

Analysis of p53-Independent Functions of the Mdm2-MdmX Complex Using Data-Independent Acquisition-Based Profiling

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
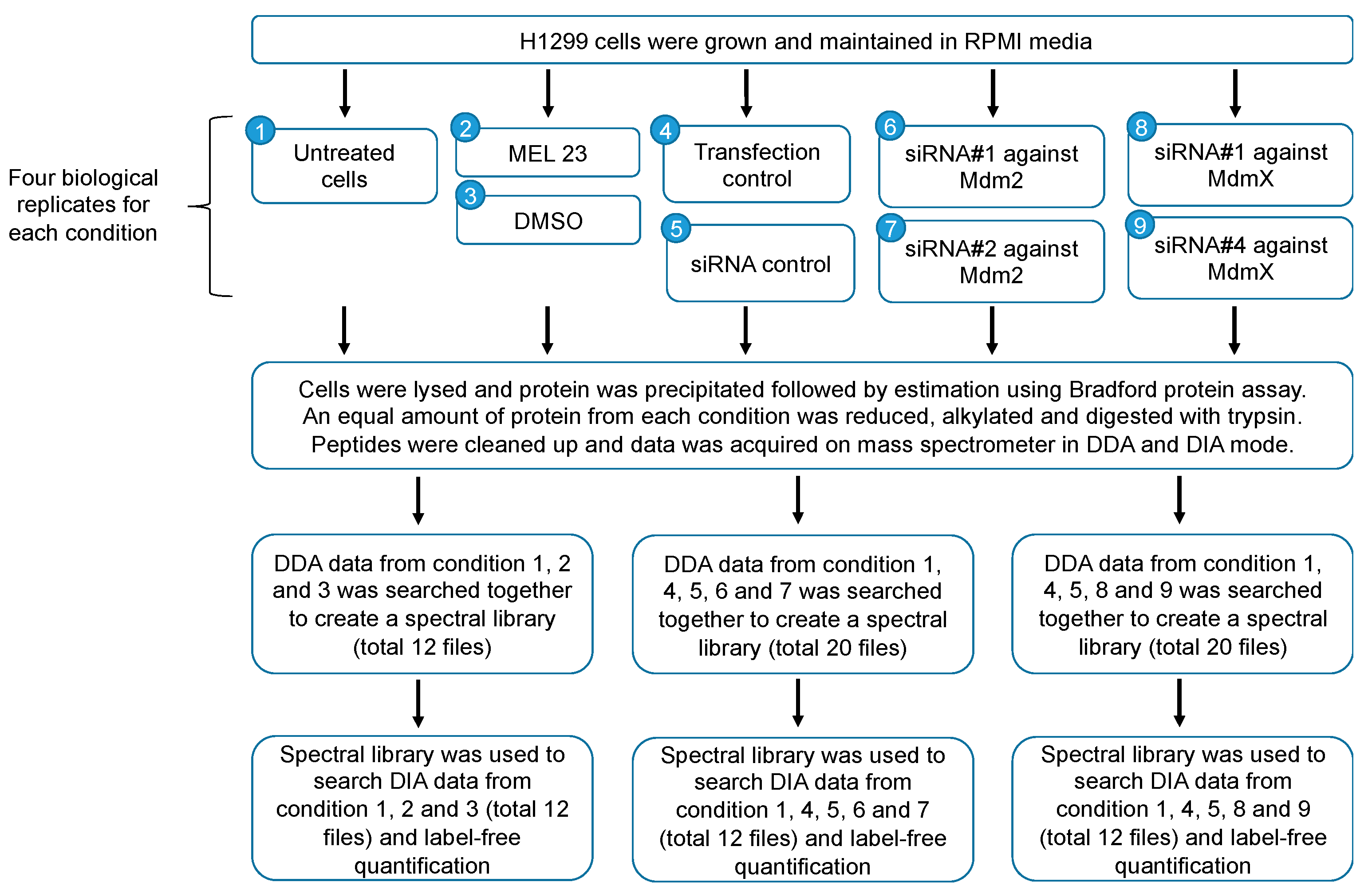
2.2. Study Design
2.3. Cell Culture
2.4. siRNA Transfection and Drug Treatment
2.5. Sample Preparation for Proteomic Analysis, Mass Spectrometry, and Data Analysis
2.6. Mass Spectral Libraries
2.7. Data-Independent Acquisition
2.8. Mass Spectrometry Data Analysis
2.9. Post Hoc Statistical Analysis
2.10. Gene Ontology and Biological Inference
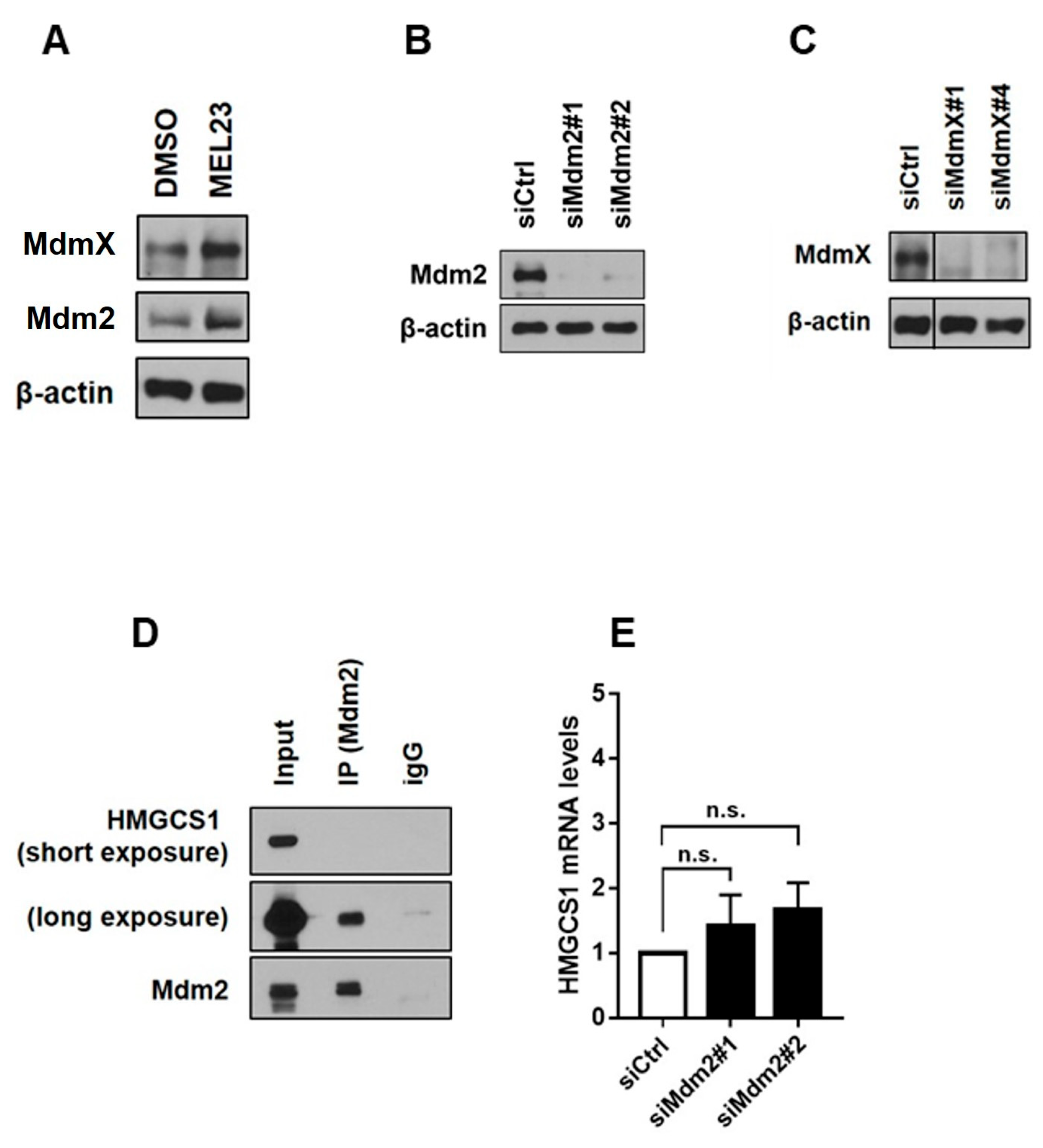
2.11. Immunoblotting
2.12. Co-Immunoprecipitation
