Constructing a Reference Genome in a Single Lab: The Possibility to Use Oxford Nanopore Technology



Abstract
:1. Introduction
2. Results
2.1. MinION Sequencing of Sorghum Accession BTx623 Genome
2.2. Assembly Results Using Canu
2.3. Assembly Results Using Minimap, Miniasm, and Racon
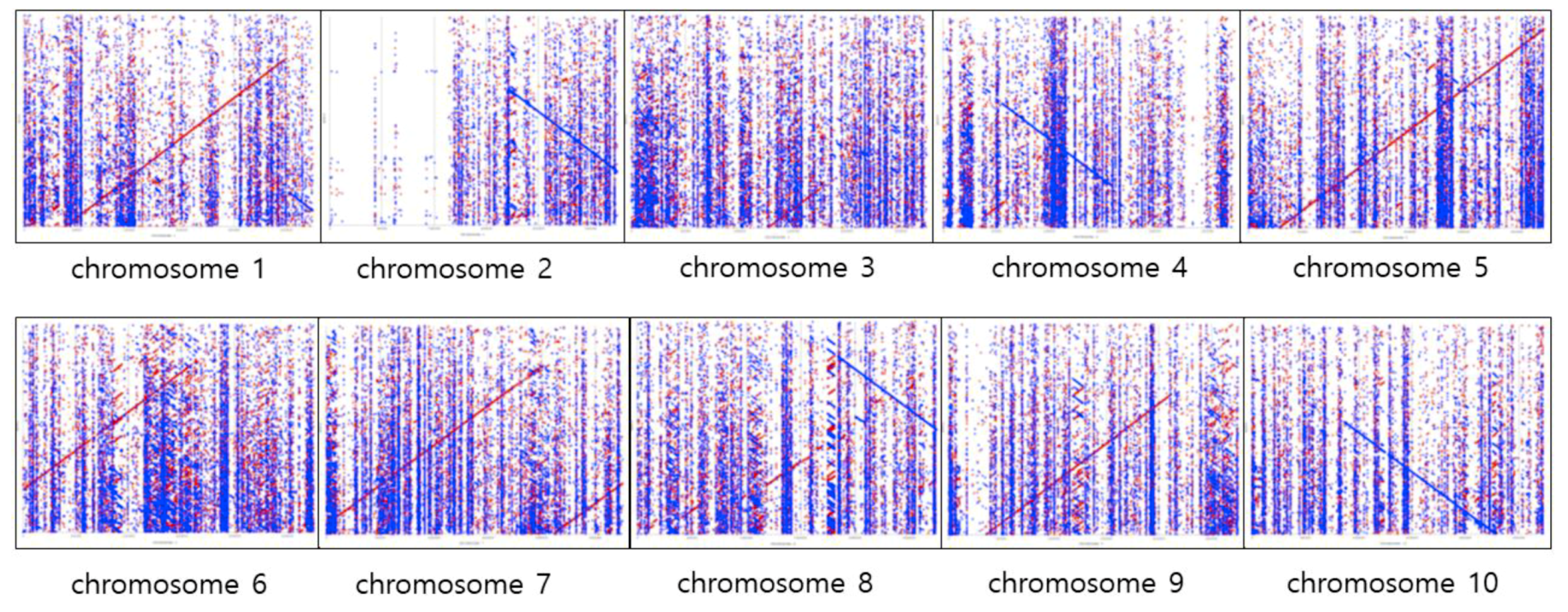
2.4. Confirmation of de Novo Assembly
3. Discussion
3.1. Optimization of Genome Assembly by Using Different Assemblers
3.2. Advantages of Current Combinational Sequencing
3.3. Improvements in the Accuracy of Long-Reads Sequencing and Assembly
3.4. DNA Fragmentation Effect on MinION Sequencing
3.5. Requirement of Effective Size Selection for Long-Reads Sequencing
4. Materials and Methods
4.1. Plant Material and Genomic DNA Extraction
4.2. Preparation of Sequencing Library and MinION Sequencing
4.3. MinION Raw Sequences Mapped against the Reference Genome
4.4. De Novo Whole Genome Assembly
4.5. Confirmation of de Novo Assembly against the Sorghum Reference Genome
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Nystrom, J.; Webster, H.; Keeble-Gagnere, G. Discoveries and advances in plant and animal genomics. Funct. Integr. Genom. 2015, 15, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome sequencing and assembly by long reads in plants. Genes 2017, 9, 6. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Bowers, J.E.; Bruggmann, R.; Dubchak, I.; Grimwood, J.; Gundlach, H.; Haberer, G.; Hellsten, U.; Mitros, T.; Poliakov, A.; et al. The Sorghum bicolor genome and the diversification of grasses. Nature 2009, 457, 551. [Google Scholar] [CrossRef]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B. The Sorghum bicolor reference genome: improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018, 93, 338–354. