
Bioassay-Guided Isolation of cis-Clerodane Diterpenoids and Monoglycerides from the Leaves of Solidago gigantea and Their Antimicrobial Activities

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results and Discussion
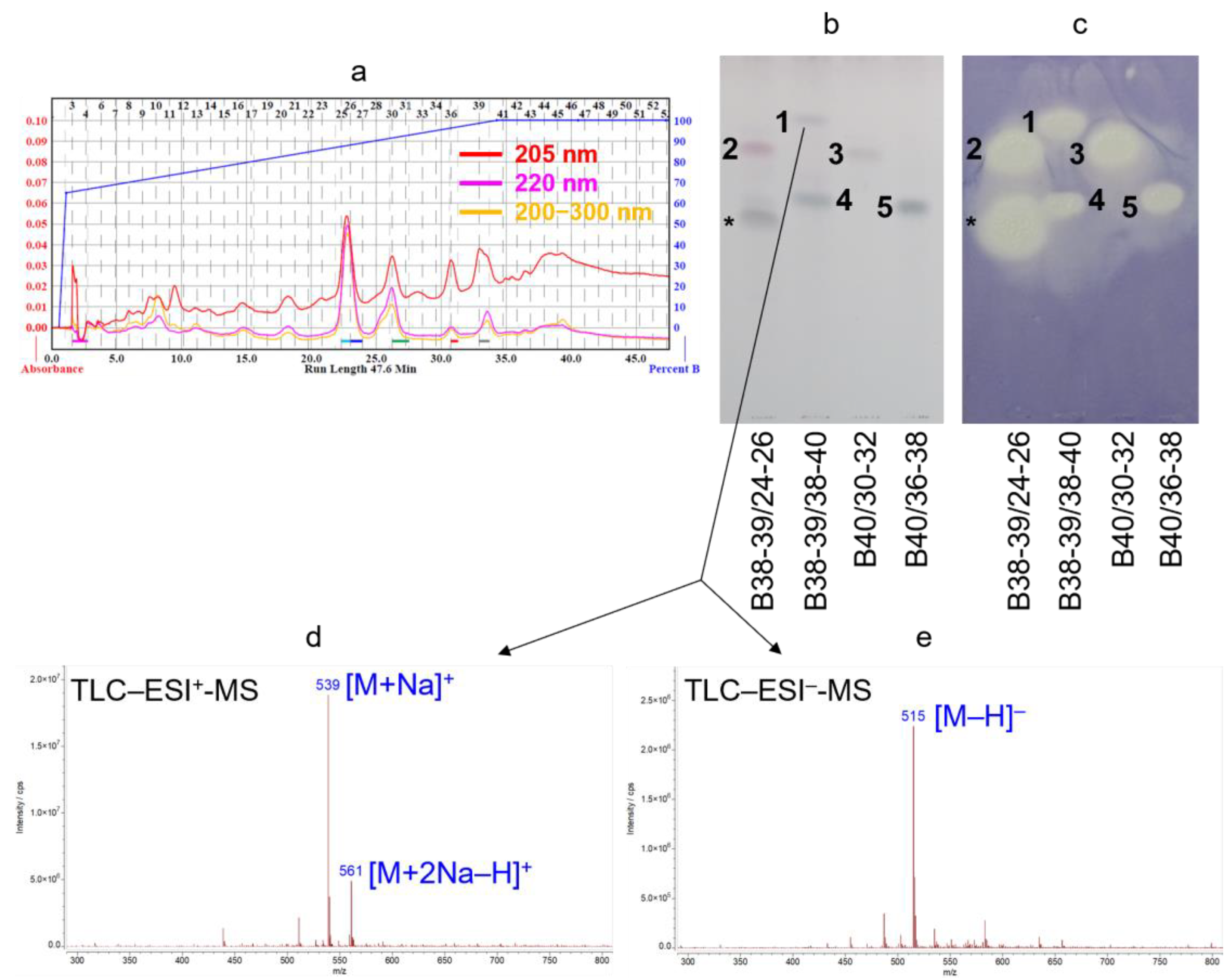
2.1. Bioassay-Guided Isolation
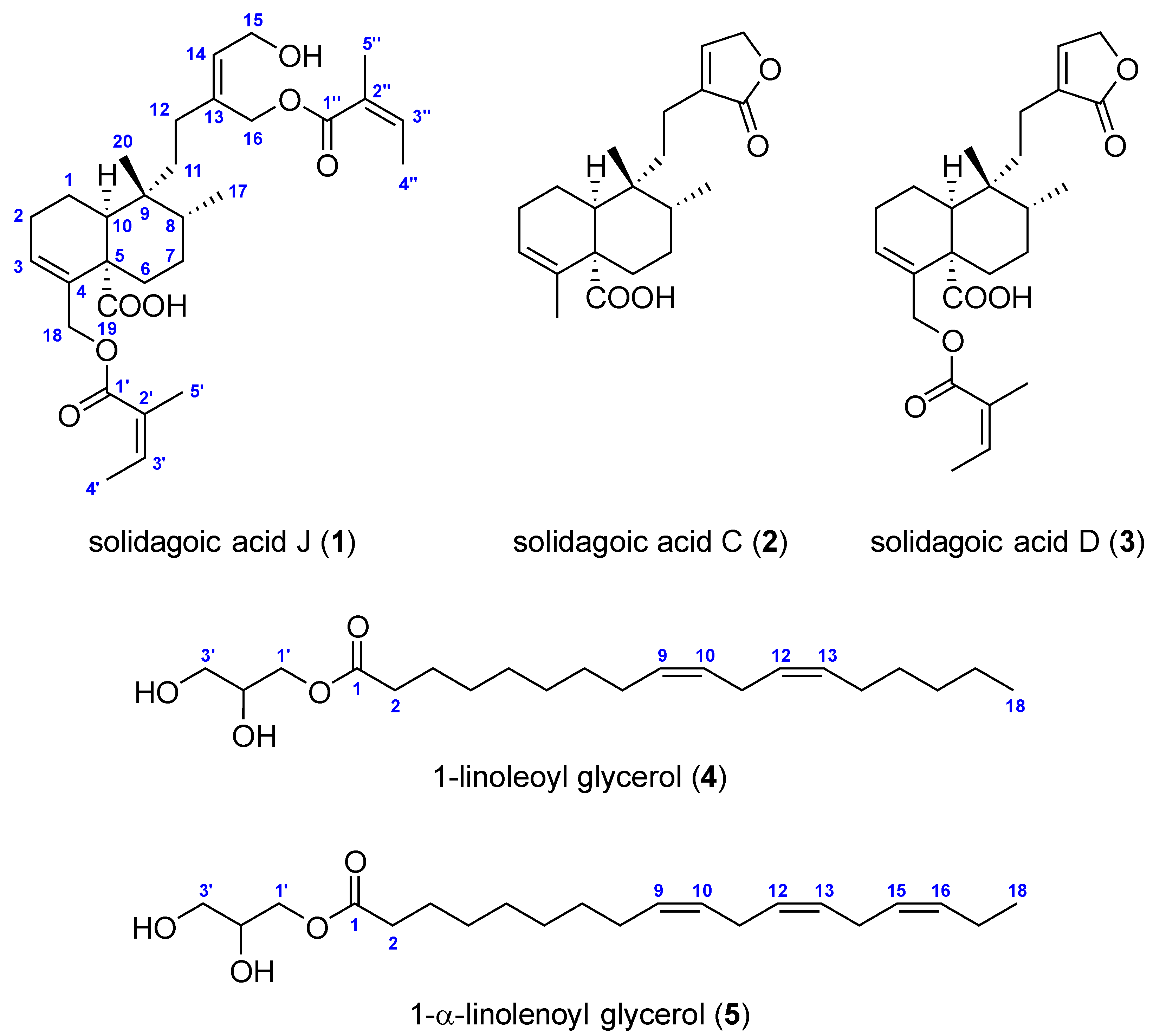
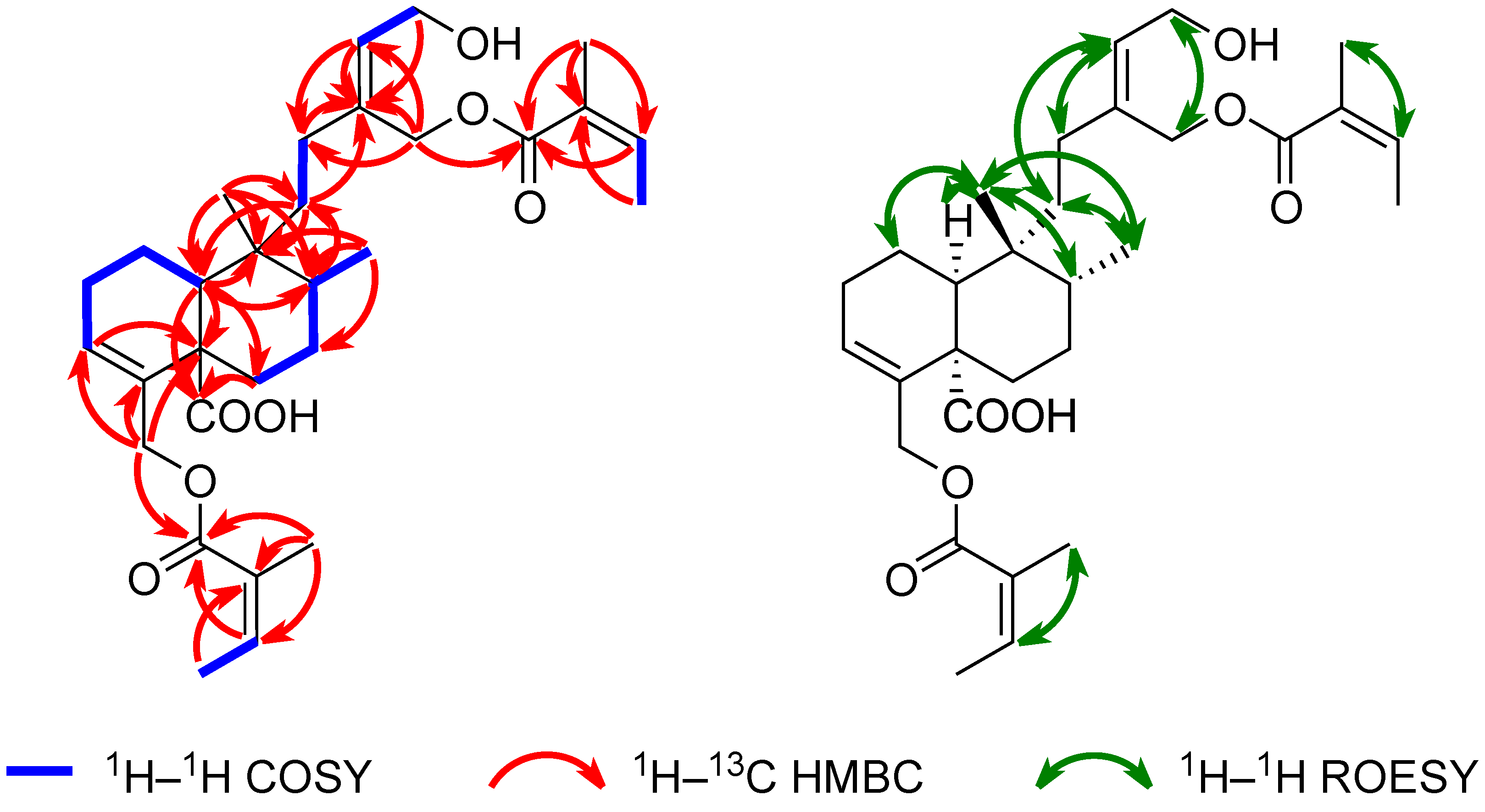
2.2. Structure Elucidation
2.3. Antimicrobial Assays
3. Materials and Methods
3.1. Materials
3.2. Plant Material
3.3. (High-Performance) TLC–UV/FLD Method
3.3.1. Detection via p-Anisaldehyde Sulfuric Acid Reagent
3.3.2. Detection via Planar B. subtilis Antibacterial Assay
3.4. Extraction and Isolation
3.5. Compound Characterization
3.6. TLC–ESI-MS
3.7. FIA–HR-HESI-MS(/MS)
3.8. NMR Spectroscopy
3.9. Polarimetry, UV, and ATR-FTIR Spectroscopy
3.9.1. Polarimetry
3.9.2. UV Spectroscopy
3.9.3. ATR-FTIR Spectroscopy
3.10. In Vitro Antimicrobial Activity Assays
3.10.1. Determination of Minimal Inhibitory Concentration (MIC) Values
3.10.2. Determination of Minimal Bactericidal Concentration (MBC) Values
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Semple, J.C.; Beck, J.B. Revised Infrageneric Classification of Solidago (Asteraceae: Astereae). Phytoneuron 2021, 10, 1–6. [Google Scholar]
- Fursenco, C.; Calalb, T.; Uncu, L.; Dinu, M.; Ancuceanu, R. Solidago virgaurea L.: A Review of Its Ethnomedicinal Uses, Phytochemistry, and Pharmacological Activities. Biomolecules 2020, 10, 1619. [Google Scholar] [CrossRef] [PubMed]
- Hejda, M.; Pyšek, P.; Jarošík, V. Impact of Invasive Plants on the Species Richness, Diversity and Composition of Invaded Communities. J. Ecol. 2009, 97, 393–403. [Google Scholar] [CrossRef]
- Goossens, E.P.; Mertens, W.; Olde Venterink, H. Solidago gigantea Invasion Homogenizes Soil Properties and Native Plant Communities. Biol. Invasions 2024, 26, 3315–3327. [Google Scholar] [CrossRef]
- Pal, R.W.; Chen, S.; Nagy, D.U.; Callaway, R.M. Impacts of Solidago gigantea on Other Species at Home and Away. Biol. Invasions 2015, 17, 3317–3325. [Google Scholar] [CrossRef]