2.13. Quantitative PCR
3. Results
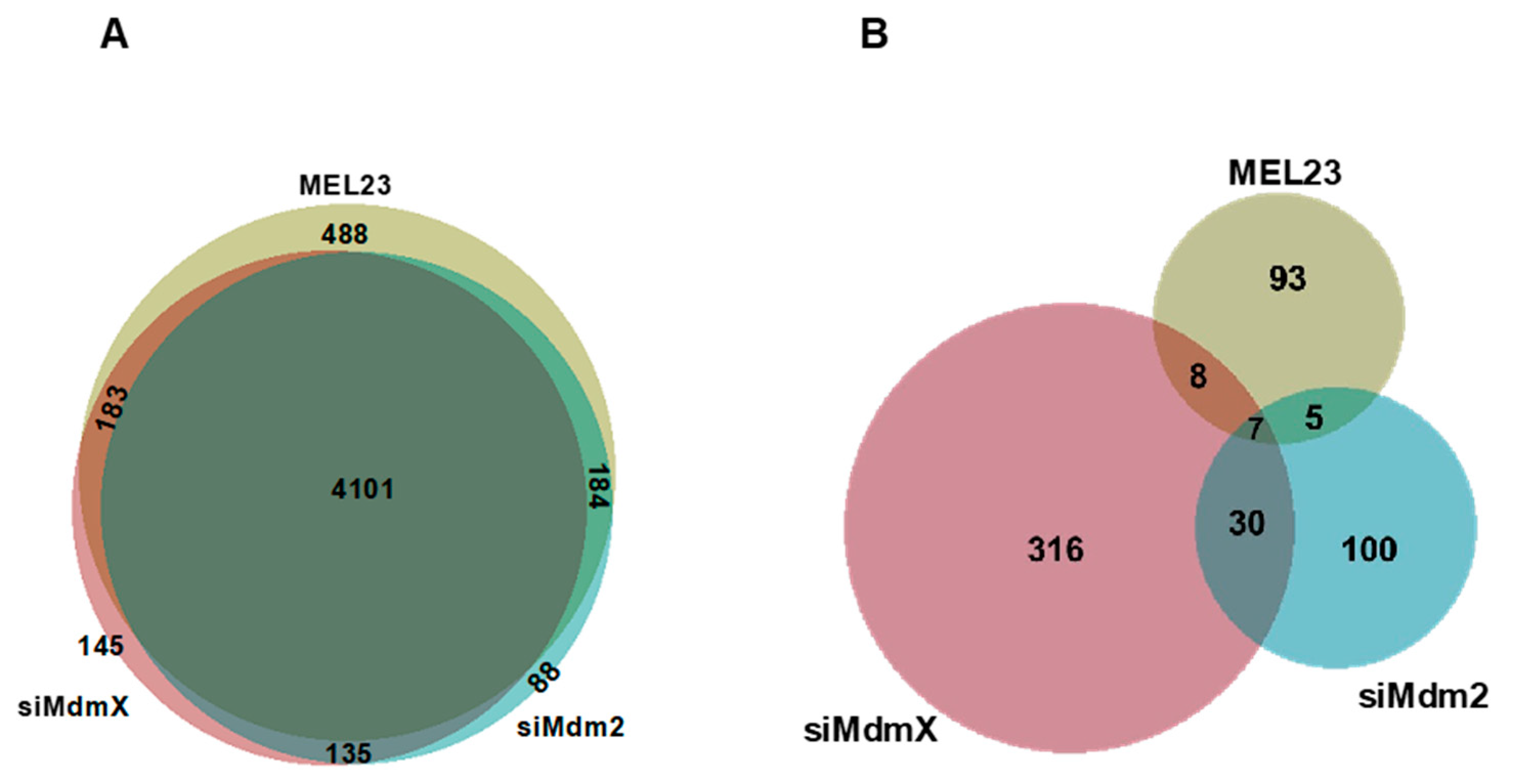
3.1. Biological Proteomics Results: H1299 Cells with Modulated Mdm2/MdmX Expression or Activity
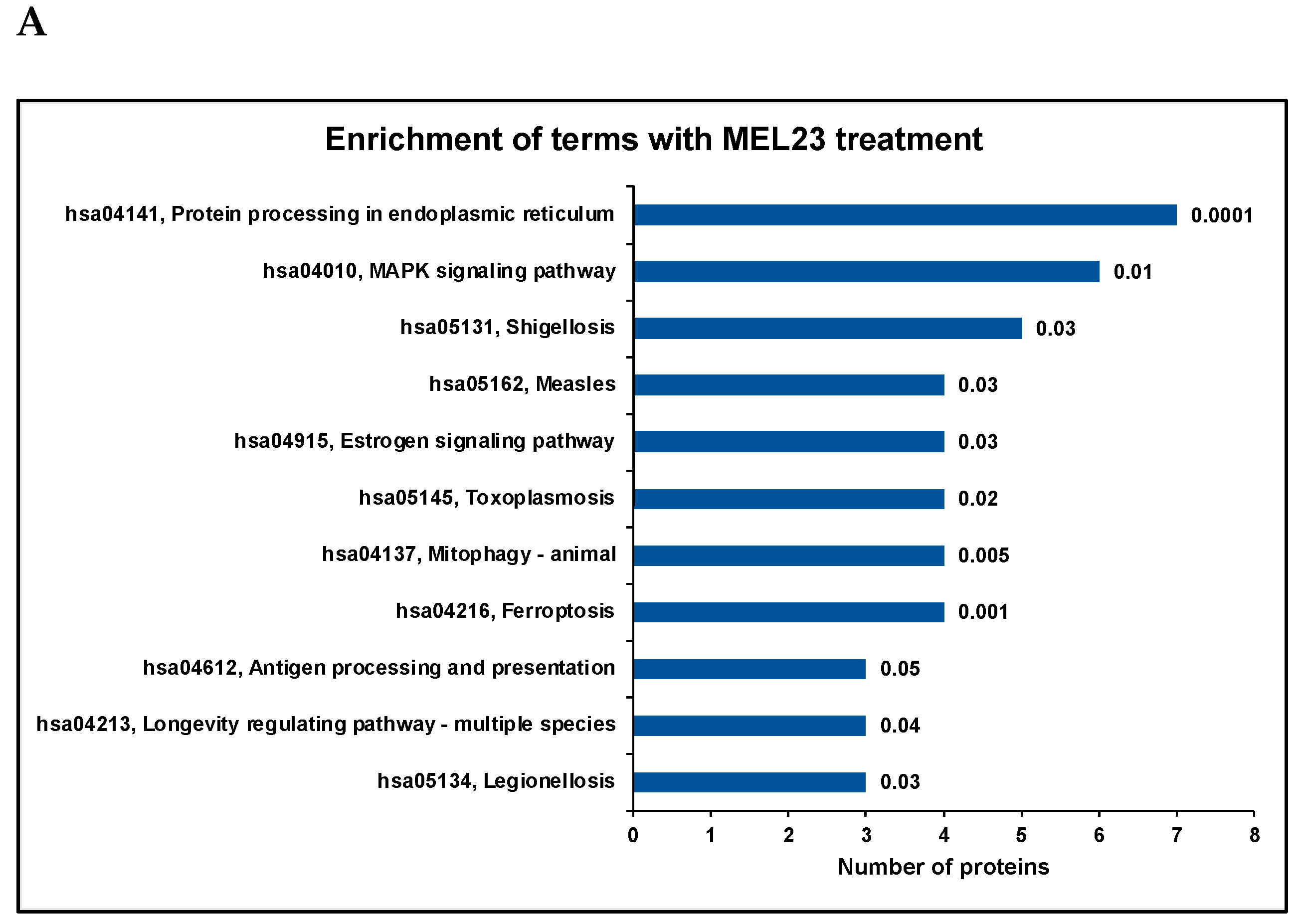
3.2. Gene Ontology and Pathway Enrichment Analysis
3.3. Differential Protein Abundance and Discovery of a New Mdm2 Binding Partner
3.4. Proteoform Listing
4. Discussion
4.1. Limitations of This Study
4.2. Summary
4.3. Future Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valente, L.J.; Tarangelo, A.; Li, A.M.; Naciri, M.; Raj, N.; Boutelle, A.M.; Li, Y.; Mello, S.S.; Bieging-Rolett, K.; DeBerardinis, R.J.; et al. p53 deficiency triggers dysregulation of diverse cellular processes in physiological oxygen. J. Cell Biol. 2020, 219, e201908212. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef]
- Gnanapradeepan, K.; Basu, S.; Barnoud, T.; Budina-Kolomets, A.; Kung, C.P.; Murphy, M.E. The p53 Tumor Suppressor in the Control of Metabolism and Ferroptosis. Front. Endocrinol. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer—P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Vousden, K.H. p53: Death star. Cell 2000, 103, 691–694. [Google Scholar] [CrossRef]
- Soussi, T. The TP53 Website. Available online: https://p53.fr/ (accessed on 27 February 2025).
