Patterns of Genetic Diversity in Highly Invasive Species: Cogongrass (Imperata cylindrica) Expansion in the Invaded Range of the Southern United States (US)


Abstract
1. Introduction
2. Results
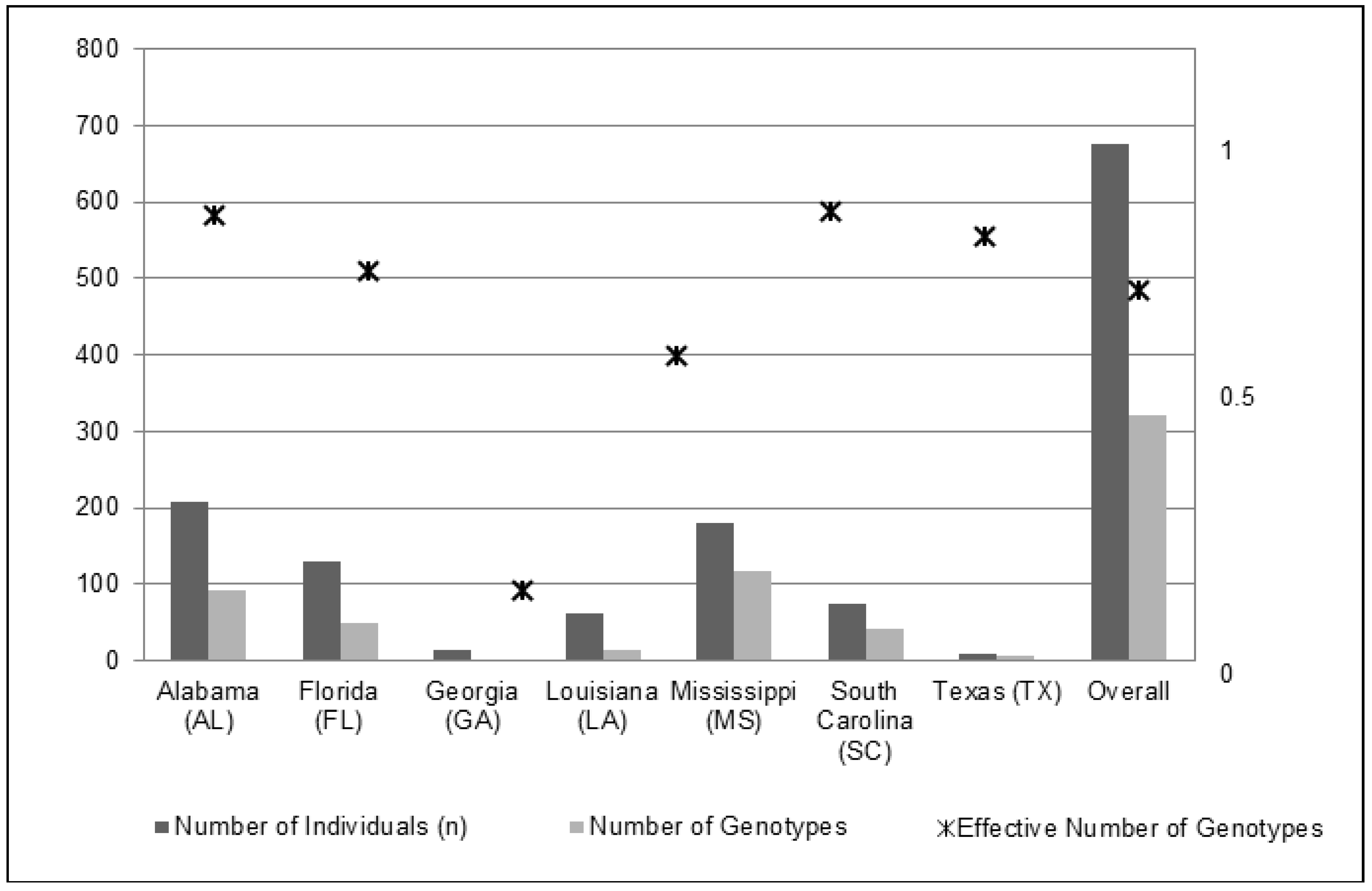
2.1. Genetic Diversity
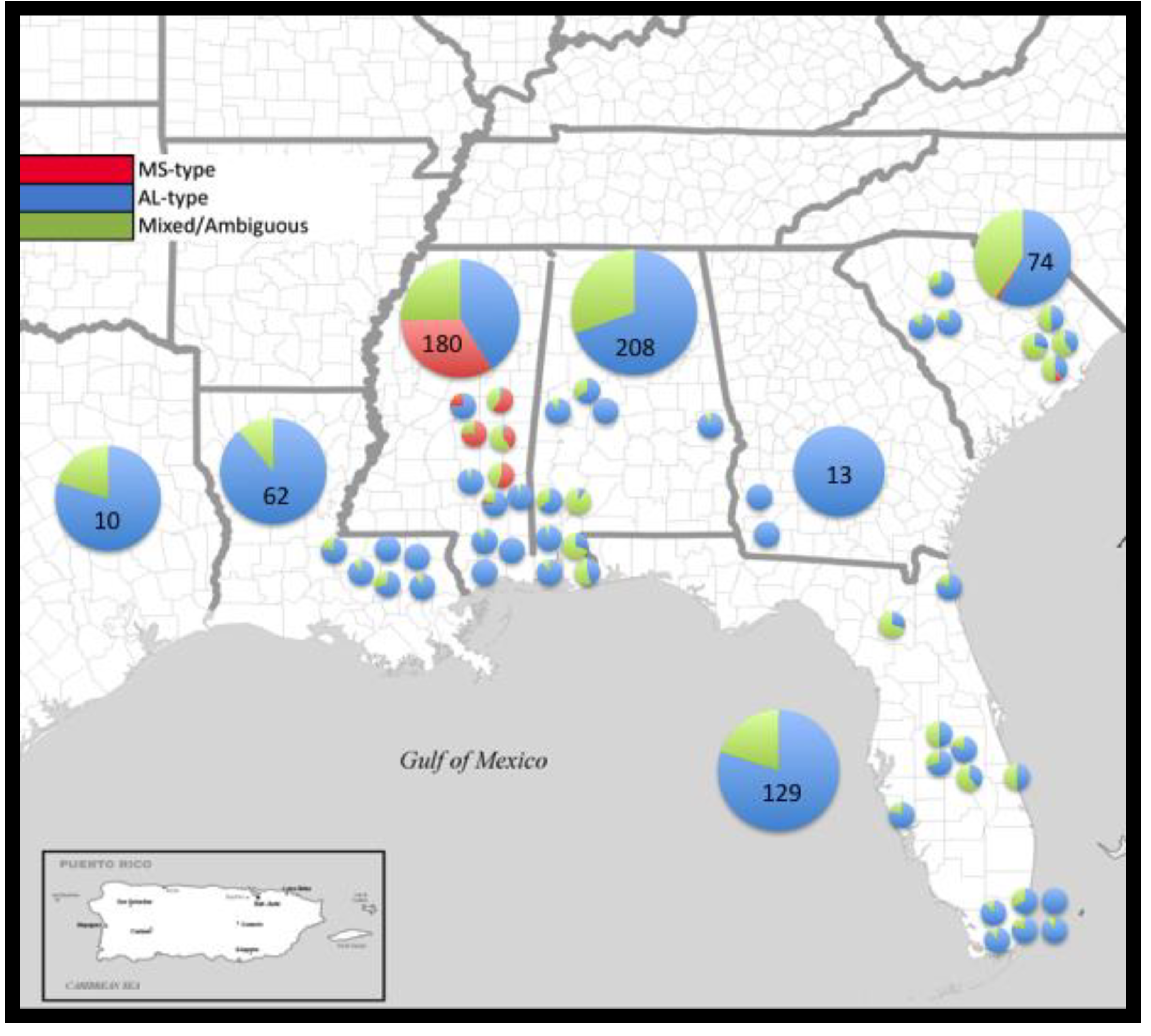
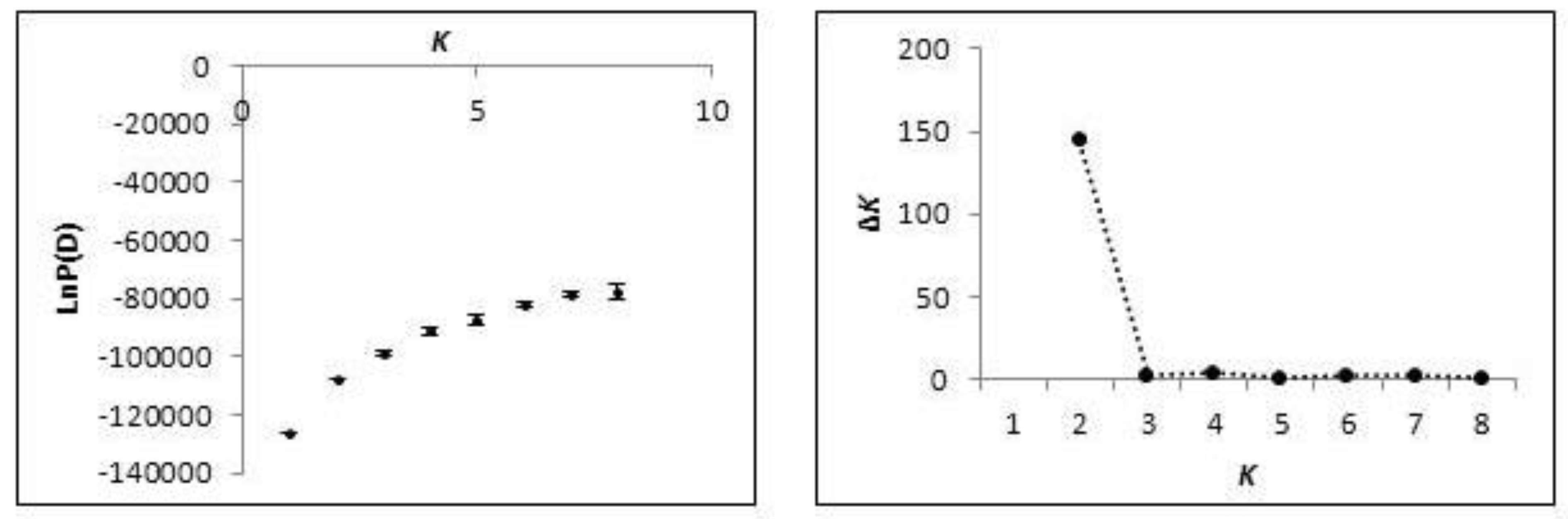
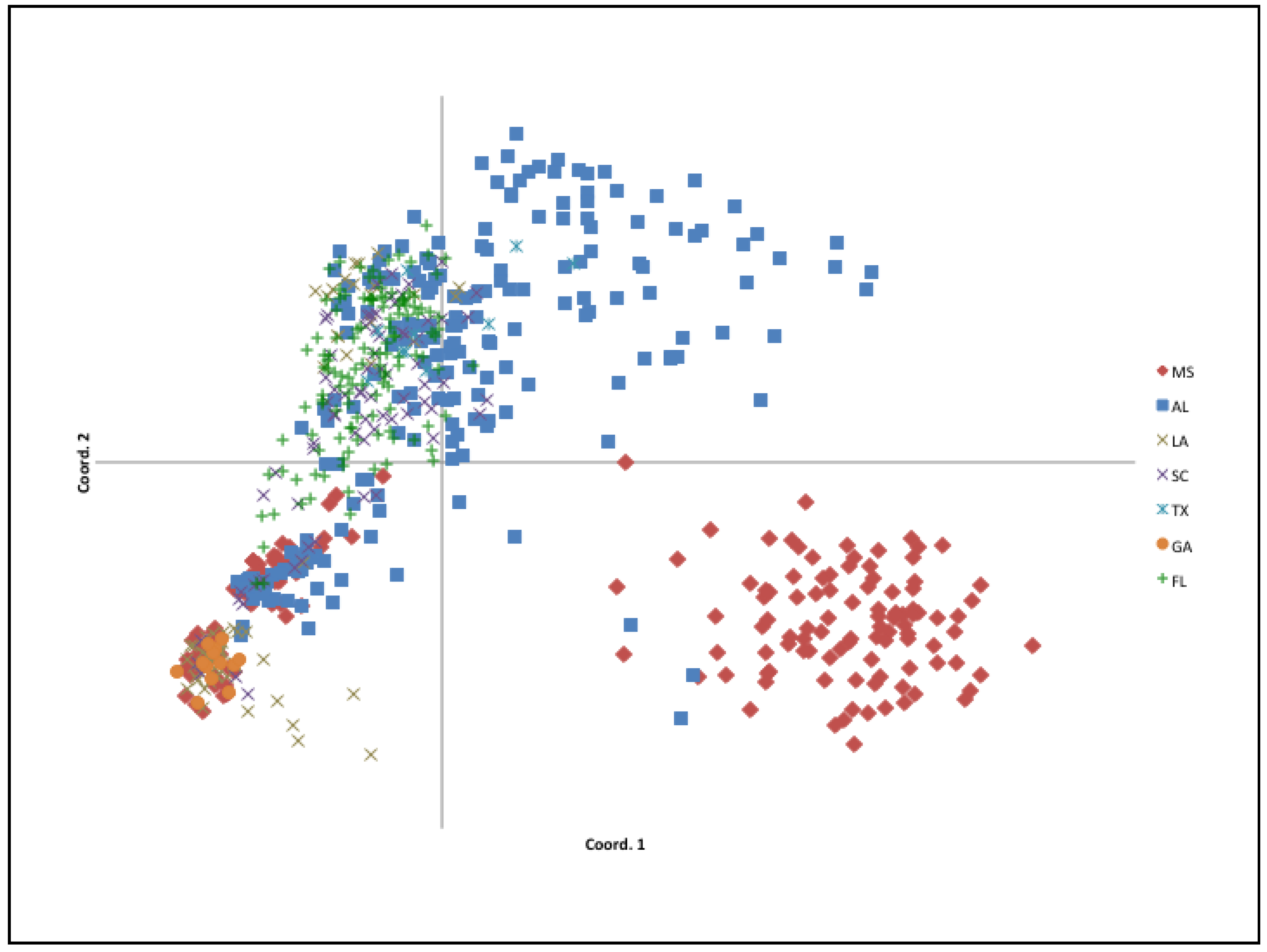
2.2. Population Structure
3. Discussion
4. Materials and Methods
4.1. Study Area and Sampling
4.2. Molecular Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Accessibility
References
- Meyerson, L.A.; Mooney, H.A. Invasive alien species in an era of globalization. Front. Ecol. Environ. 2007, 5, 199–208. [Google Scholar] [CrossRef]
- Schulz, A.N.; Lucardi, R.D.; Marsico, T.D. Successful Invasions and Failed Biocontrol: The Role of Antagonistic Species Interactions. BioScience 2019, 69, 711–724. [Google Scholar] [CrossRef]
- Lee, C.E. Evolutionary genetics of invasive species. Trends Ecol. Evol. 2002, 17, 386–391. [Google Scholar] [CrossRef]
