
Comparative Analysis of Differentially Expressed Genes and Metabolites in Waxy Maize Inbred Lines with Distinct Twin-Shoot Phenotypes

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
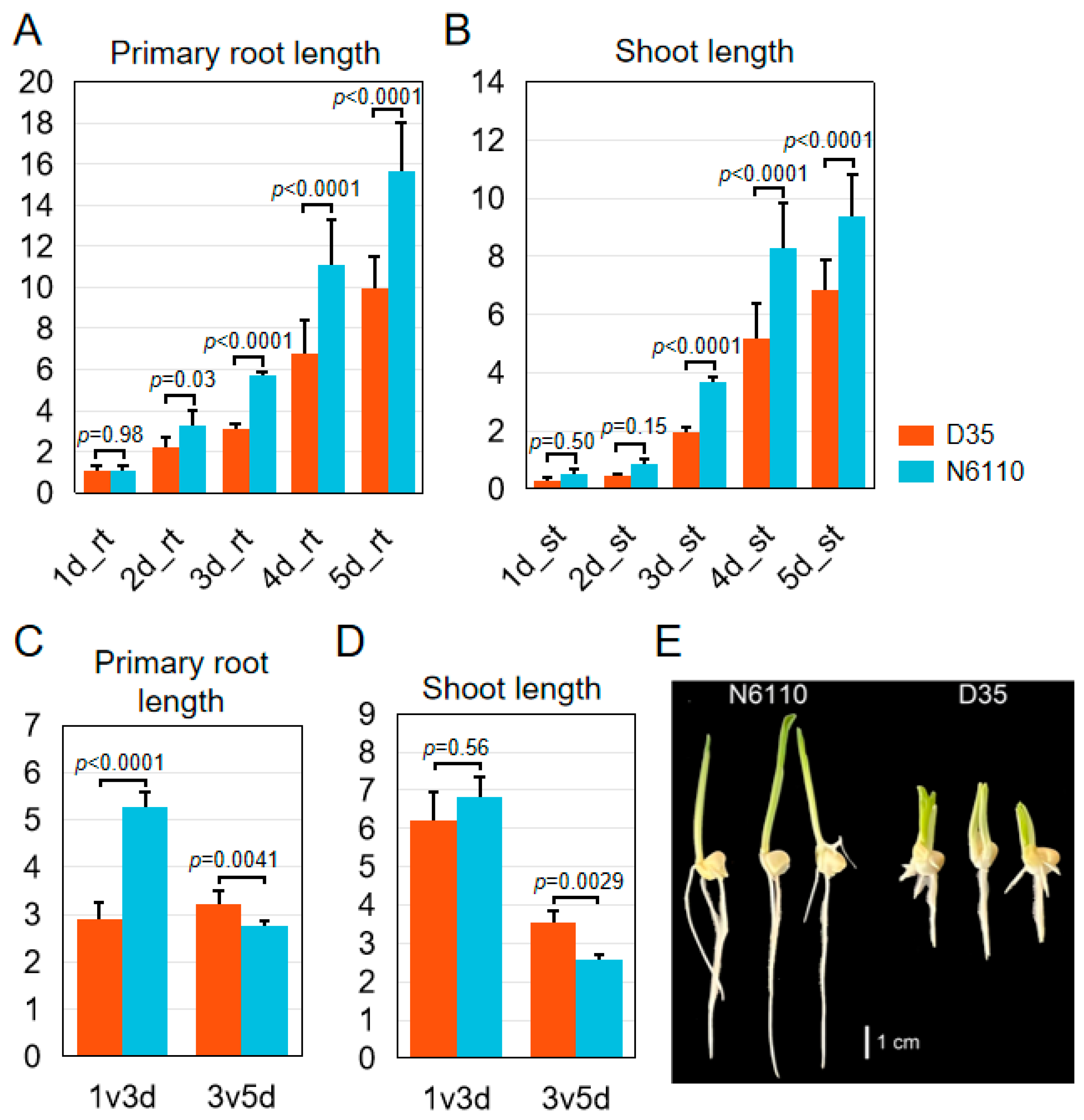
2.1. Morphology of N6110 and D35 Seedlings