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2011, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Claros, M.G.; Bautista, R.; Guerrero-Fernández, D.; Benzerki, H.; Seoane, P.; Fernández-Pozo, N. Why assembling plant genome sequences is so challenging. Biology 2012, 1, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Crow, K.D.; Wagner, G.P. What is the role of genome duplication in the evolution of complexity and diversity. Mol. Biol. Evol. 2005, 23, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Jackson, S.A.; Meyers, B.C.; Wing, R.A. Evolution of plant genome architecture. Genome Biol. 2016, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.A.; Iwata, A.; Lee, S.H.; Schmutz, J.; Shoemaker, R. Sequencing crop genomes: approaches and applications. New Phytol. 2011, 191, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Debladis, E.; Llauro, C.; Carpentier, M.-C.; Mirouze, M.; Panaud, O. Detection of active transposable elements in Arabidopsis thaliana using Oxford Nanopore Sequencing technology. BMC Genom. 2017, 18, 537. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef]
- Schmidt, M.H.-W.; Vogel, A.; Denton, A.K.; Istace, B.; Wormit, A.; van de Geest, H.; Bolger, M.E.; Alseekh, S.; Maß, J.; Pfaff, C. De novo assembly of a new Solanum pennellii accession using nanopore sequencing. Plant Cell 2017, 29, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Giolai, M.; Paajanen, P.; Verweij, W.; Witek, K.; Jones, J.D.; Clark, M.D. Comparative analysis of targeted long read sequencing approaches for characterization of a plant’s immune receptor repertoire. BMC Genom. 2017, 18, 564. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.-S. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524. [Google Scholar] [CrossRef]