- Woźniak, D.; Ślusarczyk, S.; Domaradzki, K.; Dryś, A.; Matkowski, A. Comparison of Polyphenol Profile and Antimutagenic and Antioxidant Activities in Two Species Used as Source of Solidaginis herba—Goldenrod. Chem. Biodivers. 2018, 15, e1800023. [Google Scholar] [CrossRef]
- Leuschner, J. Anti-Inflammatory, Spasmolytic and Diuretic Effects of a Commercially Available Solidago gigantea Herb. Extract. Arzneimittelforschung 1995, 45, 165–168. [Google Scholar]
- Kołodziej, B. Antibacterial and Antimutagenic Activity of Extracts Aboveground Parts of Three Solidago Species: Solidago virgaurea L., Solidago canadensis L. and Solidago gigantea Ait. J. Med. Plants Res. 2011, 5, 6770–6779. [Google Scholar] [CrossRef]
- Anžlovar, S.; Janeš, D.; Dolenc Koce, J. The Effect of Extracts and Essential Oil from Invasive Solidago spp. and Fallopia japonica on Crop-Borne Fungi and Wheat Germination. Food Technol. Biotechnol. 2020, 58, 273–283. [Google Scholar] [CrossRef]
- Nkuimi Wandjou, J.G.; Quassinti, L.; Gudžinskas, Z.; Nagy, D.U.; Cianfaglione, K.; Bramucci, M.; Maggi, F. Chemical Composition and Antiproliferative Effect of Essential Oils of Four Solidago Species (S. canadensis, S. gigantea, S. virgaurea and S. × niederederi). Chem. Biodivers. 2020, 17, e2000685. [Google Scholar] [CrossRef]
- Benelli, G.; Pavela, R.; Cianfaglione, K.; Nagy, D.U.; Canale, A.; Maggi, F. Evaluation of Two Invasive Plant Invaders in Europe (Solidago canadensis and Solidago gigantea) as Possible Sources of Botanical Insecticides. J. Pest Sci. 2019, 92, 805–821. [Google Scholar] [CrossRef]
- Zekič, J.; Vovk, I.; Glavnik, V. Extraction and Analyses of Flavonoids and Phenolic Acids from Canadian Goldenrod and Giant Goldenrod. Forests 2020, 12, 40. [Google Scholar] [CrossRef]
- Radusiene, J.; Marska, M.; Ivanauskas, L.; Jakstas, V.; Karpaviciene, B. Assessment of Phenolic Compound Accumulation in Two Widespread Goldenrods. Ind. Crops Prod. 2015, 63, 158–166. [Google Scholar] [CrossRef]
- Marksa, M.; Zymone, K.; Ivanauskas, L.; Radušienė, J.; Pukalskas, A.; Raudone, L. Antioxidant Profiles of Leaves and Inflorescences of Native, Invasive and Hybrid Solidago Species. Ind. Crops Prod. 2020, 145, 112123. [Google Scholar] [CrossRef]