- Dlugosch, K.M.; Parker, I.M. Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 2008, 17, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.M.; Gaskin, J.F.; Wilson, L.M. Ecological Genetics of Plant Invasion: What Do We Know? Invasive Plant Sci. Manag. 2008, 1, 98–109. [Google Scholar] [CrossRef]
- Catford, J.A.; Jansson, R.; Nilsson, C. Reducing redundancy in invasion ecology by integrating hypotheses into a single theoretical framework. Divers. Distrib. 2009, 15, 22–40. [Google Scholar] [CrossRef]
- Hulme, P.E. Trade, transport and trouble: Managing invasive species pathways in an era of globalization. J. Appl. Ecol. 2009, 46, 10–18. [Google Scholar] [CrossRef]
- Holzmueller, E.J.; Jose, S. Invasion success of cogongrass, an alien C4 perennial grass, in the southeastern United States: Exploration of the ecological basis. Boil. Invasions 2010, 13, 435–442. [Google Scholar] [CrossRef]
- Vitousek, P.; Mooney, H.A.; Lubchenco, J.; Melillo, J.M. Human Domination of Earth’s Ecosystems. Science 1997, 277, 494–499. [Google Scholar] [CrossRef]
- Hobbs, R.J. Land-use changes and invasions. In Invasive Species in a Changing World; Mooney, H.A., Hobbs, R.J., Eds.; Island Press: Washington, DC, USA, 2000. [Google Scholar]
- Harrison, S.; Hohn, C.; Ratay, S. Distribution of Exotic Plants along Roads in a Peninsular Nature Reserve. Boil. Invasions 2002, 4, 425–430. [Google Scholar] [CrossRef]
- Shuster, W.D.; Herms, C.P.; Frey, M.N.; Doohan, D.J.; Cardina, J. Comparison of survey methods for an invasive plant at the subwatershed level. Boil. Invasions 2005, 7, 393–403. [Google Scholar] [CrossRef]
- Christen, D.; Matlack, G.R. The role of roadsides in plant invasions: A demographic approach. Conserv. Boil. 2006, 20, 385–391. [Google Scholar] [CrossRef] [PubMed]
- With, K.A. The Landscape Ecology of Invasive Spread. Conserv. Boil. 2002, 16, 1192–1203. [Google Scholar] [CrossRef]
