
Identification of Quality-Related Genomic Regions and Candidate Genes in Silage Maize by Combining GWAS and Meta-Analysis

Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Materials and Design
2.2. Phenotypic Data Acquisition and Processing
2.3. Genome-Wide Association Analysis
2.4. Collection and Collation of Meta-Analysis Data
2.5. Meta-Analysis of Quality-Related Traits
2.6. Candidate Gene Mining of Quality-Related Traits in Silage Maize
3. Results
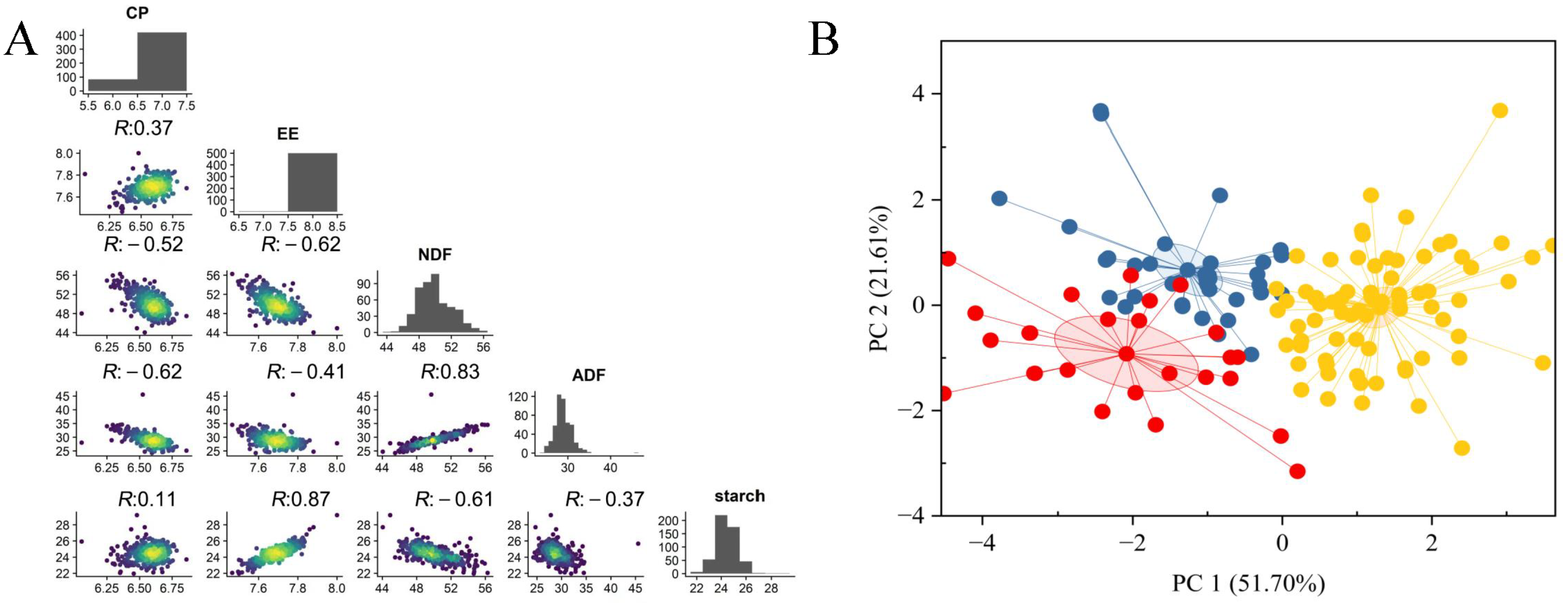
3.1. Phenotypic Analysis of Quality-Related Traits
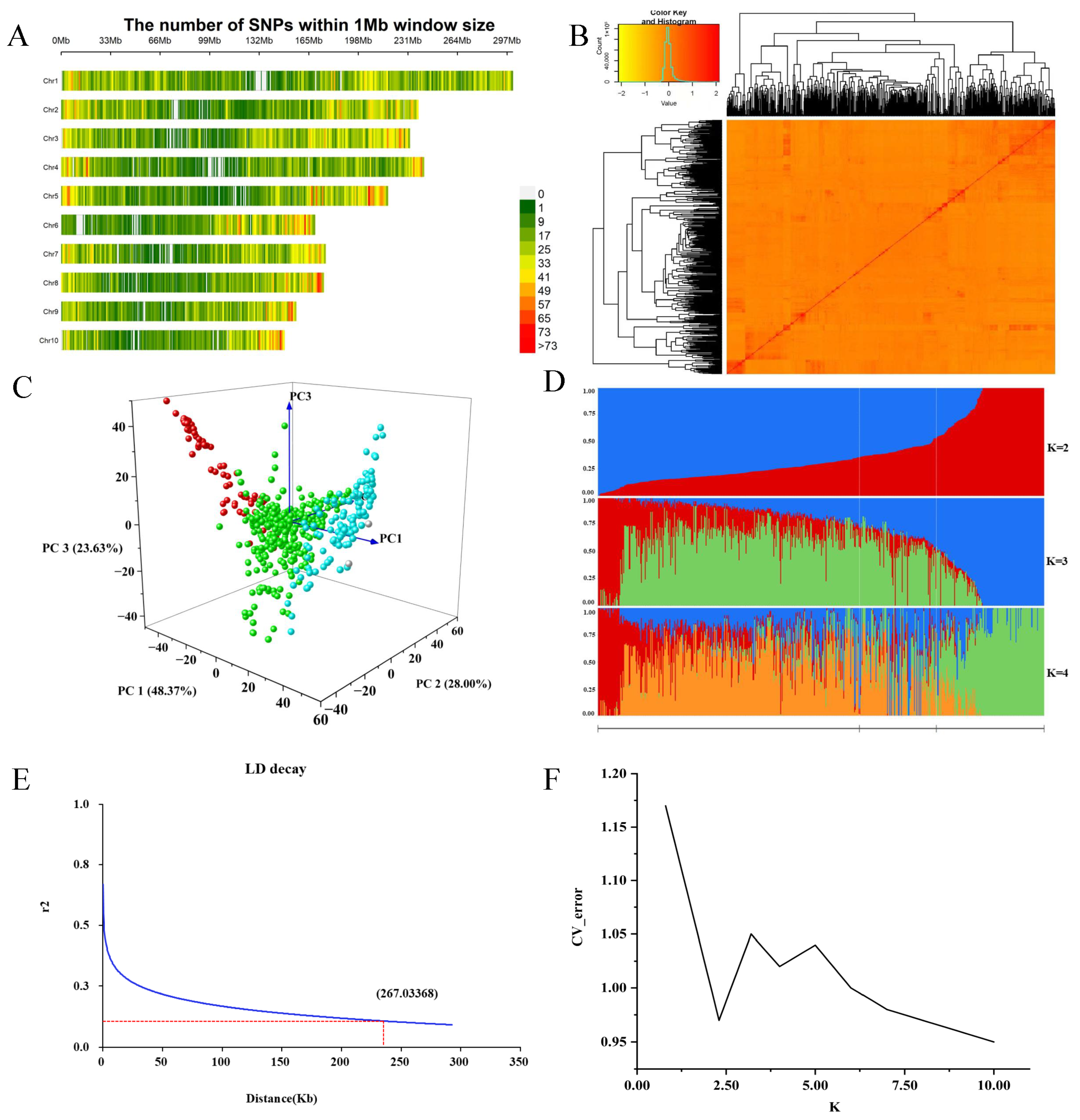
3.2. Analysis of Population Genetic Diversity and Genetic Relationships
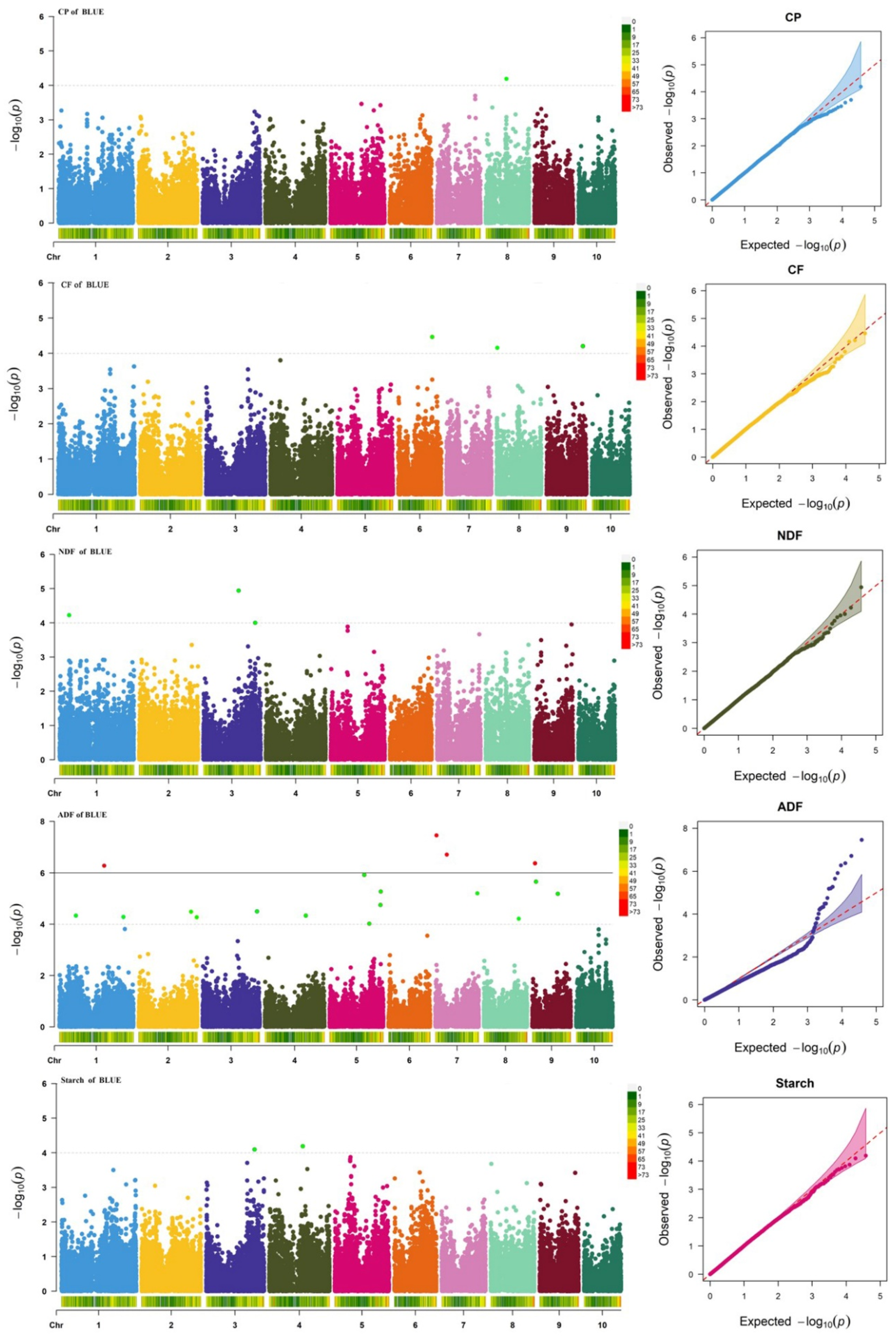
3.3. Genome-Wide Association Analysis of Quality-Related Traits
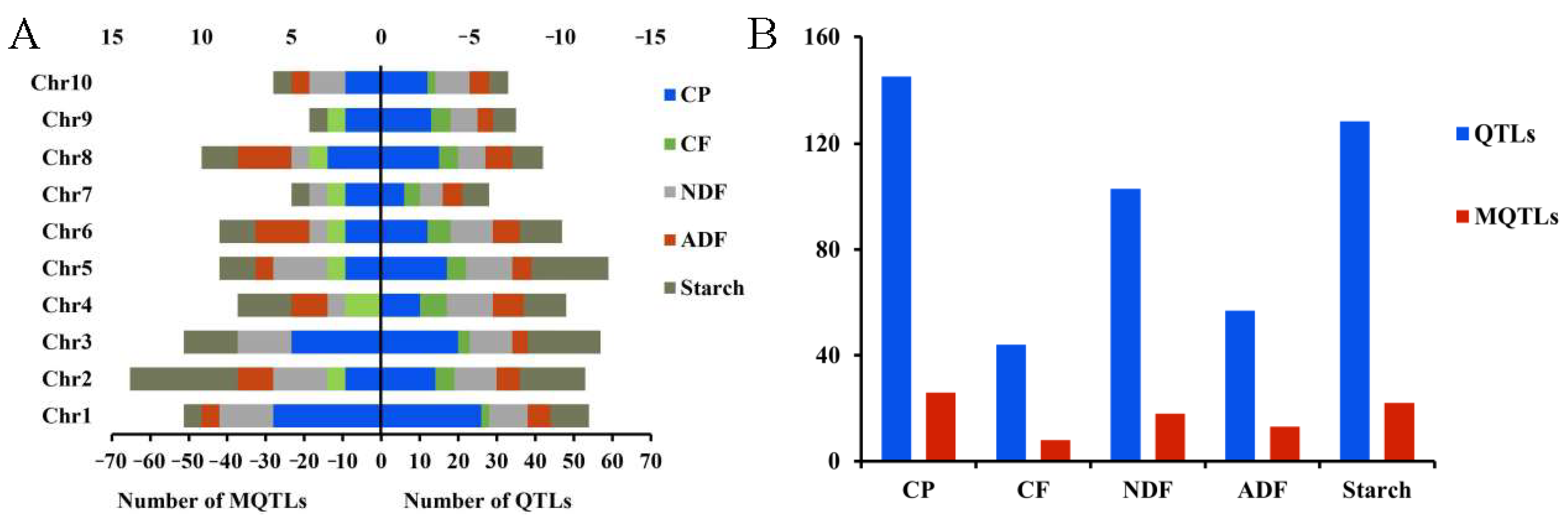
3.4. QTL Analysis of Quality-Related Traits in Silage Maize
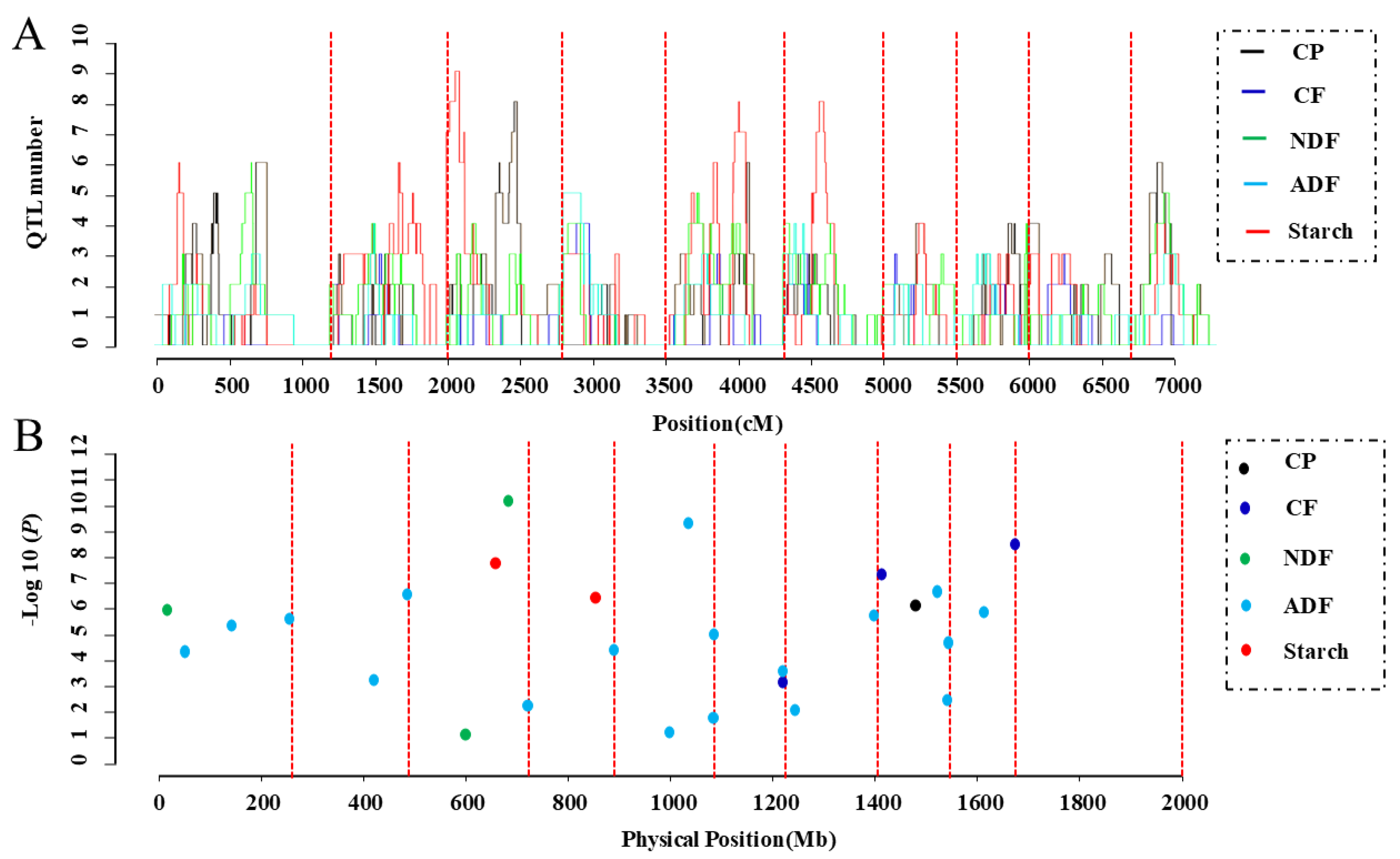
3.5. Construction of a “Consistent” Map for Quality Traits and QTL Meta-Analysis
3.6. Identification of Important Loci Using GWAS and Meta-Analysis
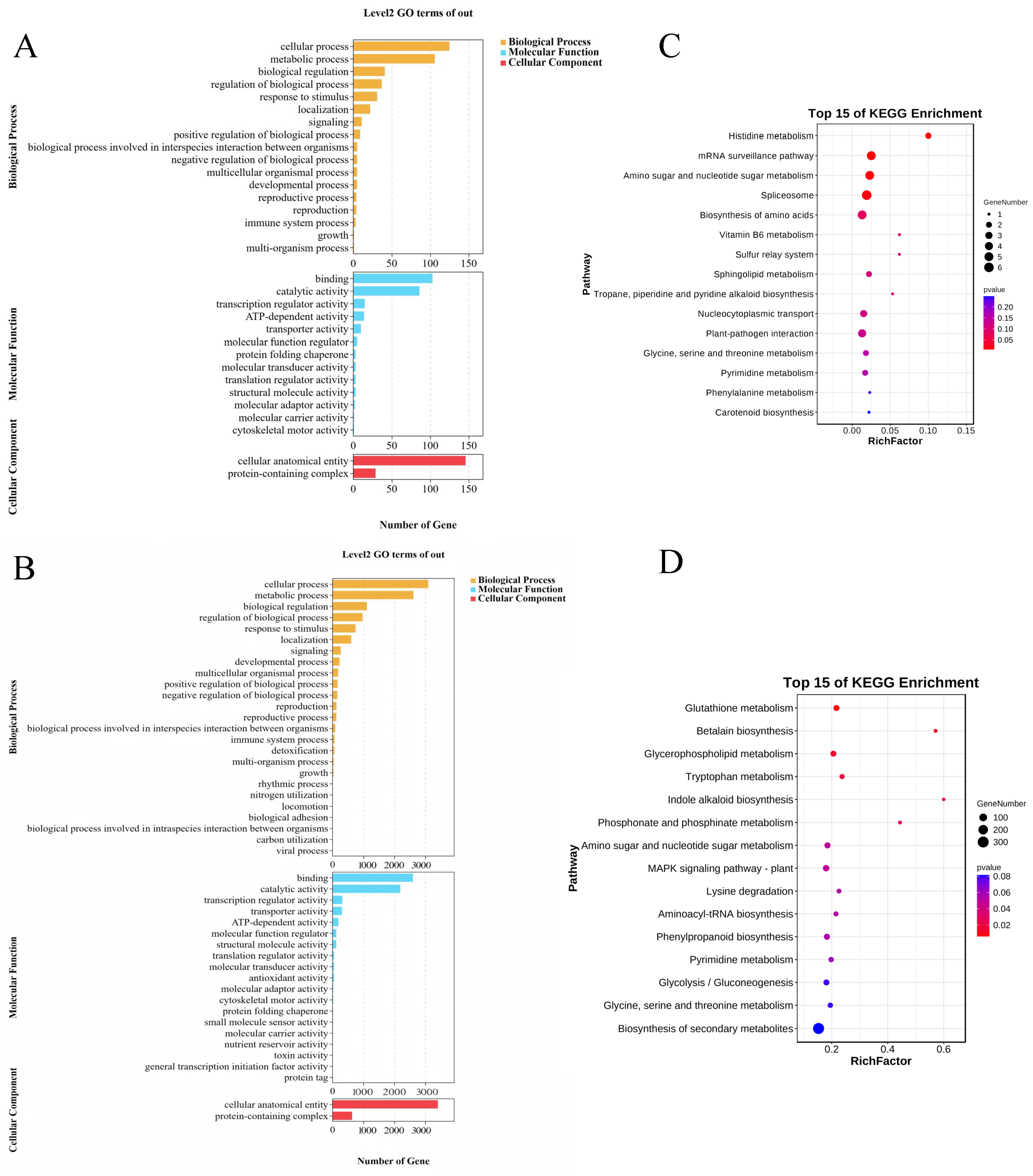
3.7. Identification and Functional Analysis of Candidate Genes
4. Discussions
4.1. Phenotypic Structure Variation in the Associated Population
4.2. Genetic Basis of Quality-Related Traits
4.3. Analysis of Genetic Basis of Quality Traits Using GWAS and Meta-Analysis
4.4. Functional Predictive Analysis of Candidate Genes
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Johnson, L.; Harrison, H.J.; Hunt, C.; Shinners, K.; Doggett, C.G.; Sapienza, D. Nutritive value of corn silage as affected by maturity and mechanical processing: A contemporary review. J. Dairy Sci. 1999, 82, 2813–2825. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.E.; Undersander, D. An Alternative to Relative Feed Value and Quality Index. In Proceedings of the 13th Annual Florida Ruminant Nutrition Symposium, Gainesville, FL, USA, 24–26 February 2002; pp. 16–32. [Google Scholar]