- Radušienė, J.; Karpavičienė, B.; Marksa, M.; Ivanauskas, L.; Raudonė, L. Distribution Patterns of Essential Oil Terpenes in Native and Invasive Solidago Species and Their Comparative Assessment. Plants 2022, 11, 1159. [Google Scholar] [CrossRef]
- Kalemba, D.; Marschall, H.; Bradesi, P. Constituents of the Essential Oil of Solidago gigantea Ait. (Giant Goldenrod). Flavour Fragr. J. 2001, 16, 19–26. [Google Scholar] [CrossRef]
- Henderson, M.S.; McCrindle, R.; McMaster, D. Constituents of Solidago Species. Part V. Non-Acidic Diterpenoids from Solidago gigantea var. serotina. Can. J. Chem. 1973, 51, 1346–1358. [Google Scholar] [CrossRef]
- Jurenitsch, J.; Maurer, J.; Rain, U.; Robien, W. Diterpenebutenolides in Solidago gigantea. Phytochemistry 1988, 27, 626–627. [Google Scholar] [CrossRef]
- Baglyas, M.; Ott, P.G.; Garádi, Z.; Glavnik, V.; Béni, S.; Vovk, I.; Móricz, Á.M. High-Performance Thin-Layer Chromatography—Antibacterial Assay First Reveals Bioactive Clerodane Diterpenes in Giant Goldenrod (Solidago gigantea Ait.). J. Chromatogr. A 2022, 1677, 463308. [Google Scholar] [CrossRef]
- Choi, S.Z.; Choi, S.U.; Lee, K.R. Pytochemical Constituents of the Aerial Parts from Solidago virga-aurea var. gigantea. Arch. Pharmacal Res. 2004, 27, 164–168. [Google Scholar] [CrossRef]
- Reznicek, G.; Jurenitsch, J.; Michl, G.; Haslinger, E. The First Structurally Confirmed Saponin from Solidago gigantea: Structure Elucidation by Modern NMR Techniques. Tetrahedron Lett. 1989, 30, 4097–4100. [Google Scholar] [CrossRef]
- Reznicek, G.; Jurenitsch, J.; Michl, G.; Haslinger, E.; Hiller, K.; Kubelka, W. Structure of Two New Saponins from Solidago gigantea. Planta Med. 1989, 55, 623–624. [Google Scholar] [CrossRef]
- Reznicek, G.; Freiler, M.; Schader, M.; Schmidt, U. Determination of the Content and the Composition of the Main Saponins from Solidago gigantea AIT. Using High-Performance Liquid Chromatography. J. Chromatogr. A 1996, 755, 133–137. [Google Scholar] [CrossRef]