- Rouget, M.; Richardson, D.M. Inferring Process from Pattern in Plant Invasions: A Semimechanistic Model Incorporating Propagule Pressure and Environmental Factors. Am. Nat. 2003, 162, 713–724. [Google Scholar] [CrossRef]
- Lázaro-Lobo, A.; Ervin, G.N. A global examination on the differential impacts of roadsides on native vs. exotic and weedy plant species. Glob. Ecol. Conserv. 2019, 17, e00555. [Google Scholar] [CrossRef]
- Hodkinson, D.J.; Thompson, K. Plant Dispersal: The Role of Man. J. Appl. Ecol. 1997, 34, 1484–1496. [Google Scholar] [CrossRef]
- Tikka, P.M.; Högmander, H.; Koski, P.S. Road and railway verges serve as dispersal corridors for grassland plants. Landsc. Ecol. 2001, 16, 659–666. [Google Scholar] [CrossRef]
- Watkins, R.Z.; Chen, J.; Pickens, J.; Brosofske, K.D. Effects of Forest Roads on Understory Plants in a Managed Hardwood Landscape. Conserv. Boil. 2003, 17, 411–419. [Google Scholar] [CrossRef]
- Lockwood, J.L.; Hoopes, M.F.; Marchetti, M.P. Invasion Ecology; Blackwell: Malden, MA, USA, 2007. [Google Scholar]
- Holly, D.C. Multi-Scale Evaluation of Mechanisms Associated with the Establishment of a Model Invasive Species in Mississippi: Imperata cylindrica. Ph.D. Thesis, Mississippi State University, Starkville, MS, USA, 2008; 140p. [Google Scholar]
- Ervin, G.N.; Holly, D.C. Examining Local Transferability of Predictive Species Distribution Models for Invasive Plants: An Example with Cogongrass (Imperata cylindrica). Invasive Plant Sci. Manag. 2011, 4, 390–401. [Google Scholar] [CrossRef]
- Bryson, C.T.; Carter, R. Cogongrass, Imperata cylindrica, in the United States. Weed Technol. 1993, 7, 1005–1009. [Google Scholar] [CrossRef]
- MacDonald, G.E. Cogongrass (Imperata cylindrica)—Biology, Ecology, and Management. Crit. Rev. Plant Sci. 2004, 23, 367–380. [Google Scholar] [CrossRef]
- The PLANTS Database. Available online: http://plants.usda.gov (accessed on 23 March 2020).