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gvuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein Mdm2 Conceals the Activation Domain of Tumor Suppressor-P53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The Mdm-2 Oncogene Product Forms a Complex with the P53 Protein and Inhibits P53-Mediated Transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Alarcon-Vargas, D.; Ronai, Z. p53-Mdm2—The affair that never ends. Carcinogenesis 2002, 23, 541–547. [Google Scholar] [CrossRef]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88–99. [Google Scholar] [CrossRef]
- Klein, A.M.; de Queiroz, R.M.; Venkatesh, D.; Prives, C. The roles and regulation of MDM2 and MDMX: It is not just about p53. Gene Dev. 2021, 35, 575–601. [Google Scholar] [CrossRef]
- Leroy, B.; Anderson, M.; Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Hum. Mutat. 2014, 35, 672–688. [Google Scholar] [CrossRef]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol-Mech. 2016, 11, 617–644. [Google Scholar] [CrossRef]
- Herman, A.G.; Hayano, M.; Poyurovsky, M.V.; Shimada, K.; Skouta, R.; Prives, C.; Stockwell, B.R. Discovery of Mdm2-MdmX E3 Ligase Inhibitors Using a Cell-Based Ubiquitination Assay. Cancer Discov. 2011, 1, 312–325. [Google Scholar] [CrossRef]
- Hansen, F.M.; Tanzer, M.C.; Bruning, F.; Bludau, I.; Stafford, C.; Schulman, B.A.; Robles, M.S.; Karayel, O.; Mann, M. Data-independent acquisition method for ubiquitinome analysis reveals regulation of circadian biology. Nat. Commun. 2021, 12, 254. [Google Scholar] [CrossRef]
- Krasny, L.; Huang, P.H. Data-independent acquisition mass spectrometry (DIA-MS) for proteomic applications in oncology. Mol. Omics 2021, 17, 29–42. [Google Scholar] [CrossRef]
- Li, K.W.; Gonzalez-Lozano, M.A.; Koopmans, F.; Smit, A.B. Recent Developments in Data Independent Acquisition (DIA) Mass Spectrometry: Application of Quantitative Analysis of the Brain Proteome. Front. Mol. Neurosci. 2020, 13, 564446. [Google Scholar] [CrossRef]
- Ludwig, C.; Gillet, L.; Rosenberger, G.; Amon, S.; Collins, B.; Aebersold, R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2018, 14, e8126. [Google Scholar] [CrossRef]
- Ma, B.; Zhang, K.Z.; Hendrie, C.; Liang, C.Z.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide sequencing by tandem mass spectrometry. Rapid Commun. Mass. Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef]
- Zhang, J.; Xin, L.; Shan, B.Z.; Chen, W.W.; Xie, M.J.; Yuen, D.; Zhang, W.M.; Zhang, Z.F.; Lajoie, G.A.; Ma, B. PEAKS DB: Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Mol. Cell. Proteom. 2012, 11, M111.010587. [Google Scholar] [CrossRef] [PubMed]
- Seddighi, S.; Qi, Y.A.; Brown, A.L.; Wilkins, O.G.; Bereda, C.; Belair, C.; Zhang, Y.J.; Prudencio, M.; Keuss, M.J.; Khandeshi, A.; et al. Mis-spliced transcripts generate de novo proteins in TDP-43-related ALS/FTD. Sci. Transl. Med. 2024, 16, eadg7162. [Google Scholar] [CrossRef] [PubMed]
- Abid, M.S.R.; Bredahl, E.C.; Clifton, A.D.; Qiu, H.W.; Andrews, M.T.; Checco, J.W. Proteomic Identification of Seasonally Expressed Proteins Contributing to Heart Function and the Avoidance of Skeletal Muscle Disuse Atrophy in a Hibernating Mammal. J. Proteome Res. 2023, 23, 215–225. [Google Scholar] [CrossRef]
- Trithavisup, T.; Krobthong, S.; Yingehutrakul, Y.; Sanpinit, P.; Malila, Y. Impact of Wooden Breast myopathy on protein digestibility, metabolomic profile, and cell cytotoxicity of cooked chicken breast meat. Poult. Sci. 2024, 103, 103261. [Google Scholar] [CrossRef]
- Mays, S.; Parker, G.; Johns, C.; Stark, S.; Young, A.J.; Reich, D.; Buikstra, J.; Sawyer, K.; Hale, K. Sex identification of a Late Iron Age sword and mirror cist burial from Hillside Farm, Bryher, Isles of Scilly, England. J. Archaeol. Sci.-Rep. 2023, 52, 104099. [Google Scholar] [CrossRef]
- Demeuse, J.; Massonnet, P.; Schoumacher, M.; Grifnée, E.; Huyghebaert, L.; Dubrowski, T.; Peeters, S.; Le Goff, C.; Cavalier, E. Innovative workflow for the identification of cathepsin K cleavage sites in type I collagen. J. Chromatogr. B 2023, 1228, 123864. [Google Scholar] [CrossRef]