- Baglyas, M.; Ott, P.G.; Schwarczinger, I.; Nagy, J.K.; Darcsi, A.; Bakonyi, J.; Móricz, Á.M. Antimicrobial Diterpenes from Rough Goldenrod (Solidago rugosa Mill.). Molecules 2023, 28, 3790. [Google Scholar] [CrossRef]
- Móricz, Á.M.; Krüzselyi, D.; Ott, P.G.; Garádi, Z.; Béni, S.; Morlock, G.E.; Bakonyi, J. Bioactive Clerodane Diterpenes of Giant Goldenrod (Solidago gigantea Ait.) Root Extract. J. Chromatogr. A 2021, 1635, 461727. [Google Scholar] [CrossRef]
- Krüzselyi, D.; Bakonyi, J.; Ott, P.G.; Darcsi, A.; Csontos, P.; Morlock, G.E.; Móricz, Á.M. Goldenrod Root Compounds Active against Crop Pathogenic Fungi. J. Agric. Food Chem. 2021, 69, 12686–12694. [Google Scholar] [CrossRef]
- Móricz, Á.M.; Ott, P.G.; Häbe, T.T.; Darcsi, A.; Böszörményi, A.; Alberti, Á.; Krüzselyi, D.; Csontos, P.; Béni, S.; Morlock, G.E. Effect-Directed Discovery of Bioactive Compounds Followed by Highly Targeted Characterization, Isolation and Identification, Exemplarily Shown for Solidago virgaurea. Anal. Chem. 2016, 88, 8202–8209. [Google Scholar] [CrossRef]
- Nishidono, Y.; Tanaka, K. New Clerodane Diterpenoids from Solidago altissima and Stereochemical Elucidation via 13C NMR Chemical Shift Analysis. Tetrahedron 2022, 110, 132691. [Google Scholar] [CrossRef]
- Nogueira, R.T.; Shepherd, G.J.; Laverde, A., Jr.; Marsaioli, A.J.; Imamura, P.M. Clerodane-Type Diterpenes from the Seed Pods of Hymenaea courbaril var. stilbocarpa. Phytochemistry 2001, 58, 1153–1157. [Google Scholar] [CrossRef]
- Manabe, S.; Nishino, C. Stereochemistry of cis-Clerodane Diterpenes. Tetrahedron 1986, 42, 3461–3470. [Google Scholar] [CrossRef]
- Miyata, O.; Shinada, T.; Ninomiya, I.; Naito, T. A Facile Conversion of (Z)-2-Alkenoic Esters into the (E)-Isomers with Diphenyl Disulfide. Synthesis 1990, 1990, 1123–1125. [Google Scholar] [CrossRef]
- Anthonsen, T.; Henderson, M.S.; Martin, A.; Murray, R.D.H.; McCrindle, R.; McMaster, D. Constituents of Solidago Species. Part IV. Solidagoic Acids A and B, Diterpenoids from Solidago gigantea var. serotina. Can. J. Chem. 1973, 51, 1332–1345. [Google Scholar] [CrossRef]