- Hubbard, C.E.; Gray, A.P.; Brown, D.; Whyte, R.O. Imperata Cylindrica: Taxonomy, Distribution, Economic Significance, and Control; Imperial Agricultural Bureaux Joint Publication: Oxford, UK, 1944; Volume 7, 63p. [Google Scholar]
- Holm, L.G.; Donald, P.; Pancho, J.V.; Herberger, J.P. The World’s Worst Weeds: Distribution and Biology; The University Press of Hawaii: Honolulu, HI, USA, 1977; p. 609. [Google Scholar]
- Gabel, M.L. A Biosystematic Study of the Genus Imperata (Gramineae: Andropogoneae). Ph.D. Thesis, Iowa State University, Ames, IA, USA, 1982; 94p. [Google Scholar]
- Tabor, P. Cogon grass, Imperata cylindrica (L.) Beauir in the South-eastern United States. Agron. J. 1949, 41, 270. [Google Scholar] [CrossRef]
- Tabor, P. Cogongrass in Mobile County, Alabama. Agron. J. 1952, 44, 50. [Google Scholar] [CrossRef]
- Lucardi, R.D.; Wallace, L.E.; Ervin, G.N. Invasion Success in Cogongrass (Imperata cylindrica): A Population Genetic Approach Exploring Genetic Diversity and Historical Introductions. Invasive Plant Sci. Manag. 2014, 7, 59–75. [Google Scholar] [CrossRef]
- Dickens, R. Cogongrass in Alabama after Sixty Years. Weed Sci. 1974, 22, 177–179. [Google Scholar] [CrossRef]
- Dozier, H.; Gaffney, J.F.; McDonald, S.K.; Johnson, E.R.; Shilling, D.G. Cogongrass in the United States: History, Ecology, Impacts, and Management. Weed Technol. 1998, 12, 737–743. [Google Scholar] [CrossRef]
- Howard, J. Imperata brasiliensis, I. cylindrical. In Fire Effects Information System; U.S. Department of Agriculture, Forest Service, Rocky Mountain Research Station, Fire Science Laboratory (Producer): Bozeman, MT, USA, 2005; Available online: https://www.fs.fed.us/database/feis/plants/graminoid/impspp/all.html (accessed on 29 March 2020).
- Hall, D.W. Is cogon grass really an exotic? Wildland Weeds 1998, 1, 14–15. [Google Scholar]
- Lucardi, R.D.; Wallace, L.E.; Ervin, G.N. Evaluating hybridization as a potential facilitator of successful cogongrass (Imperata cylindrica) invasion in Florida, USA. Boil. Invasions 2014, 16, 2147–2161. [Google Scholar] [CrossRef]
- Amsellem, L.; Noyer, J.L.; LeBourgeois, T.; Hossaert-McKey, M. Comparison of genetic diversity of the invasive weed Rubus alcefolius Poir. (Rosaceae) in its native range and in areas of introduction, using amplified fragment length polymorphism (AFLP) markers. Mol. Ecol. 2000, 9, 443–455. [Google Scholar] [CrossRef]
- Campbell, D.; Duchesne, P.; Bernatchez, L. AFLP utility for population assignment studies: Analytical investigation and empirical comparison with microsatellites. Mol. Ecol. 2003, 12, 1979–1991. [Google Scholar] [CrossRef]
- Meudt, H.; Clarke, A. Almost Forgotten or Latest Practice? AFLP applications, analyses and advances. Trends Plant Sci. 2007, 12, 106–117. [Google Scholar] [CrossRef]
- Capo-Chichi, L.J.A.; Faircloth, W.H.; Williamson, A.G.; Patterson, M.G.; Miller, J.H.; Van Santen, E. Invasion Dynamics and Genotypic Diversity of Cogongrass (Imperata cylindrica) at the Point of Introduction in the Southeastern United States. Invasive Plant Sci. Manag. 2008, 1, 133–141. [Google Scholar] [CrossRef]
- Keller, S.R.; Fields, P.D.; Berardi, A.E.; Taylor, D.R. Recent admixture generates heterozygosity-fitness correlations during the range expansion of an invading species. J. Evol. Boil. 2014, 27, 616–627. [Google Scholar] [CrossRef]