- Osorio, J.; Villa-Arias, S.; Camargo, C.; Ramírez-Sánchez, L.F.; Barrientos, L.M.; Bedoya, C.; Rúa-Uribe, G.; Dorus, S.; Alfonso-Parra, C.; Avila, F.W. wMel alters female post-mating behaviors and physiology in the dengue vector mosquito. Commun. Biol. 2023, 6, 865. [Google Scholar] [CrossRef]
- Azizov, V.; Hubner, M.; Frech, M.; Hofmann, J.; Kubankova, M.; Lapuente, D.; Tenbusch, M.; Guck, J.; Schett, G.; Zaiss, M.M. Alcohol-sourced acetate impairs T cell function by promoting cortactin acetylation. Iscience 2023, 26, 107230. [Google Scholar] [CrossRef]
- Neves, L.X.; Wilson, R.A.; Brownridge, P.; Holman, S.W.; Harman, V.M.; Eyers, C.E.; Beynon, R.J.; Castro-Borges, W. Dissection of schistosome tissues under LC-MS compatible preservative conditions for quantitative proteomics. Rapid Commun. Mass. Spectrom. 2023, 38, e9523. [Google Scholar] [CrossRef]
- Nishimura, T.; Végvari, A.; Nakamura, H.; Kato, H.; Saji, H. Mutant Proteomics of Lung Adenocarcinomas Harboring Different Mutations. Front. Oncol. 2020, 10, 1494. [Google Scholar] [CrossRef]
- Chen, B.; Foo, J.L.; Ling, H.; Chang, M.W. Mechanism-Driven Metabolic Engineering for Bio-Based Production of Free R-Lipoic Acid in Saccharomyces cerevisiae Mitochondria. Front. Bioeng. Biotechnol. 2020, 8, 965. [Google Scholar] [CrossRef]
- Nuevo-Tapioles, C.; Santacatterina, F.; Stamatakis, K.; de Arenas, C.N.; de Cedrón, M.G.; Formentini, L.; Cuezva, J.M. Coordinate β-adrenergic inhibition of mitochondrial activity and angiogenesis arrest tumor growth. Nat. Commun. 2020, 11, 3606. [Google Scholar] [CrossRef] [PubMed]
- Dalla, P.V.; Santos, J.; Milthorpe, B.K.; Padula, M.P. Selectively-Packaged Proteins in Breast Cancer Extracellular Vesicles Involved in Metastasis. Int. J. Mol. Sci. 2020, 21, 4990. [Google Scholar] [CrossRef]
- Kaddour, H.; Lyu, Y.; Welch, J.L.; Paromov, V.; Mandape, S.N.; Sakhare, S.S.; Pandhare, J.; Stapleton, J.T.; Pratap, S.; Dash, C.; et al. Proteomics Profiling of Autologous Blood and Semen Exosomes from HIV-infected and Uninfected Individuals Reveals Compositional and Functional Variabilities. Mol. Cell. Proteom. 2020, 19, 78–100. [Google Scholar] [CrossRef]
- Macheroni, C.; Leite, G.G.F.; Souza, D.S.; Vicente, C.M.; Lacerda, J.T.; Moraes, M.N.; Julianoc, M.A.; Porto, C.S. Activation of estrogen receptor induces differential proteomic responses mainly involving migration, invasion, and tumor development pathways in human testicular embryonal carcinoma NT2/D1 cells. J. Steroid Biochem. 2024, 237, 106443. [Google Scholar] [CrossRef]
- Lane, S.; White, T.L.A.; Walsh, E.E.; Cattley, R.T.; Cumberland, R.; Hawse, W.F.; Delgoffe, G.M.; Badylak, S.F.; Bomberger, J.M. Antiviral epithelial-macrophage crosstalk permits secondary bacterial infections. Mbio 2023, 14, e0086323. [Google Scholar] [CrossRef]
- Lesage, A.; Lorenzini, M.; Burel, S.; Sarlandie, M.; Bibault, F.; Lindskog, C.; Maloney, D.; Silva, J.R.; Townsend, R.R.; Nerbonne, J.M.; et al. Determinants of iFGF13-mediated regulation of myocardial voltage-gated sodium (Na) channels in mouse. J. Gen. Physiol. 2023, 155, e202213293. [Google Scholar] [CrossRef]
- Xin, L.; Qiao, R.; Chen, X.; Tran, H.; Pan, S.Y.; Rabinoviz, S.; Bian, H.B.; He, X.L.; Morse, B.; Shan, B.Z.; et al. A streamlined platform for analyzing tera-scale DDA and DIA mass spectrometry data enables highly sensitive immunopeptidomics. Nat. Commun. 2022, 13, 3108. [Google Scholar] [CrossRef]
- Nishiyama, M.J.r.; Tashima, A.K.; Nishiduka, E.S.; Abreu, T.F.; Abukawa, F.M.; Oliveira, U.C.; Tardivo, C.E.O.; Nascimento, S.M.; Meissner, G.O.; Chaim, O.M.; et al. Multiomics Profiling of Toxins in the Venom of the Amazonian Spider Acanthoscurria juruenicola. J. Proteome Res. 2022, 21, 2783–2797. [Google Scholar] [CrossRef]
- Makepeace, K.A.T.; Rookyard, A.W.; Das, L.; Vardarajan, B.N.; Chakrabarty, J.K.; Jain, A.; Kang, M.S.; Werth, E.G.; Reyes-Dumeyer, D.; Zerlin-Esteves, M.; et al. Data-Independent Acquisition and Label-Free Quantification for Quantitative Proteomics Analysis of Human Cerebrospinal Fluid. Curr. Protoc. 2024, 4, e1014. [Google Scholar] [CrossRef]