- Starks, C.M.; Williams, R.B.; Goering, M.G.; O’Neil-Johnson, M.; Norman, V.L.; Hu, J.-F.; Garo, E.; Hough, G.W.; Rice, S.M.; Eldridge, G.R. Antibacterial Clerodane Diterpenes from Goldenrod (Solidago virgaurea). Phytochemistry 2010, 71, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Jang, H.-J.; Kim, Y.; Oh, H.-M.; Lee, S.; Jung, K.; Kim, Y.-H.; Lee, W.-S.; Lee, S.-W.; Rho, M.-C. Inhibitory Effects of IL-6-Induced STAT3 Activation of Bio-Active Compounds Derived from Salvia plebeia R.Br. Process Biochem. 2016, 51, 2222–2229. [Google Scholar] [CrossRef]
- Gläser, P.; Dawid, C.; Meister, S.; Bader-Mittermaier, S.; Schott, M.; Eisner, P.; Hofmann, T. Molecularization of Bitter Off-Taste Compounds in Pea-Protein Isolates (Pisum sativum L.). J. Agric. Food Chem. 2020, 68, 10374–10387. [Google Scholar] [CrossRef]
- Oku, N.; Hayashi, S.; Yamaguchi, Y.; Takenaka, H.; Igarashi, Y. Nostochopcerol, a New Antibacterial Monoacylglycerol from the Edible Cyanobacterium Nostochopsis lobatus. Beilstein J. Org. Chem. 2023, 19, 133–138. [Google Scholar] [CrossRef]
- Ogihara, T.; Amano, N.; Mitsui, Y.; Fujino, K.; Ohta, H.; Takahashi, K.; Matsuura, H. Determination of the Absolute Configuration of a Monoglyceride Antibolting Compound and Isolation of Related Compounds from Radish Leaves (Raphanus sativus). J. Nat. Prod. 2017, 80, 872–878. [Google Scholar] [CrossRef]
- Li, R.; Morris-Natschke, S.L.; Lee, K.-H. Clerodane Diterpenes: Sources, Structures, and Biological Activities. Nat. Prod. Rep. 2016, 33, 1166–1226. [Google Scholar] [CrossRef]
- Herrera-Mayorga, V.; Guerrero-Sánchez, J.A.; Méndez-Álvarez, D.; Paredes-Sánchez, F.A.; Rodríguez-Duran, L.V.; Niño-García, N.; Paz-González, A.D.; Rivera, G. Insecticidal Activity of Organic Extracts of Solidago graminifolia and Its Main Metabolites (Quercetin and Chlorogenic Acid) against Spodoptera frugiperda: An In Vitro and In Silico Approach. Molecules 2022, 27, 3325. [Google Scholar] [CrossRef]
- Kabara, J.J. Antimicrobial Agents Derived from Fatty Acids. J. Am. Oil Chem. Soc. 1984, 61, 397–403. [Google Scholar] [CrossRef]