- Ertiro, B.T.; Zeleke, H.; Friesen, D.; Blummel, M.; Twumasi-Afriyie, S. Relationship between the performance of parental inbred lines and hybrids for food-feed traits in maize (Zea mays L.) in Ethiopia. Field Crop. Res. 2013, 153, 86–93. [Google Scholar] [CrossRef]
- Sharma, J.; Sharma, S.; Karnatam, K.S.; Raigar, O.P.; Lahkar, C.; Saini, D.K.; Kumar, S.; Singh, A.; Das, A.K.; Sharma, P. Surveying the opgenomic landscape of silage-quality traits in maize (Zea mays L.). Crop J. 2023, 11, 1893–1901. [Google Scholar] [CrossRef]
- Lübberstedt, T.; Melchinger, A.E.; Chris, C.; Schn, H.F.; Klein, U.D. QTL mapping in test crosses of European flint lines of maize: I. comparison of different testers for forage yield traits. Crop Sci. 1997, 37, 921–931. [Google Scholar] [CrossRef]
- Wang, Q.; Li, K.; Shi, H.; Liu, Z.; Wu, Y.; Wang, H.; Huang, C. Genetic analysis and QTL mapping of stalk cell wall components and digestibility in maize recombinant inbred lines from B73 × By804. Crop J. 2020, 8, 132–139. [Google Scholar] [CrossRef]
- Li, K.; Yang, X.; Liu, X.G.; Jiao, H.X.; Jin, W.Y.; Wang, Q.; Qian, M.F.; Qiang, L.S.; Wang, H.W.; Liu, Z.F. QTL analysis of the developmental changes in cell wall components and forage digestibility in maize (Zea mays L.). Integr. Agric. 2022, 21, 3501–3513. [Google Scholar] [CrossRef]
- Saini, D.K.; Chopra, Y.; Singh, J.; Sandhu, K.S.; Kumar, A.; Bazzer, S.; Srivastava, P. A comprehensive evaluation of mapping complex traits in wheat using genome-wide association studies. Mol. Breed. 2022, 42, 1–52. [Google Scholar] [CrossRef]
- Wang, H.W.; Li, K.; Hu, X.J.; Liu, Z.F.; Wu, Y.J.; Huang, C.L. Genome-wide association analysis of forage quality in maize mature stalk. BMC Plant Biol. 2016, 16, 227. [Google Scholar] [CrossRef]