- Parker, J.; Helmstetter, A.J.; Devey, D.; Wilkinson, T.; Papadopulos, A.S. Field-based species identification of closely-related plants using real-time nanopore sequencing. Sci. Rep. 2017, 7, 8345. [Google Scholar] [CrossRef]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225. [Google Scholar] [CrossRef] [PubMed]
- Zimin, A.V.; Puiu, D.; Hall, R.; Kingan, S.; Clavijo, B.J.; Salzberg, S.L. The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. Gigascience 2017, 6, gix097. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Sovic, I.; Nagarajan, N.; Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, B.S.; Quinlan, A.R. Mosdepth: quick coverage calculation for genomes and exomes. Bioinformatics 2017, 34, 867–868. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- de Lannoy, C.; de Ridder, D.; Risse, J. The long reads ahead: De novo genome assembly using the MinION. F1000 Res. 2017, 6. [Google Scholar] [CrossRef]
- Wee, Y.; Bhyan, S.B.; Liu, Y.; Lu, J.; Li, X.; Zhao, M. The bioinformatics tools for the genome assembly and analysis based on third-generation sequencing. Brief Funct Genomics 2019, 18, 1–12. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Bouri, L.; Lavenier, D.; Gibrat, J.-F.; del Angel, V.F.D. Evaluation of genome assembly software based on long reads. Fr. Genomique 2017. [Google Scholar] [CrossRef]
- Gill, B.S.; Appels, R.; Botha-Oberholster, A.-M.; Buell, C.R.; Bennetzen, J.L.; Chalhoub, B.; Chumley, F.; Dvořák, J.; Iwanaga, M.; Keller, B.; et al. A workshop report on wheat genome sequencing: International Genome Research on Wheat Consortium. Genetics 2004, 168, 1087–1096. [Google Scholar] [CrossRef] [PubMed]
- The International Wheat Genome Sequencing Consortium. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, V.; Sakakibara, Y. Comprehensive evaluation of non-hybrid genome assembly tools for third-generation PacBio long-read sequence data. Brief. Bioinform. 