- Ellstrand, N.C.; Schierenbeck, K.A. Hybridization as a stimulus for the evolution of invasiveness in plants? Proc. Natl. Acad. Sci. USA 2000, 97, 7043–7050. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, J.; Keller, S.R.; Lavergne, S.; Kaproth, M.A.; Eppinga, M.B. Human-aided admixture may fuel ecosystem transformation during biological invasions: Theoretical and experimental evidence. Ecol. Evol. 2014, 4, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, T.M.; Duncan, R.P. Determinants of establishment success in introduced birds. Nature 2001, 414, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Genton, B.J.; Shykoff, J.A.; Giraud, T. High genetic diversity in French invasive populations of common ragweed, Ambrosia artemisiifolia, as a result of multiple sources of introduction. Mol. Ecol. 2005, 14, 4275–4285. [Google Scholar] [CrossRef]
- Walls, R. Hybridization and Plasticity Contribute to Divergence among Coastal and Wetland Populations of Invasive Hybrid Japanese Knotweed s.l. (Fallopia spp.). Chesap. Sci. 2009, 33, 902–918. [Google Scholar] [CrossRef]
- Mothapo, N.P.; Wossler, T.C. Behavioural and chemical evidence for multiple colonisation of the Argentine ant, Linepithema humile, in the Western Cape, South Africa. BMC Ecol. 2011, 11, 6. [Google Scholar] [CrossRef]
- Garbelotto, M.; Guglielmo, F.; Mascheretti, S.; Croucher, P.J.P.; Gonthier, P. Population genetic analyses provide insights on the introduction pathway and spread patterns of the North American forest pathogen Heterobasidion irregulare in Italy. Mol. Ecol. 2013, 22, 4855–4869. [Google Scholar] [CrossRef]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Sakai, A.K.; Allendorf, F.W.; Holt, J.S.; Lodge, D.M.; Molofsky, J.; With, K.A.; Baughman, S.; Cabin, R.J.; Cohen, J.E.; Ellstrand, N.C.; et al. The Population Biology of Invasive Species. Annu. Rev. Ecol. Syst. 2001, 32, 305–332. [Google Scholar] [CrossRef]
- Vellend, M.; Drummond, E.B.M.; Tomimatsu, H. Effects of genotype identity and diversity on the invasiveness and invisibility of plant populations. Oecologia 2010, 162, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, R.A.; Facon, B.; Ravigné, V.; Turgeon, J.; Foucaud, J.; Lee, C.E.; Rey, O.; Estoup, A. Anthropogenically induced adaptation to invade (AIAI): Contemporary adaptation to human-altered habitats within the native range can promote invasions. Evol. Appl. 2011, 5, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; Van De Lee, T.; Hornes, M.; Friters, A.; Pot, J.; Paleman, J.; Kuiper, M.; et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef] [PubMed]
- Ehrich, D. aflpdat: A collection of r functions for convenient handling of AFLP data. Mol. Ecol. Notes 2006, 6, 603–604. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. genalex 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Mariette, S.; Le Corre, V.; Austerlitz, F.; Kremer, A. Sampling within the genome for measuring within-population diversity: Trade-offs between markers. Mol. Ecol. 2002, 11, 1145–1156. [Google Scholar] [CrossRef]