- Reiner, D.S.; Wang, C.-S.; Gillin, F.D. Human Milk Kills Giardia Lamblia by Generating Toxic Lipolytic Products. J. Infect. Dis. 1986, 154, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.K.; Skurrie, I.J.; May, J.T. Use of Semliki Forest Virus to Identify Lipid-Mediated Antiviral Activity and Anti-Alphavirus Immunoglobulin A in Human Milk. Infect. Immun. 1978, 19, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Nie, L.; Wang, Q.; Ouyang, D.; Zhang, L.; Yuan, Y.; Hong, Y.; Wang, J.; Hu, X. Phytochemical Constituents, Chemotaxonomic Significance and Anti-Arthritic Effect of Eucommia ulmoides Oliver Staminate Flowers. Nat. Prod. Res. 2020, 36, 3455–3459. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Wang, S.; Pan, K.; Luo, H.; Zou, K.; Wang, H. Ligulariatinside A, a New Sesquiterpene Glycoside from Roots of Ligularia veitchiana. Nat. Prod. Res. 2025, 39, 262–268. [Google Scholar] [CrossRef]
- Ko, H.N.; Kim, J.E.; Jo, Y.J.; Hong, S.H.; Yang, D.W.; Kim, G.O.; Lee, N.H. Antimelanogenic Effects of Raphanus sativus L. var. niger Roots on α-MSH Stimulated B16F10 Melanoma Cells. Bull. Korean Chem. Soc. 2018, 39, 1254–1258. [Google Scholar] [CrossRef]
- Jiang, J.; Yu, X.; Fang, Y.; Zhang, Y.; Li, N.; Wang, K. Chemical Constituents of the Roots of Patrinia scabiosaefolia and the Cytotoxicity of Patrineolignans A and B. Chem. Nat. Compd. 2017, 53, 143–146. [Google Scholar] [CrossRef]
- Reuk-ngam, N.; Chimnoi, N.; Khunnawutmanotham, N.; Techasakul, S. Antimicrobial Activity of Coronarin D and Its Synergistic Potential with Antibiotics. BioMed Res. Int. 2014, 2014, 581985. [Google Scholar] [CrossRef]
- Yu, R.; Li, X.; Yi, P.; Wen, P.; Wang, S.; Liao, C.; Song, X.; Wu, H.; He, Z.; Li, C. Isolation and Identification of Chemical Compounds from Agaricus blazei Murrill and Their In Vitro Antifungal Activities. Molecules 2023, 28, 7321. [Google Scholar] [CrossRef]
- Höller, U.; Wright, A.D.; Matthee, G.F.; Konig, G.M.; Draeger, S.; Aust, H.-J.; Schulz, B. Fungi from Marine Sponges: Diversity, Biological Activity and Secondary Metabolites. Mycol. Res. 2000, 104, 1354–1365. [Google Scholar] [CrossRef]