- Rookyard, A.W.; Chakrabarty, J.K.; Jain, A.; Vardarajan, B.N.; Kang, M.S.; Das, L.; Werth, E.G.; Zerlin-Esteves, M.; Honig, L.S.; Brown, L.M.; et al. Preliminary analyses from a planned cohort of 1,000 individuals: Cerebrospinal fluid proteomics from a multi-omic study of Alzheimer’s disease. In Proceedings of the 71st Annual ASMS Conference on Mass Spectrometry and Allied Topics, American Society for Mass Spectrometry, Houston, TX, USA, 4–8 June 2023. [Google Scholar]
- Jia, W.; Peng, J.; Zhang, Y.; Zhu, J.; Qiang, X.; Zhang, R.; Shi, L. Exploring novel ANGICon-EIPs through ameliorated peptidomics techniques: Can deep learning strategies as a core breakthrough in peptide structure and function prediction? Food Res. Int. 2023, 174 Pt 1, 113640. [Google Scholar] [CrossRef]
- Shahbazy, M.; Ramarathinam, S.H.; Illing, P.T.; Jappe, E.C.; Faridi, P.; Croft, N.P.; Purcell, A.W. Benchmarking Bioinformatics Pipelines in Data-Independent Acquisition Mass Spectrometry for Immunopeptidomics. Mol. Cell Proteom. 2023, 22, 100515. [Google Scholar] [CrossRef] [PubMed]
- Wobma, H.M.; Tamargo, M.A.; Goeta, S.; Brown, L.M.; Duran-Struuck, R.; Vunjak-Novakovic, G. The influence of hypoxia and IFN-gamma on the proteome and metabolome of therapeutic mesenchymal stem cells. Biomaterials 2018, 167, 226–234. [Google Scholar] [CrossRef]
- Werth, E.G.; Rajbhandari, P.; Stockwell, B.R.; Brown, L.M. Time Course of Changes in Sorafenib-Treated Hepatocellular Carcinoma Cells Suggests Involvement of Phospho-Regulated Signaling in Ferroptosis Induction. Proteomics 2020, 20, e2000006. [Google Scholar] [CrossRef]
- Mi, H.Y.; Muruganujan, A.; Huang, X.S.; Ebert, D.; Mills, C.; Guo, X.Y.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.L.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W.Z. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Qing, J.; Zhang, Y.; Derynck, R. Structural and functional characterization of the transforming growth factor-beta -induced Smad3/c-Jun transcriptional cooperativity. J. Biol. Chem. 2000, 275, 38802–38812. [Google Scholar] [CrossRef]
- Jo, U.; Cai, W.; Wang, J.; Kwon, Y.; D’Andrea, A.D.; Kim, H. PCNA-Dependent Cleavage and Degradation of SDE2 Regulates Response to Replication Stress. PLOS Genet. 2016, 12, e1006465. [Google Scholar] [CrossRef]
- Kato, T., Jr.; Gotoh, Y.; Hoffmann, A.; Ono, Y. Negative regulation of constitutive NF-kappaB and JNK signaling by PKN1-mediated phosphorylation of TRAF1. Genes. Cells 2008, 13, 509–520. [Google Scholar] [CrossRef]
- Carbone, M.; Melino, G. Stearoyl CoA Desaturase Regulates Ferroptosis in Ovarian Cancer Offering New Therapeutic Perspectives. Cancer Res. 2019, 79, 5149–5150. [Google Scholar] [CrossRef]
- Paton, C.M.; Ntambi, J.M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol.-Endocrinol. Metab. 2009, 297, E28–E37. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Yan, W.; Chen, X. RNPC1, an RNA-binding protein and a target of the p53 family, is required for maintaining the stability of the basal and stress-induced p21 transcript. Gene Dev. 2006, 20, 2961–2972. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Hidaka, K.; Shirai, M.; Morisaki, T. RNA-binding motif protein 24 regulates myogenin expression and promotes myogenic differentiation. Genes. Cells 2010, 15, 1158–1167. [Google Scholar] [CrossRef]
- Xu, E.; Zhang, J.; Zhang, M.; Jiang, Y.; Cho, S.J.; Chen, X. RNA-binding protein RBM24 regulates p63 expression via mRNA stability. Mol. Cancer Res. 2014, 12, 359–369. [Google Scholar] [CrossRef]
- Park, J.Y.; Yoo, H.W.; Kim, B.R.; Park, R.; Choi, S.Y.; Kim, Y. Identification of a novel human Rad51 variant that promotes DNA strand exchange. Nucleic Acids Res. 2008, 36, 3226–3234. [Google Scholar] [CrossRef]
- Shimada, K.; Skouta, R.; Kaplan, A.; Yang, W.S.; Hayano, M.; Dixon, S.J.; Brown, L.M.; Valenzuela, C.A.; Wolpaw, A.J.; Stockwell, B.R. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 2016, 12, 497–503. [Google Scholar] [CrossRef]