- Bonin, A.; Ehrich, D.; Manel, S. Statistical analysis of amplified fragment length polymorphism data: A toolbox for molecular ecologists and evolutionists. Mol. Ecol. 2007, 16, 3737–3758. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. Available online: http://pritch.bsd.uchicago.edu/structure.html (accessed on 29 March 2020). [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of Molecular Variance Inferred from Metric Distances among DNA Haplotypes: Application to Human Mitochondrial DNA Restriction Data. Genetics 1992, 131, 479–491. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Counties Sampled | Other Location Information | Number of Individuals (n) | Shannon’s Information Index (I) | Nei’s Gene Diversity | Number of Genotypes | Effective Number of Genotypes |
|---|---|---|---|---|---|---|---|
| Alabama (AL) | Baldwin, Hale, Lee, Mobile, Washington, Sumter | Talladega NF, Frank Boykin WMA | 208 | 0.033 ± 0.002 | 0.023 | 92 | 0.935 |
| Florida (FL) | Alachua, Duval, Indian River, Miami-Dade, Osceola, Sarasota | Disney Wilderness Preserve (TNC), Miami-Dade municipal parks | 129 | 0.033 ± 0.002 | 0.028 | 49 | 0.816 |
| Georgia (GA) | Baker, Crawford, Decatur, Mitchell, Thomas, Worth | Georgia Forestry Commission | 13 | 0.009 ± 0.001 | 0.006 | 2 | 0.154 |
| Louisiana (LA) | St. Tammany, Washington | Benscreek WMA | 62 | 0.024 ± 0.002 | 0.029 | 14 | 0.64 |
| Mississippi (MS) | Greene, Harrison, Jasper, Jones, Scott, Smith, Wayne | Desoto NF, Bienville NF | 180 | 0.038 ± 0.003 | 0.023 | 117 | 0.94 |
| South Carolina (SC) | Berkeley, Greenwood, Saluda, Union | Frances Marion NF, Sumter NF | 74 | 0.063 ± 0.002 | 0.042 | 41 | 0.888 |
| Texas (TX) | Tyler | Texas Forest Service | 10 | 0.012 ± 0.002 | 0.011 | 6 | 0.778 |
| Overall | 676 | 321 | |||||
| Mean (±SE) | 0.030 ± 0.001 | 0.023 ± 0.001 | 0.736 ± 0.105 |
| MS-Coast | AL | LA | SC | TX | GA | FL | |
|---|---|---|---|---|---|---|---|
| 0.132 | * | AL | |||||
| 0.096 | 0.175 | * | LA | ||||
| 0.098 | 0.100 | 0.120 | * | SC | |||
| 0.217 | 0.090 | 0.264 | 0.083 | * | TX | ||
| 0.150 | 0.277 | 0.050 | 0.158 | 0.553 | * | GA | |
| 0.146 | 0.094 | 0.158 | 0.060 | 0.083 | 0.281 | * | FL |
| 0.330 | 0.320 | 0.444 | 0.314 | 0.348 | 0.478 | 0.385 | MS-Central |
| Source of Variation | d.f. | Sum of Squares | Percentage of Variation | P-Value |
|---|---|---|---|---|
| Among Groups | 1 | 1325.57 | 27.43 | <0.001 |
| Among populations within groups | 6 | 1283.18 | 8.88 | 0.110 |
| Within populations | 668 | 12,151.01 | 63.68 | <0.001 |
| Total | 675 | 14,776.76 | ||
| FST = 0.363 (P < 0.001), FSC = 0.122 (P < 0.001), FCT = 0.274 (P = 0.11) | ||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucardi, R.D.; Wallace, L.E.; Ervin, G.N. Patterns of Genetic Diversity in Highly Invasive Species: Cogongrass (Imperata cylindrica) Expansion in the Invaded Range of the Southern United States (US). Plants 2020, 9, 423. https://doi.org/10.3390/plants9040423
Lucardi RD, Wallace LE, Ervin GN. Patterns of Genetic Diversity in Highly Invasive Species: Cogongrass (Imperata cylindrica) Expansion in the Invaded Range of the Southern United States (US). Plants. 2020; 9(4):423. https://doi.org/10.3390/plants9040423
Chicago/Turabian StyleLucardi, Rima D., Lisa E. Wallace, and Gary N. Ervin. 2020. "Patterns of Genetic Diversity in Highly Invasive Species: Cogongrass (Imperata cylindrica) Expansion in the Invaded Range of the Southern United States (US)" Plants 9, no. 4: 423. https://doi.org/10.3390/plants9040423
APA StyleLucardi, R. D., Wallace, L. E., & Ervin, G. N. (2020). Patterns of Genetic Diversity in Highly Invasive Species: Cogongrass (Imperata cylindrica) Expansion in the Invaded Range of the Southern United States (US). Plants, 9(4), 423. https://doi.org/10.3390/plants9040423