- Truntzler, M.; Barrière, Y.; Sawkins, M.C.; Lespinasse, D.; Betran, J.; Charcosset, A.; Moreau, L. Meta-analysis of QTL involved in silage quality of maize and comparison with the position of candidate genes. Theor. Appl. Genet. 2010, 121, 1465–1482. [Google Scholar] [CrossRef]
- Zhang, H. Identification of quantitative trait loci underlying the protein, oil, and starch contents of maize in multiple environments. Euphytica Int. J. Plant Breed. 2015, 205, 169–183. [Google Scholar] [CrossRef]
- Goffinet, B.; Gerber, S. Quantitative trait loci: A meta-analysis. Genetics 2000, 155, 463–473. [Google Scholar] [CrossRef]
- Gopinath, I.; Muthusamy, V.; Katral, A.; Zunjare, R.U.; Madhavan, J.; Yathish, K.R.; Sekhar, J.C.; Hossain, F. Meta-QTL analysis and identification of candidate genes governing popping quality attributes in maize. S. Afr. J. Bot. 2023, 159, 461–471. [Google Scholar] [CrossRef]
- Darvasi, A.; Soller, M. A Simple Method to Calculate Resolving Power and Confidence Interval of QTL Map Location. Behav. Genet. 1997, 27, 125–132. [Google Scholar] [CrossRef]
- Gupta, M.; Choudhary, S. Meta-QTL analysis for mining of candidate genes and constitutive gene network development for fungal disease resistance in maize (Zea mays L.). Crop J. 2023, 11, 511–522. [Google Scholar] [CrossRef]
- Guillet-Claude, C.; Birolleau-Touchard, C.; Manicacci, D.; Fourmann, D.; Barraud, S.; Carret, V.; Martinant, J.P.; Barrière, Y. Genetic diversity associated with variation in silage corn digestibility for three o-methyltransferase genes involved in lignin biosynthesis. Theor. Appl. Genet. 2004, 110, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Guillet-Claude, C.; Birolleau-Touchard, C.; Manicacci, D.; Rogowsky, P.M.; Barrière, Y. Nucleotide diversity of the zmpox3 maize peroxidase gene: Relationships between a mite insertion in exon 2 and variation in forage maize digestibility. BMC Genet. 2004, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.R.; Zein, I.; Wenzel, G.; Krützfeldt, B.; Eder, J.; Ouzunova, M.; Lübberstedt, T. High levels of linkage disequilibrium and associations with forage quality at a phenylalanine ammonia-lyase locus in European maize (Zea mays L.) inbreds. Theor. Appl. Genet. 2007, 114, 307–319. [Google Scholar] [CrossRef]
