Libault, M.; Brechenmacher, L.; Wan, J.; May, G.D. Genetics and functional genomics of legume nodulation. Curr. Opin. Plant Biol. 2006, 9, 110–121. [Google Scholar] [CrossRef]
- Nouri, M.Z.; Hiraga, S.; Yanagawa, Y.; Sunohara, Y.; Matsumoto, H.; Komatsu, S. Characterization of calnexin in soybean roots and hypocotyls under osmotic stress. Phytochemistry 2012, 74, 20–29. [Google Scholar] [CrossRef]
- Brockmeier, A.; Williams, D.B. Potent lectin-independent chaperone function of calnexin under conditions prevalent within the lumen of the endoplasmic reticulum. Biochemistry 2006, 45, 12906–12916. [Google Scholar] [CrossRef]
- Smaczniak, C.; Li, N.; Boeren, S.; America, T.; van Dongen, W.; Goerdayal, S.S.; de Vries, S.; Angenent, G.C.; Kaufmann, K. Proteomics-based identification of low-abundance signaling and regulatory protein complexes in native plant tissues. Nat. Protoc. 2012, 7, 2144–2158. [Google Scholar] [CrossRef]
- Dembinsky, D.; Woll, K.; Saleem, M.; Liu, Y.; Fu, Y.; Borsuk, L.A.; Lamkemeyer, T.; Fladerer, C.; Madlung, J.; Barbazuk, B.; et al. Transcriptomic and proteomic analyses of pericycle cells of the maize primary root. Plant Physiol. 2007, 145, 575–588. [Google Scholar] [CrossRef]
- Gil-Quintana, E.; Larrainzar, E.; Seminario, A.; Díaz-Leal, J.L.; Alamillo, J.M.; Pineda, M.; Arrese-Igor, C.; Wienkoop, S.; González, E.M. Local inhibition of nitrogen fixation and nodule metabolism in drought-stressed soybean. J. Exp. Bot. 2013, 64, 2171–2182. [Google Scholar] [CrossRef]
- Larrainzar, E.; Wienkoop, S.; Weckwerth, W.; Ladrera, R.; Arrese-Igor, C.; González, E.M. Medicago truncatula root nodule proteome analysis reveals differential plant and bacteroid responses to drought stress. Plant Physiol. 2007, 144, 1495–1507. [Google Scholar] [CrossRef]
- Herman, E.M.; Helm, R.M.; Jung, R.; Kinney, A.J. Genetic modification removes an immunodominant allergen from soybean. Plant Physiol. 2003, 132, 36–43. [Google Scholar] [CrossRef]
- Cho, J.H.; Hwang, H.; Cho, M.H.; Kwon, Y.K.; Jeon, J.S.; Bhoo, S.H.; Hahn, T.R. The effect of DTT in protein preparations for proteomic analysis: Removal of a highly abundant plant enzyme, ribulose bisphosphate carboxylase/oxygenase. J. Plant Biol. 2008, 51, 297–301. [Google Scholar] [CrossRef]
- Widjaja, I.; Naumann, K.; Roth, U.; Wolf, N.; Mackey, D.; Dangl, J.L.; Scheel, D.; Lee, J. Combining subproteome enrichment and Rubisco depletion enables identification of low abundance proteins differentially regulated during plant defense. Proteomics 2009, 9, 138–147. [Google Scholar] [CrossRef]
- Ahsan, N.; Lee, D.G.; Lee, S.H.; Kang, K.Y.; Bahk, J.D.; Choi, M.S.; Lee, I.J.; Renaut, J.; Lee, B.H. A comparative proteomic analysis of tomato leaves in response to waterlogging stress. Physiol. Plant 2007, 131, 555–570. [Google Scholar] [CrossRef]
- Hashimoto, M.; Komatsu, S. Proteomic analysis of rice seedlings during cold stress. Proteomics 2007, 7, 1293–1302. [Google Scholar] [CrossRef]
- Krishnan, H.B.; Natarajan, S.S. A rapid method for depletion of Rubisco from soybean (Glycine max) leaf for proteomic analysis of lower abundance proteins. Phytochemistry 2009, 70, 1958–1964. [Google Scholar] [CrossRef]
- Khan, N.A.; Komatsu, S.; Sawada, H.; Nouri, M.Z.; Kohno, Y. Analysis of proteins associated with ozone stress response in soybean cultivars. Protein Pept. Lett. 2013, 20, 1144–1152. [Google Scholar] [CrossRef]
- Lee, Y.H.; Tan, H.T.; Chung, M.C. Subcellular fractionation methods and strategies for proteomics. Proteomics 2010, 10, 3935–3956. [Google Scholar] [CrossRef]
- Ackermann, B.L.; Berna, M.J. Coupling immunoaffinity techniques with MS for quantitative analysis of low abundance protein biomarkers. Exp. Rev. Proteomics 2007, 4, 175–186. [Google Scholar] [CrossRef]
- Cossarizza, A.; Ceccarelli, D.; Masini, A. Functional heterogeneity of an isolated mitochondrial population revealed by cytofluorometric analysis at the single organelle level. Exp. Cell Res. 1996, 222, 84–94. [Google Scholar] [CrossRef]