- Peng, B.; Cai, J.; Xiao, Z.; Liu, M.; Li, X.; Yang, B.; Fang, W.; Huang, Y.-Y.; Chen, C.; Zhou, X.; et al. Bioactive Polyketides and Benzene Derivatives from Two Mangrove Sediment-Derived Fungi in the Beibu Gulf. Mar. Drugs 2023, 21, 327. [Google Scholar] [CrossRef]
- Kusumah, D.; Wakui, M.; Murakami, M.; Xie, X.; Yukihito, K.; Maeda, I. Linoleic Acid, α-Linolenic Acid, and Monolinolenins as Antibacterial Substances in the Heat-Processed Soybean Fermented with Rhizopus oligosporus. Biosci. Biotechnol. Biochem. 2020, 84, 1285–1290. [Google Scholar] [CrossRef]
- Móricz, Á.M.; Ott, P.G.; Yüce, I.; Darcsi, A.; Béni, S.; Morlock, G.E. Effect-Directed Analysis via Hyphenated High-Performance Thin-Layer Chromatography for Bioanalytical Profiling of Sunflower Leaves. J. Chromatogr. A 2018, 1533, 213–220. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Solidagoic Acid J (1) | |
|---|---|---|
| δH (ppm), Multiplicity, J (Hz) | δC (ppm), Type | |
| 1a | 1.75, m | 19.6, CH2 |
| 1b | 1.54, ov. | |
| 2a | 2.17, m | 26.5, CH2 |
| 2b | ||
| 3 | 5.91, t (4.0) | 128.1, CH |
| 4 | – | 136.2, C |
| 5 | – | 50.0, C |
| 6a | 2.41, dt (13.9, 2.8) | 30.1, CH2 |
| 6b | 1.51, ov. | |
| 7a | 1.68, ov. | 28.0, CH2 |
| 7b | 1.33, m | |
| 8 | 1.65, ov. | 37.0, CH |
| 9 | – | 38.7, C |
| 10 | 2.32, dd (12.9, 1.5) | 42.6, CH |
| 11a | 1.59, ov. | 30.0, CH2 |
| 11b | 1.22, td (13.7, 4.8) | |
| 12a | 2.26, td (14.3, 2.4) | 29.4, CH2 |
| 12b | 1.94, td (13.6, 4.7) | |
| 13 | – | 139.1, C |
| 14 | 5.72, t (7.1) | 128.7, CH |
| 15 | 4.26, d (7.1) | 58.7, CH2 |
| 16a | 4.80, d (12.3) | 61.7, CH2 |
| 16b | 4.66, d (12.3) | |
| 17 | 0.79, d (6.4) | 15.8, CH3 |
| 18 | 4.51, m | 64.5, CH2 |
| 19 | – | 178.8, C |
| 20 | 0.92, s | 27.0, CH3 |
| 1′ | – | 167.7, C |
| 2′ | – | 128.0, C |
| 3′ | 6.04, qq (7.2, 1.3) | 138.3, CH |
| 4′ | 1.97, ov. | 15.9, CH3 |
| 5′ | 1.88, ov. | 20.8, CH3 |
| 1″ | – | 168.7, C |
| 2″ | – | 127.7, C |
| 3″ | 6.10, qq (7.2, 1.3) | 139.3, CH |
| 4″ | 1.97, ov. | 16.0, CH3 |
| 5″ | 1.86, ov. | 20.7, CH3 |
| Compounds | Bs (G+) | Cff (G+) | Cm (G+) | Rf (G+) | Pstom (G−) | Xap (G−) | Bip | Fg | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MIC | |