- Andersen, J.R.; Zein, I.; Wenzel, G.; Darnhofer, B.; Eder, J.; Ouzunova, M.; Lübberstedt, T. Characterization of phenylpropanoid pathway genes within European maize (Zea mays L.) inbreds. BMC Plant Biol. 2008, 8, 2. [Google Scholar] [CrossRef]
- Barriere, Y.; Gibelin, C.; Argillier, O.; Mechin, V. Genetic analysis in recombinant inbred lines of early dent forage maize. QTL mapping for yield, earliness, starch, and crude protein contents from per se value and top cross experiments. Maydica 2001, 46, 253–266. [Google Scholar]
- Cardinal, A.; Lee, M.; Moore, K. Genetic mapping and analysis of quantitative trait loci affecting fiber and lignin content in maize. Theor. Appl. Genet. 2003, 106, 866–874. [Google Scholar] [CrossRef]
- Krakowsky, M.D.; Lee, M.; Coors, J.G. Quantitative trait loci for cell wall components in recombinant inbred lines of maize (Zea mays L.) ii: Leaf sheath tissue. Theor. Appl. Genet. 2006, 112, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, J.; Bian, Y.; Zhang, H.; Li, X.; Wu, Z.; Guo, G.; Lv, G. Identification of Genetic Variations and Candidate Genes Responsible for Stalk Sugar Content and Agronomic Traits in Fresh Corn via GWAS across Multiple Environments. Int. J. Mol. Sci. 2022, 23, 13490. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Warburton, M.; Crouch, J. Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci. 2011, 51, 433–449. [Google Scholar] [CrossRef]
- Kump, K.L.; Bradbury, P.J.; Wisser, R.J.; Buckler, E.S.; Belcher, A.R.; Oropeza-Rosas, M.A.; Zwonitzer, J.C.; Kresovich, S.; Mcmullen, M.D.; Ware, D. Genome-wide association study of quantitative resistance to southern leaf blight in the maize nested association mapping population. Nat. Genet. 2011, 43, 163–168. [Google Scholar] [CrossRef]