- Kitata, R.B.; Dimayacyac-Esleta, B.R.; Choong, W.K.; Tsai, C.F.; Lin, T.D.; Tsou, C.C.; Weng, S.H.; Chen, Y.J.; Yang, P.C.; Arco, S.D.; et al. Mining Missing Membrane Proteins by High-pH Reverse Phase StageTip Fractionation and Multiple Reaction Monitoring Mass Spectrometry. J. Proteome Res. 2015, 14, 3658–3669. [Google Scholar] [CrossRef]
- Guo, Z.; Yuan, J.; Tang, W.; Chen, X.; Gu, X.; Luo, K.; Wang, Y.; Wan, B.; Yu, L. Cloning and characterization of the human gene RAP2C, a novel member of Ras family, which activates transcriptional activities of SRE. Mol. Biol. Rep. 2007, 34, 137–144. [Google Scholar] [CrossRef]
- Nakayama, K.; Nakayama, N.; Davidson, B.; Sheu, J.J.; Jinawath, N.; Santillan, A.; Salani, R.; Bristow, R.E.; Morin, P.J.; Kurman, R.J.; et al. A BTB/POZ protein, NAC-1, is related to tumor recurrence and is essential for tumor growth and survival. Proc. Natl. Acad. Sci. USA 2006, 103, 18739–18744. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, C.; Xing, G.; Chen, Q.; He, F. Functional characterization of novel human ARFGAP3. FEBS Lett. 2001, 490, 79–83. [Google Scholar] [CrossRef]
- Royle, S.J.; Bright, N.A.; Lagnado, L. Clathrin is required for the function of the mitotic spindle. Nature 2005, 434, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Jani, D.; Lutz, S.; Hurt, E.; Laskey, R.A.; Stewart, M.; Wickramasinghe, V.O. Functional and structural characterization of the mammalian TREX-2 complex that links transcription with nuclear messenger RNA export. Nucleic Acids Res. 2012, 40, 4562–4573. [Google Scholar] [CrossRef]
- Ni, C.; Schmitz, D.A.; Lee, J.; Pawlowski, K.; Wu, J.; Buszczak, M. Labeling of heterochronic ribosomes reveals C1ORF109 and SPATA5 control a late step in human ribosome assembly. Cell Rep. 2022, 38, 110597. [Google Scholar] [CrossRef]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.T.; Young, J.T.F.; Tkác, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef]
- Martinho, M.S.; Nancarrow, D.J.; Lawrence, T.S.; Beer, D.G.; Ray, D. Chaperones and Ubiquitin Ligases Balance Mutant p53 Protein Stability in Esophageal and Other Digestive Cancers. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 449–464. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233. [Google Scholar] [CrossRef]
- Schilling, D.; Duwel, M.; Molls, M.; Multhoff, G. Radiosensitization of wildtype p53 cancer cells by the MDM2-inhibitor PXN727 is associated with altered heat shock protein 70 (Hsp70) levels. Cell Stress. Chaperones 2013, 18, 183–191. [Google Scholar] [CrossRef]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular Mechanism of Mutant p53 Stabilization: The Role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef]
- Kook, S.; Zhan, X.Z.; Thibeault, K.; Ahmed, M.R.; Gurevich, V.V.; Gurevich, E.V. Mdm2 enhances ligase activity of parkin and facilitates mitophagy. Sci. Rep. 2020, 10, 5028. [Google Scholar] [CrossRef]
- Klein, A.M.; Biderman, L.; Tong, D.; Alaghebandan, B.; Plumber, S.A.; Mueller, H.S.; van Vlimmeren, A.; Katz, C.; Prives, C. MDM2, MDMX, and p73 regulate cell-cycle progression in the absence of wild-type p53. Proc. Natl. Acad. Sci. USA 2021, 118, e2102420118. [Google Scholar] [CrossRef]
- Riscal, R.; Schrepfer, E.; Arena, G.; Cisse, M.Y.; Bellvert, F.; Heuillet, M.; Rambow, F.; Bonneil, E.; Sabourdy, F.; Vincent, C.; et al. Chromatin-Bound MDM2 Regulates Serine Metabolism and Redox Homeostasis Independently of p53. Mol. Cell 2016, 62, 890–902. [Google Scholar] [CrossRef] [PubMed]
- Wienken, M.; Dickmanns, A.; Nemajerova, A.; Kramer, D.; Najafova, Z.; Weiss, M.; Karpiuk, O.; Kassem, M.; Zhang, Y.P.; Lozano, G.; et al. MDM2 Associates with Polycomb Repressor Complex 2 and Enhances Stemness-Promoting Chromatin Modifications Independent of p53. Mol. Cell 2016, 61, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Wienken, M.; Moll, U.M.; Dobbelstein, M. Mdm2 as a chromatin modifier. J. Mol. Cell Biol. 2017, 9, 74–80. [Google Scholar] [CrossRef]
- Klusmann, I.; Wohlberedt, K.; Magerhans, A.; Teloni, F.; Korbel, J.O.; Altmeyer, M.; Dobbelstein, M. Chromatin modifiers Mdm2 and RNF2 prevent RNA: DNA hybrids that impair DNA replication. Proc. Natl. Acad. Sci. USA 2018, 115, E11311–E11320. [Google Scholar] [CrossRef]