2017, 20, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M.; Zywicki, M.; Twardowski, T.; Karlowski, W.M. Efficiency of PacBio long read correction by 2nd generation Illumina sequencing. Genomics 2017, 111, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalay, T.; Golovchenko, J.A. De novo sequencing and variant calling with nanopores using PoreSeq. Nat. Biotechnol. 2015, 33, 1087. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.R.; O’Neil, N.J.; Jain, M.; Olsen, H.E.; Hieter, P.; Snutch, T.P. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res. 2018, 28, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Corless, S.; Gilbert, N. Investigating DNA supercoiling in eukaryotic genomes. Brief. Funct. Genom. 2017, 16, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Carlson, J.; Tulsieram, L.; Glaubitz, J.; Luk, V.; Kauffeldt, C.; Rutledge, R. Segregation of random amplified DNA markers in F 1 progeny of conifers. Theor. Appl. Genet. 1991, 83, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Mayjonade, B.; Gouzy, J.; Donnadieu, C.; Pouilly, N.; Marande, W.; Callot, C.; Langlade, N.; Muños, S. Extraction of high-molecular-weight genomic DNA for long-read sequencing of single molecules. BioTechniques 2016, 61, 203–205. [Google Scholar] [CrossRef] [PubMed]
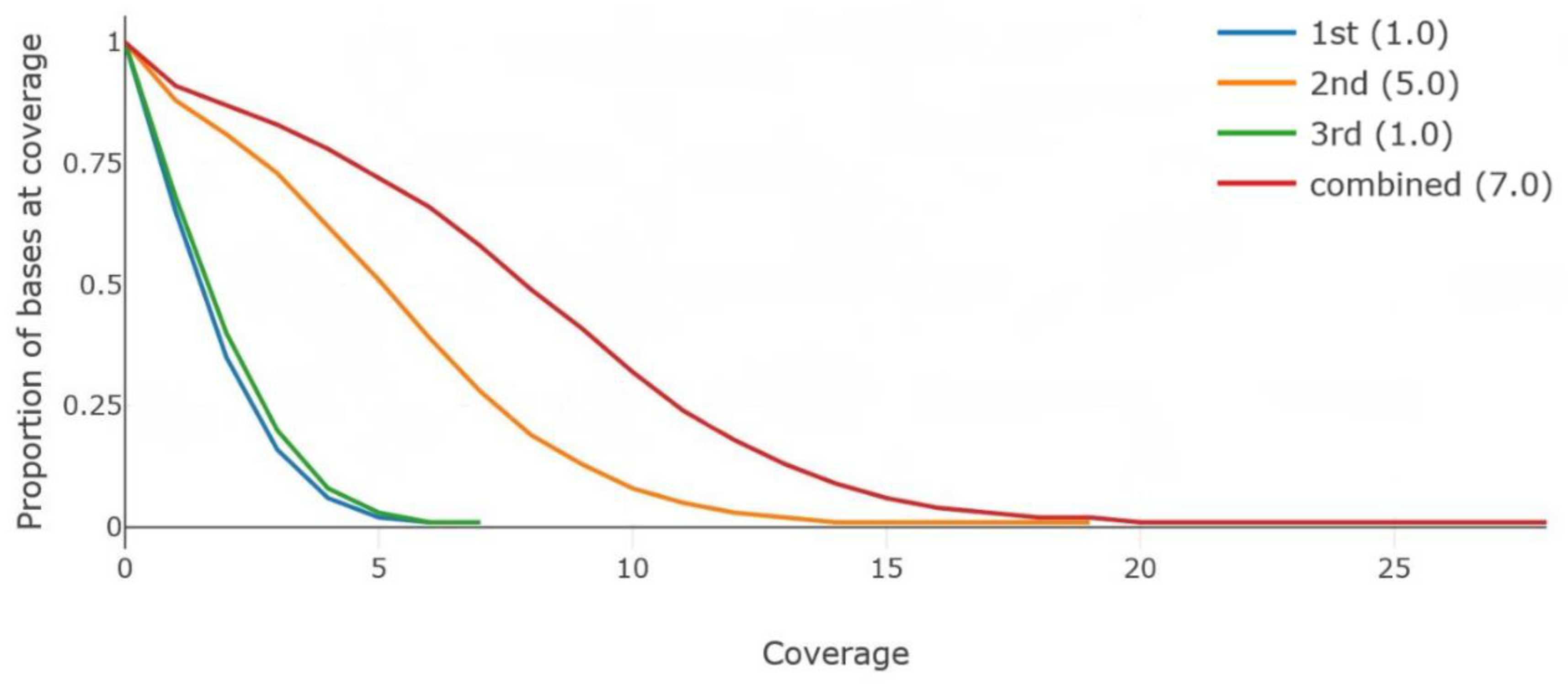

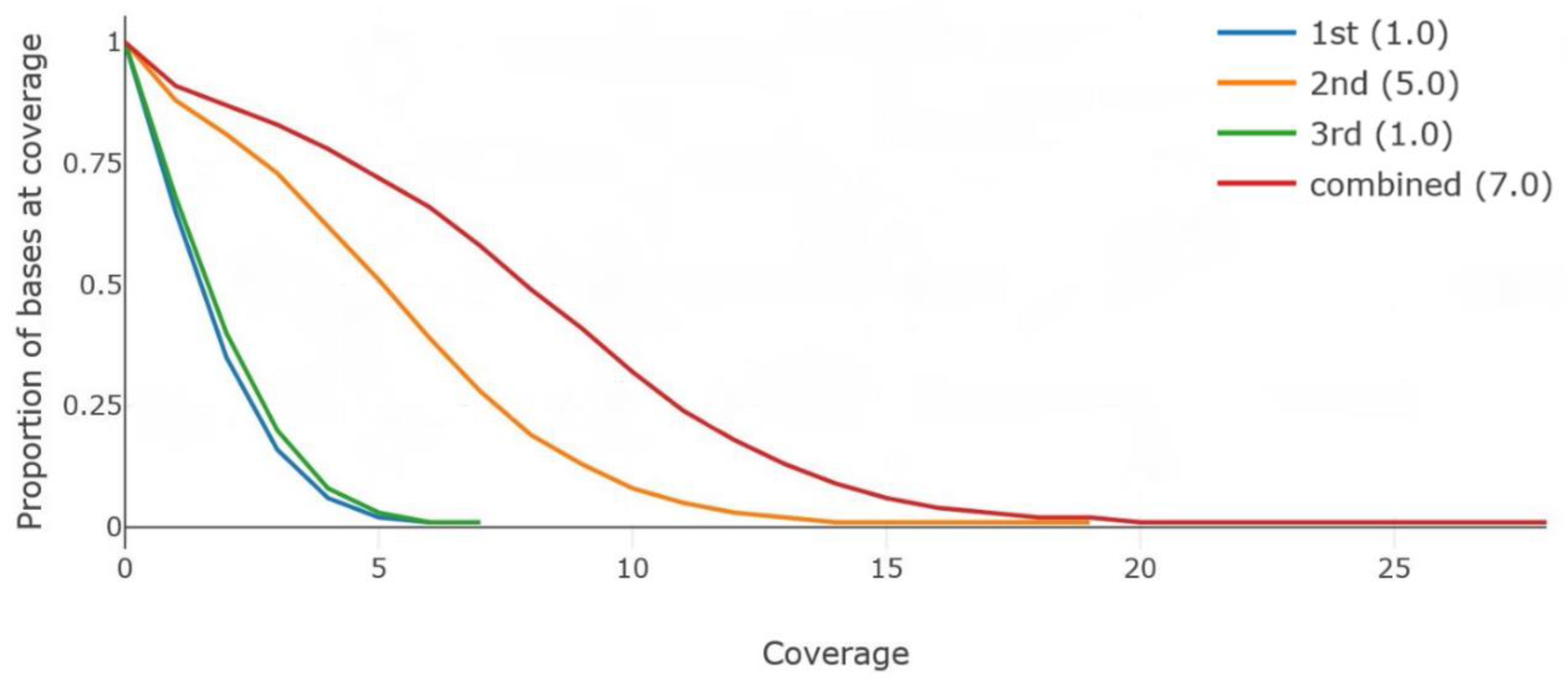{kind=link}
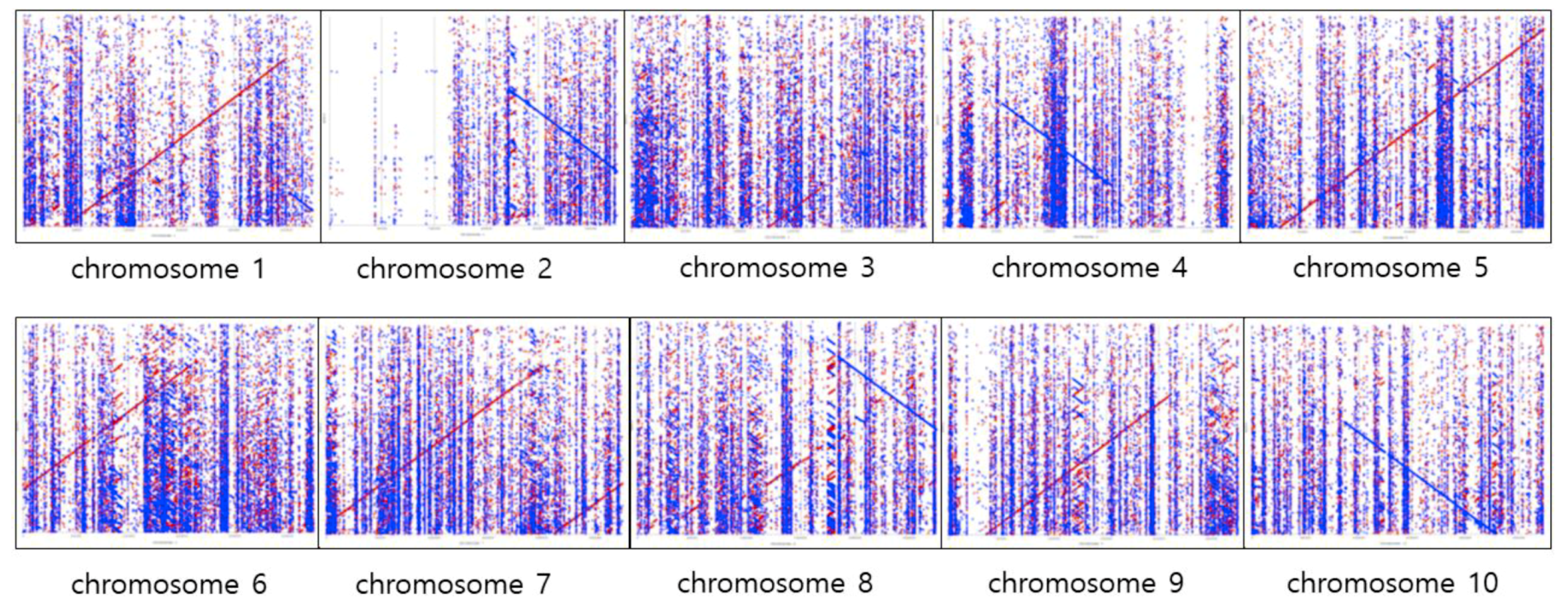{kind=link}
| Result | 1st | 2nd | 3rd |
|---|---|---|---|
| Total generated file size (Gb) | 2.83 | 11.71 | 3.34 |
| Total number of fastq files | 35 | 170 | 37 |
| Total read numbers | 136,769 | 679,658 | 146,883 |
| The shortest read length (bp) | 167 | 74 | 38 |
| The longest read length (bp) | 190,250 | 110,486 | 217,000 |
| The most abundant read length (bp) (no. of reads) | 908 (61) | 947 (111) | 1028 (69) |
| Q-score | 11.2 | 10.7 | 10.9 |