- Samayoa, L.F.; Rosa, A.M.; Bode, A.O.; James, B.H.; Ana, B. Genome-wide association study reveals a set of genes associated with resistance to the Mediterranean corn borer (Sesamia nonagrioides L.) in a maize diversity panel. BMC Plant Biol. 2015, 5, 35. [Google Scholar] [CrossRef]
- Riboulet, C.; Fabre, F.; Dénoue, D.; Martinantä, J.P. QTL mapping and candidate gene research for lignin content and cell wall digestibility in a top-cross of a flint maize recombinant inbred line progeny harvested at silage stage. Maydica 2008, 53, 1–9. [Google Scholar]
- Leng, P.F.; Ouzunova, M.; Landbeck, M.; Wenzel, G.; Eder, J.; Darnhofer, B.; Lübberstedt, T. Quantitative trait loci mapping of forage stover quality traits in six mapping populations derived from European elite maize germplasm. Plant Breed. 2018, 137, 139–147. [Google Scholar] [CrossRef]
- Edwards, M.D.; Stuber, C.W.; Wendel, J.F. Molecular-marker-facilitated investigations of quantitative-trait loci in maize. i. numbers, genomic distribution and types of gene action. Genetics 1987, 116, 113–125. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Liu, D.C.; Guo, X.L.; Yang, W.L.; Sun, J.Z.; Wang, D.W.; Zhang, A. Genomic distribution of quantitative trait loci for yield and yield-related traits in common wheat. J. Integr. Plant Biol. 2010, 52, 996–1007. [Google Scholar] [CrossRef]
- Dalware, A.V.; Rishi, S.; Singh, A.K.; Parida, S.K.; Tyagi, A.K. Regional association analysis of Meta QTLs delineates candidate grain size genes in rice. Front. Plant Sci. 2017, 8, 807. [Google Scholar] [CrossRef]
- Zhou, M.; Shabala, L.; Zhang, X.; Shabala, S.; Koutoulis, A. Meta-analysis of major QTL for abiotic stress tolerance in barley and implications for barley breeding. Planta 2017, 245, 283–295. [Google Scholar]