- Saadatzadeh, M.R.; Elmi, A.N.; Pandya, P.H.; Bijangi-Vishehsaraei, K.; Ding, J.X.; Stamatkin, C.W.; Cohen-Gadol, A.A.; Pollok, K.E. The Role of MDM2 in Promoting Genome Stability versus Instability. Int. J. Mol. Sci. 2017, 18, 2216. [Google Scholar] [CrossRef]
- Bouska, A.; Eischen, C.M. Mdm2 Affects Genome Stability Independent of p53. Cancer Res. 2009, 69, 1697–1701. [Google Scholar] [CrossRef]
- Lehman, J.A.; Mayo, L.D. Integration of DNA Damage and Repair with Murine Double-Minute 2 (Mdm2) in Tumorigenesis. Int. J. Mol. Sci. 2012, 13, 16373–16386. [Google Scholar] [CrossRef]
- Min, X.; Zhang, X.; Sun, N.; Acharya, S.; Kim, K.M. Mdm2-mediated ubiquitination of PKCbetaII in the nucleus mediates clathrin-mediated endocytic activity. Biochem. Pharmacol. 2019, 170, 113675. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, M.; Kim, K.M. GRK2-mediated receptor phosphorylation and Mdm2-mediated beta-arrestin2 ubiquitination drive clathrin-mediated endocytosis of G protein-coupled receptors. Biochem. Biophys. Res. Commun. 2020, 533, 383–390. [Google Scholar] [CrossRef]
- Deisenroth, C.; Zhang, Y. Ribosome biogenesis surveillance: Probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 2010, 29, 4253–4260. [Google Scholar] [CrossRef]
- Lindstrom, M.S.; Jin, A.; Deisenroth, C.; White Wolf, G.; Zhang, Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol. Cell. Biol. 2007, 27, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Itahana, K.; O’Keefe, K.; Zhang, Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell. Biol. 2004, 24, 7669–7680. [Google Scholar] [CrossRef]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef]
- Wang, C.; Shi, M.; Ji, J.; Cai, Q.; Zhao, Q.; Jiang, J.; Liu, J.; Zhang, H.; Zhu, Z.; Zhang, J. Stearoyl-CoA desaturase 1 (SCD1) facilitates the growth and anti-ferroptosis of gastric cancer cells and predicts poor prognosis of gastric cancer. Aging 2020, 12, 15374–15391. [Google Scholar] [CrossRef]
- Luis, G.; Godfroid, A.; Nishiumi, S.; Cimino, J.; Blacher, S.; Maquoi, E.; Wery, C.; Collignon, A.; Longuespee, R.; Montero-Ruiz, L.; et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol. 2021, 43, 102006. [Google Scholar] [CrossRef]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis. Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar] [CrossRef]
- Venkatesh, D.; O’Brien, N.A.; Zandkarimi, F.; Tong, D.R.; Stokes, M.E.; Dunn, D.E.; Kengmana, E.S.; Aron, A.T.; Klein, A.M.; Csuka, J.M.; et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Gene Dev. 2020, 34, 526–543. [Google Scholar] [CrossRef]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar] [CrossRef]
- Neumeier, J.; Meister, G. siRNA Specificity: RNAi Mechanisms and Strategies to Reduce Off-Target Effects. Front. Plant Sci. 2021, 11, 526455. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, A.; Queiroz, R.M.d.; Chakrabarty, J.K.; Makepeace, K.A.T.; Prives, C.; Brown, L.M. Analysis of p53-Independent Functions of the Mdm2-MdmX Complex Using Data-Independent Acquisition-Based Profiling. Proteomes 2025, 13, 18. https://doi.org/10.3390/proteomes13020018
Jain A, Queiroz RMd, Chakrabarty JK, Makepeace KAT, Prives C, Brown LM. Analysis of p53-Independent Functions of the Mdm2-MdmX Complex Using Data-Independent Acquisition-Based Profiling. Proteomes. 2025; 13(2):18. https://doi.org/10.3390/proteomes13020018
Chicago/Turabian StyleJain, Anu, Rafaela Muniz de Queiroz, Jayanta K. Chakrabarty, Karl A. T. Makepeace, Carol Prives, and Lewis M. Brown. 2025. "Analysis of p53-Independent Functions of the Mdm2-MdmX Complex Using Data-Independent Acquisition-Based Profiling" Proteomes 13, no. 2: 18. https://doi.org/10.3390/proteomes13020018
APA StyleJain, A., Queiroz, R. M. d., Chakrabarty, J. K., Makepeace, K. A. T., Prives, C., & Brown, L. M. (2025). Analysis of p53-Independent Functions of the Mdm2-MdmX Complex Using Data-Independent Acquisition-Based Profiling. Proteomes, 13(2), 18. https://doi.org/10.3390/proteomes13020018