- Cao, Z.; Li, C.; Higginbotham, J.N.; Franklin, J.L.; Tabb, D.L.; Graves-Deal, R.; Hill, S.; Cheek, K.; Jerome, W.G.; Lapierre, L.A.; et al. se of fluorescence-activated vesicle sorting for isolation of Naked2-associated, basolaterally targeted exocytic vesicles for proteomics analysis. Mol. Cell. Proteomics 2008, 7, 1651–1667. [Google Scholar] [CrossRef]
- Zhou, Z.; Licklider, L.J.; Gygi, S.P.; Reed, R. Comprehensive proteomic analysis of the human spliceosome. Nature 2002, 419, 182–185. [Google Scholar] [CrossRef]
- Matros, A.; Kaspar, S.; Witzel, K.; Mock, H.P. Recent progress in liquid chromatography-based separation and label-free quantitative plantproteomics. Phytochemistry 2011, 72, 963–974. [Google Scholar] [CrossRef]
- Bentem, S.; Roitinger, E.; Anrather, D.; Csaszar, E.; Hirt, H. Phosphoproteomics as a tool to unravel plant regulatory mechanisms. Physiol. Plant. 2006, 126, 110–119. [Google Scholar] [CrossRef]
- Ballif, B.A.; Villen, J.; Beausoleil, S.A.; Schwartz, D.; Gygi, S.P. Phosphoproteomic analysis of the developing mouse brain. Mol. Cell Proteomics 2004, 3, 1093–1101. [Google Scholar] [CrossRef]
- Nuhse, T.S.; Stensballe, A.; Jensen, O.N.; Peck, S.C. Large scale analysis of in vivo phosphorylated membrane proteins by immobilized metal ion affinity chromatography and mass spectrometry. Mol. Cell Proteomics 2003, 2, 1234–1243. [Google Scholar] [CrossRef]
- Nuhse, T.S.; Stensballe, A.; Jensen, O.N.; Peck, S.C. Phosphoproteomics of the Arabidopsis plasma membrane and a new phosphorylation site database. Plant Cell 2004, 16, 2394–2405. [Google Scholar] [CrossRef]
- Hsu, J.L.; Wang, L.Y.; Wang, S.Y.; Lin, C.H.; Ho, K.C.; Shi, F.K.; Chang, I.F. Functional phosphoproteomic profiling of phosphorylation sites in membrane fractions of salt-stressed Arabidopsis thaliana. Proteome Sci. 2009, 7, 1–16. [Google Scholar] [CrossRef]
- Navrot, N.; Finnie, C.; Svensson, B.; Hägglund, P. Plant redox proteomics. J. Proteomics 2011, 74, 1450–1462. [Google Scholar] [CrossRef]
- Dietz, K.J.; Pfannschmidt, T. Novel regulators in photosynthetic redox control of plant metabolism and gene expression. Plant Physiol. 2011, 155, 1477–1485. [Google Scholar] [CrossRef]
- Bykova, N.V.; Rampitsch, C. Modulating protein function through reversible oxidation: Redox-mediated processes in plants revealed through proteomics. Proteomics 2013, 13, 579–596. [Google Scholar] [CrossRef]
- Braconi, D.; Bernardini, G.; Santucci, A. Linking protein oxidation to environmental pollutants: redox proteomic approaches. J. Proteomics 2011, 74, 2324–2337. [Google Scholar] [CrossRef]
- Hu, W.; Tedesco, S.; McDonagh, B.; Sheehan, D. Shotgun redox proteomics in sub-proteomes trapped on functionalized beads: Identification of proteins targeted by oxidative stress. Mar. Environ. Res. 2010, 69, S25–S27. [Google Scholar] [CrossRef]
- Madian, A.G.; Regnier, F.E. Proteomic identification of carbonylated proteins and their oxidation sites. J. Proteome Res. 2010, 9, 3766–3780. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Sobkowiak, R.; Deckert, J. Proteins induced by cadmium in soybean cells. J. Plant Physiol. 2006, 163, 1203–1206. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hossain, Z.; Komatsu, S. Potentiality of Soybean Proteomics in Untying the Mechanism of Flood and Drought Stress Tolerance. Proteomes 2014, 2, 107-127. https://doi.org/10.3390/proteomes2010107
Hossain Z, Komatsu S. Potentiality of Soybean Proteomics in Untying the Mechanism of Flood and Drought Stress Tolerance. Proteomes. 2014; 2(1):107-127. https://doi.org/10.3390/proteomes2010107
Chicago/Turabian StyleHossain, Zahed, and Setsuko Komatsu. 2014. "Potentiality of Soybean Proteomics in Untying the Mechanism of Flood and Drought Stress Tolerance" Proteomes 2, no. 1: 107-127. https://doi.org/10.3390/proteomes2010107
APA StyleHossain, Z., & Komatsu, S. (2014). Potentiality of Soybean Proteomics in Untying the Mechanism of Flood and Drought Stress Tolerance. Proteomes, 2(1), 107-127. https://doi.org/10.3390/proteomes2010107