- Khahani, B.; Tavakol, E.; Shariati, J.V. Genome-wide meta-analysis on yield and yield-related QTLs in barley (Hordeum vulgare L.). Mol. Breed. 2019, 39, 56. [Google Scholar] [CrossRef]
- Camus-Kulandaivelu, L.; Veyrieras, J.B.; Madur, D.; Combes, V. Maize adaptation to temperate climate: Relationship between population structure and polymorphism in the dwarf8 gene. Genetics 2006, 172, 2449–2463. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, C.; Zhang, X.; Li, Y.; Wang, T. Transcriptome and GWAS analyses reveal candidate genes for seminal root length of maize seedlings under drought stress. Plant Sci. 2019, 292, 110380. [Google Scholar] [CrossRef]
- Urrutia, M.; Blein-Nicolas, M.; Prigent, S.; Bernillon, S.; Deborde, C.; Balliau, T.; Maucourt, M.; Jacob, D.; Ballias, P.; Bénard, C. Maize metabolome and proteome responses to controlled cold stress partly mimic early-sowing effects in the field and differ from those of Arabidopsis. Plant Cell Environ. 2021, 44, 1504–1521. [Google Scholar] [CrossRef]
- Zhang, H.W.; Lu, Y.; Ma, Y.; Fu, J.; Wang, G. Genetic and molecular control of grain yield in maize. Mol. Breed. 2021, 41, 1–15. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Crooks, P.A.; Sonar, V.N.; Friedman, D.B.; Michael, L.F. NADPH oxidase activity is essential for keap1/nrf2-mediated induction of gclc in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003, 63, 5636–5645. [Google Scholar]
- Hunt, L.; Lerner, F.; Ziegler, M. Nad-new roles in signalling and gene regulation in plants. New Phytol. 2004, 163, 31–44. [Google Scholar] [CrossRef]
- Hu, X. Abscisic acid-induced apoplastic H2O2 accumulation up-regulates the activities of chloroplastic and cytosolic antioxidant enzymes in maize leaves. Planta 2005, 223, 57–68. [Google Scholar] [CrossRef]