| 1 | 67 | >133 | 133 | >133 | 67 | >133 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| 2 | >133 | >133 | >133 | >133 | 133 | >133 | >133 | >133 | >133 | >133 | >133 | >133 | >333 b | >333 c |
| 3 | 133 | >133 | >133 | >133 | 33 | >133 | 133 | >133 | >133 | >133 | >133 | >133 | >333 c | >333 d |
| 4 | >133 | >133 | >133 | >133 | 33 | >133 | >133 | >133 | >133 | >133 | >133 | >133 | >167 | >167 |
| 5 | >133 | >133 | >133 | >133 | 17 | >133 | 33 | >133 | >133 | >133 | >133 | >133 | >167 | >167 |
| Gent a | 0.8 | 1.7 | 0.8 | 0.8 | 1.7 | 1.7 | 0.8 | 1.7 | 0.4 | 0.8 | 1.7 | 1.7 | ||
| Ben a | 1042 | 521 | ||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baglyas, M.; Ott, P.G.; Bozsó, Z.; Schwarczinger, I.; Bakonyi, J.; Dlauchy, D.; Darcsi, A.; Varga, S.; Móricz, Á.M. Bioassay-Guided Isolation of cis-Clerodane Diterpenoids and Monoglycerides from the Leaves of Solidago gigantea and Their Antimicrobial Activities. Plants 2025, 14, 2152. https://doi.org/10.3390/plants14142152
Baglyas M, Ott PG, Bozsó Z, Schwarczinger I, Bakonyi J, Dlauchy D, Darcsi A, Varga S, Móricz ÁM. Bioassay-Guided Isolation of cis-Clerodane Diterpenoids and Monoglycerides from the Leaves of Solidago gigantea and Their Antimicrobial Activities. Plants. 2025; 14(14):2152. https://doi.org/10.3390/plants14142152
Chicago/Turabian StyleBaglyas, Márton, Péter G. Ott, Zoltán Bozsó, Ildikó Schwarczinger, József Bakonyi, Dénes Dlauchy, András Darcsi, Szilárd Varga, and Ágnes M. Móricz. 2025. "Bioassay-Guided Isolation of cis-Clerodane Diterpenoids and Monoglycerides from the Leaves of Solidago gigantea and Their Antimicrobial Activities" Plants 14, no. 14: 2152. https://doi.org/10.3390/plants14142152
APA StyleBaglyas, M., Ott, P. G., Bozsó, Z., Schwarczinger, I., Bakonyi, J., Dlauchy, D., Darcsi, A., Varga, S., & Móricz, Á. M. (2025). Bioassay-Guided Isolation of cis-Clerodane Diterpenoids and Monoglycerides from the Leaves of Solidago gigantea and Their Antimicrobial Activities. Plants, 14(14), 2152. https://doi.org/10.3390/plants14142152