| Result | 1st | 2nd | 3rd | Combined a |
|---|---|---|---|---|
| Average depth | 2.01 | 5.64 | 2.10 | 8.56 |
| Mapping rate (%) | 97.93 | 96.87 | 97.14 | 97.08 |
| Result | 1st | 2nd | 3rd | Combined | |
|---|---|---|---|---|---|
| Total loaded reads | No. of reads | 119,022 | 649,003 | 127,653 | 895,678 |
| Total length (bp) | 1,495,987,647 | 6,216,312,936 | 1,767,114,081 | 9,479,414,664 | |
| Coverage | 2.04 | 8.51 | 2.42 | 12.63 | |
| Expected corrected reads | No. of reads | 117,932 | 647,151 | 125,836 | 893,520 |
| Total length (bp) | 1,333,102,902 | 6,187,551,664 | 1,359,065,630 | 8,029,184,425 | |
| Mean read length (bp) | 11,304 | 7,900 | 10,800 | 8,986 | |
| N50 length (bp) | 49,358 | 23,337 | 53,805 | 72,703 | |
| After correction/Before trimming | No. of reads | 110,540 | 607,805 | 116,100 | 845,774 |
| Total length (bp) | 1,235,198,760 | 5,658,532,542 | 1,549,842,529 | 8,673,782,926 | |
| Coverage | 1.68 | 7.75 | 2.12 | 11.56 | |
| After trimming a | No. of reads | 68,176 | 403,755 | 56,719 | 566,533 |
| Total bases (bp) | 411,454,770 | 2,794,594,634 | 376,245,841 | 4,739,533,665 | |
| UniTigging/READs | No. of reads | 68,176 | 410,746 | 56,719 | 577,103 |
| Total length (bp) | 424,463,809 | 2,844,670,276 | 381,679,172 | 4,833,385,452 | |
| Coverage | 0.58 | 3.89 | 0.52 | 6.44 | |
| UniTigging/concensus | No. of sequences | 159 | 5,740 | 127 | 6,124 |
| No. of repeats | 28 | 692 | 26 | 712 | |
| Length of repeats (bp) | 573,105 | 10,509,344 | 472,695 | 14,815,759 | |
| Total length (bp) | 3,088,777 | 178,246,454 | 3,256,717 | 344,366,012 | |
| Coverage | 0.004 | 0.237 | 0.004 | 0.459 | |
| Unassembled | No. of sequences | 38,897 | 168,888 | 32,340 | 216,120 |
| Total length (bp) | 259,436,098 | 1,180,881,063 | 252,418,869 | 1,832,920,246 | |
| Result | No. of Round | 1st | 2nd | 3rd | Combined | |