- Hu, X.L.; Jiang, M.Y.; Zhang, J.H.; Zhang, A.Y.; Lin, F.; Tan, M.P. Calcium/calmodulin is required for abscisic acid-induced antioxidant defense and functions both upstream and downstream of H2O2 production in leaves of maize plants. New Phytol. 2007, 173, 27–38. [Google Scholar] [CrossRef]
- Zhang, A.Y.; Jiang, M.Y.; Zhang, J.H.; Tan, M.P.; Hu, X.L. Mitogen-activated protein kinase is involved in abscisic acid-induced antioxidant defense and acts downstream of reactive oxygen species production in leaves of maize plants. Plant Physiol. 2006, 141, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Shevell, D.E.; Kunkel, T.; Chua, N.H. Cell wall alterations in the Arabidopsis emb30 mutant. Plant Cell 2000, 12, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Orfila, C.; Seymour, G.B.; Willats, W.G.T.; Huxham, I.M.; Knox, J.P. Altered middle lamella homogalacturonan and disrupted deposition of (1→5)-alpha-l-arabinan in the pericarp of cnr, a ripening mutant of tomato. Plant Physiol. 2001, 126, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Fomale, R.; Humphreys, J.M.; Hemm, M.R.; Denault, J.W. The ArabidopsisREF8 gene encodes the 3-hydroxylase of phenylpropanoid metabolism. Plant J. 2002, 30, 33–45. [Google Scholar]
- Formal, S. Cell wall modifications triggered by the down-regulation of coumarate 3-hydroxylase-1 in maize. Plant Sci. 2015, 236, 272–282. [Google Scholar] [CrossRef]
- Reddy, M.S.; Chen, F.; Shadle, G.; Jackson, L.; Aljoe, H.; Dixon, R.A. Targeted down-regulation of cytochrome p450 enzymes for forage quality improvement in alfalfa (Medicago sativa L.). Proc. Natl. Acad. Sci. USA 2005, 102, 16573–16578. [Google Scholar] [CrossRef]
- Ralph, J.; Akiyama, T.; Coleman, H.D.; Mansfield, S.D. Effects on lignin structure of coumarate 3-hydroxylase downregulation in poplar. Bioenerg. Res. 2012, 5, 1009–1019. [Google Scholar] [CrossRef]
- Fontaine, A.S.; Barrière, Y. Caffeic acid o-methyltransferase allelic polymorphism characterization and analysis in different maize inbred lines. Mol. Breed. 2003, 11, 69–75. [Google Scholar] [CrossRef]
- Zein, I.; Wenzel, G.; Andersen, J.R.; Lübberstedt, T. Low level of linkage disequilibrium at the cost (caffeic acid o-methyl transferase) locus in European maize (Zea mays L.). Genet. Resour. Crop Evol. 2007, 54, 139–148. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Minimum Value | Maximum Value | Average Value | CV (%) | Skewness | Kurtosis |
|---|---|---|---|---|---|---|
| CP | 6.06 | 6.77 | 6.57 ± 0.099 | 1.51 | −1.173 | 2.556 |
| CF | 7.47 | 8.00 | 7.69 ± 0.092 | 1.20 | −0.017 | 0.501 |
| NDF | 44.05 | 56.27 | 49.92 + 2.098 | 4.20 | 0.437 | 0.01 |
| ADF | 24.27 | 45.55 | 29.05 ± 2.074 | 7.14 | 1.573 | 0.497 |
| Starch | 21.97 | 29.21 | 24.51 ± 0.906 | 3.70 | 0.367 | 1.865 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Ding, Y.; Xu, C.; Chen, S.; Xia, C.; Zhang, L.; Sang, Z.; Zhang, Z. Identification of Quality-Related Genomic Regions and Candidate Genes in Silage Maize by Combining GWAS and Meta-Analysis. Plants 2025, 14, 2250. https://doi.org/10.3390/plants14152250
Lu Y, Ding Y, Xu C, Chen S, Xia C, Zhang L, Sang Z, Zhang Z. Identification of Quality-Related Genomic Regions and Candidate Genes in Silage Maize by Combining GWAS and Meta-Analysis. Plants. 2025; 14(15):2250. https://doi.org/10.3390/plants14152250
Chicago/Turabian StyleLu, Yantian, Yongfu Ding, Can Xu, Shubin Chen, Chunlan Xia, Li Zhang, Zhiqing Sang, and Zhanqin Zhang. 2025. "Identification of Quality-Related Genomic Regions and Candidate Genes in Silage Maize by Combining GWAS and Meta-Analysis" Plants 14, no. 15: 2250. https://doi.org/10.3390/plants14152250
APA StyleLu, Y., Ding, Y., Xu, C., Chen, S., Xia, C., Zhang, L., Sang, Z., & Zhang, Z. (2025). Identification of Quality-Related Genomic Regions and Candidate Genes in Silage Maize by Combining GWAS and Meta-Analysis. Plants, 14(15), 2250. https://doi.org/10.3390/plants14152250