|---|---|---|---|---|---|---|
| Raw file | Total size (Gb) | 2.83 | 11.71 | 3.34 | 17.9 | |
| Minimap | File size (byte) | 607,227,298 | 5,089,824,937 | 546,909,744 | 13,226,110,131 | |
| Miniasm | File size (byte) | 1,282,822 | 177,933,354 | 2,126,525 | 368,271,934 | |
| Total length (bp) | 1,286,782 | 176,978,175 | 2,139,682 | 370,303,449 | ||
| Racon | Total length (bp) | 1 | 1,289,492 | 177,650,167 | 2,145,749 | 373,675,134 |
| 2 | 1,278,467 | 177,931,139 | 2,141,277 | 374,668,365 | ||
| 3 | 1,262,947 | 177,915,228 | 2,127,089 | 374,934,532 | ||
| 4 | 1,247,138 | 177,805,838 | 2,112,239 | 375,048,732 | ||
| 5 | 1,232,808 | 177,683,528 | 2,097,341 | 375,105,174 |
| Canu | Miniasm | |
|---|---|---|
| Number of Conigs | 6124 | 2661 |
| Assembled read length (bp) | 344,366,012 | 375,105,174 |
| N50 (bp) | 98,000,000 | 199,000,000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.G.; Choi, S.C.; Kang, Y.; Kim, K.M.; Kang, C.-S.; Kim, C. Constructing a Reference Genome in a Single Lab: The Possibility to Use Oxford Nanopore Technology. Plants 2019, 8, 270. https://doi.org/10.3390/plants8080270
Lee YG, Choi SC, Kang Y, Kim KM, Kang C-S, Kim C. Constructing a Reference Genome in a Single Lab: The Possibility to Use Oxford Nanopore Technology. Plants. 2019; 8(8):270. https://doi.org/10.3390/plants8080270
Chicago/Turabian StyleLee, Yun Gyeong, Sang Chul Choi, Yuna Kang, Kyeong Min Kim, Chon-Sik Kang, and Changsoo Kim. 2019. "Constructing a Reference Genome in a Single Lab: The Possibility to Use Oxford Nanopore Technology" Plants 8, no. 8: 270. https://doi.org/10.3390/plants8080270
APA StyleLee, Y. G., Choi, S. C., Kang, Y., Kim, K. M., Kang, C.-S., & Kim, C. (2019). Constructing a Reference Genome in a Single Lab: The Possibility to Use Oxford Nanopore Technology. Plants, 8(8), 270. https://doi.org/10.3390/plants8080270