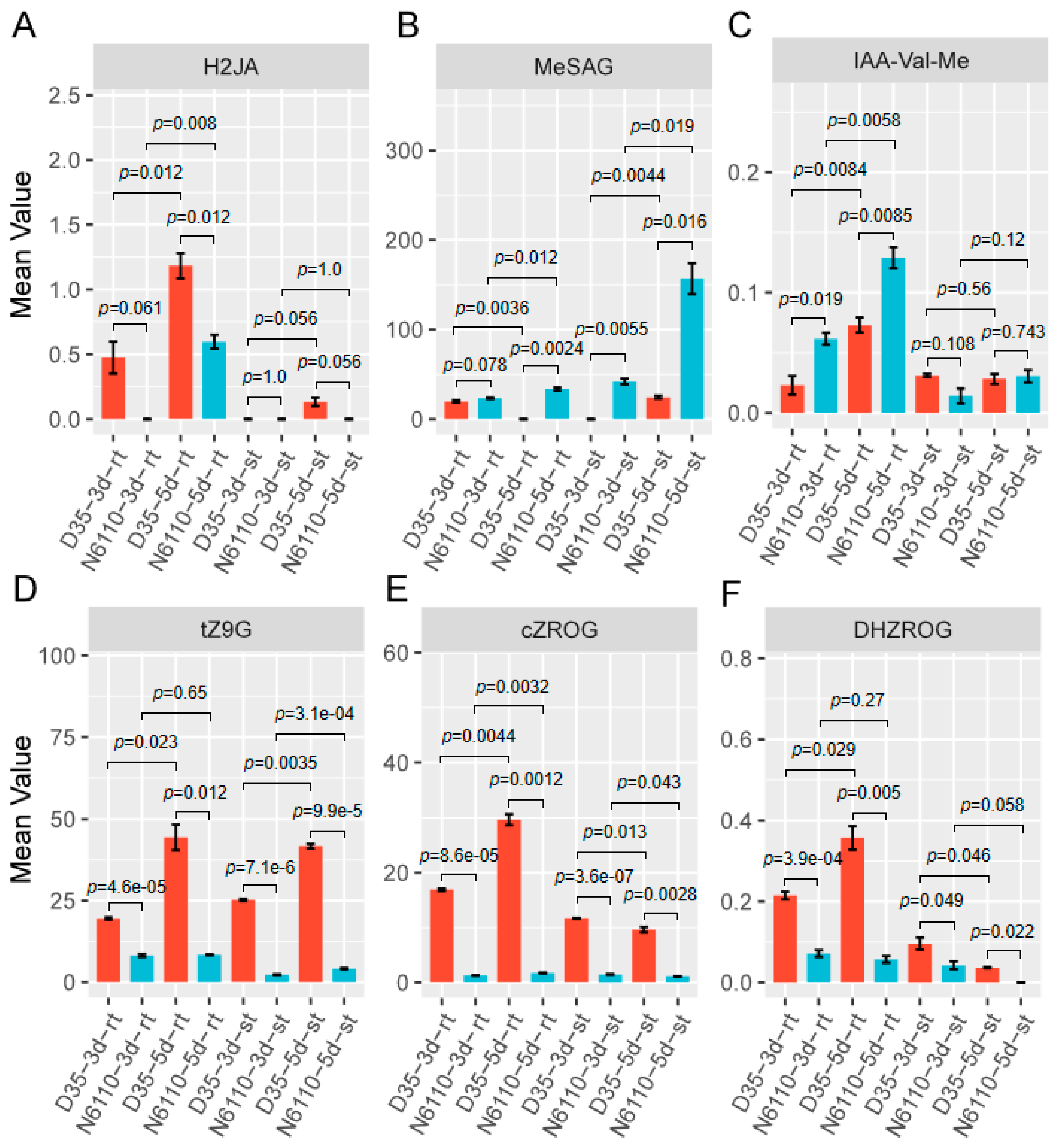
2.2. Analysis of Different Hormone-Related Compounds
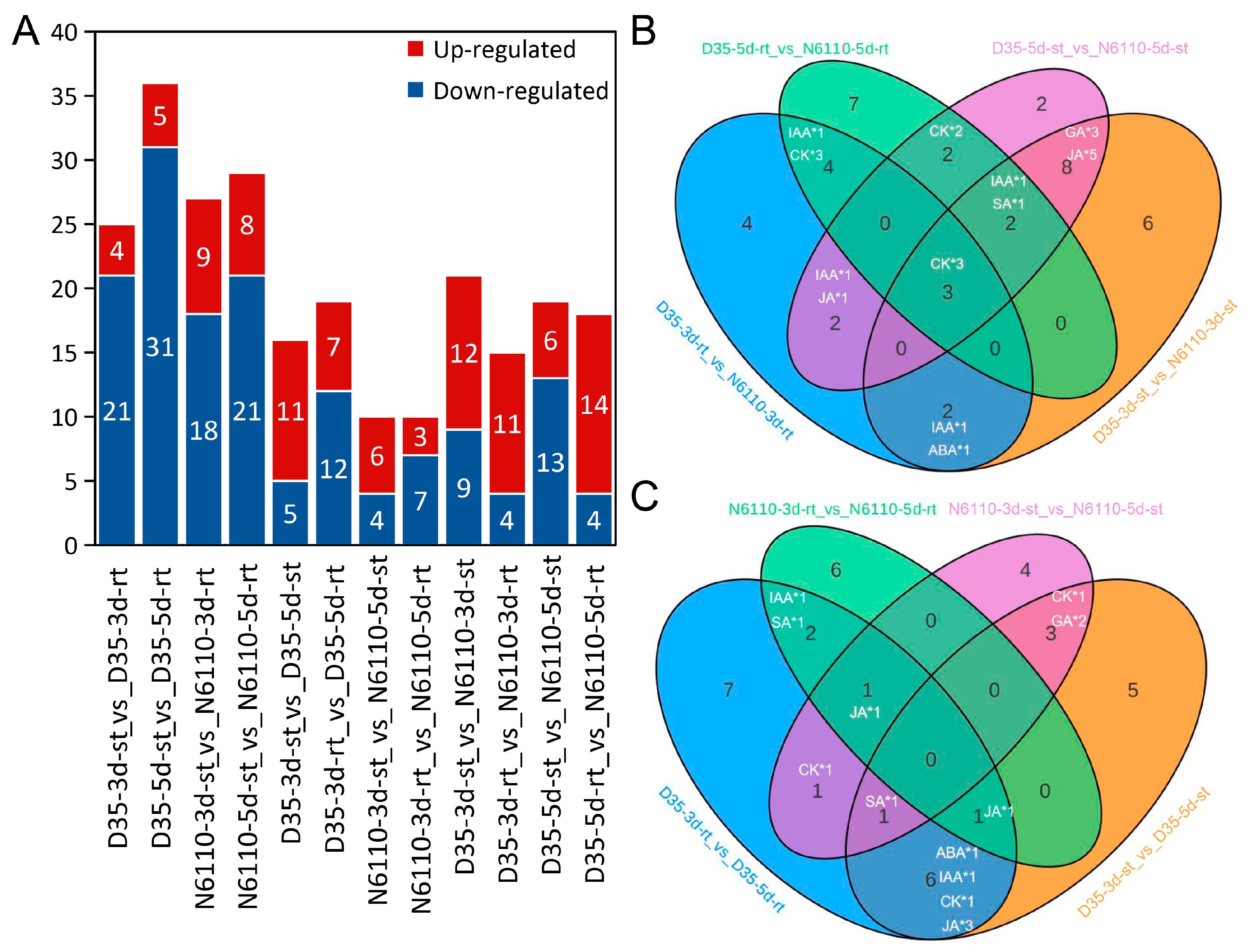
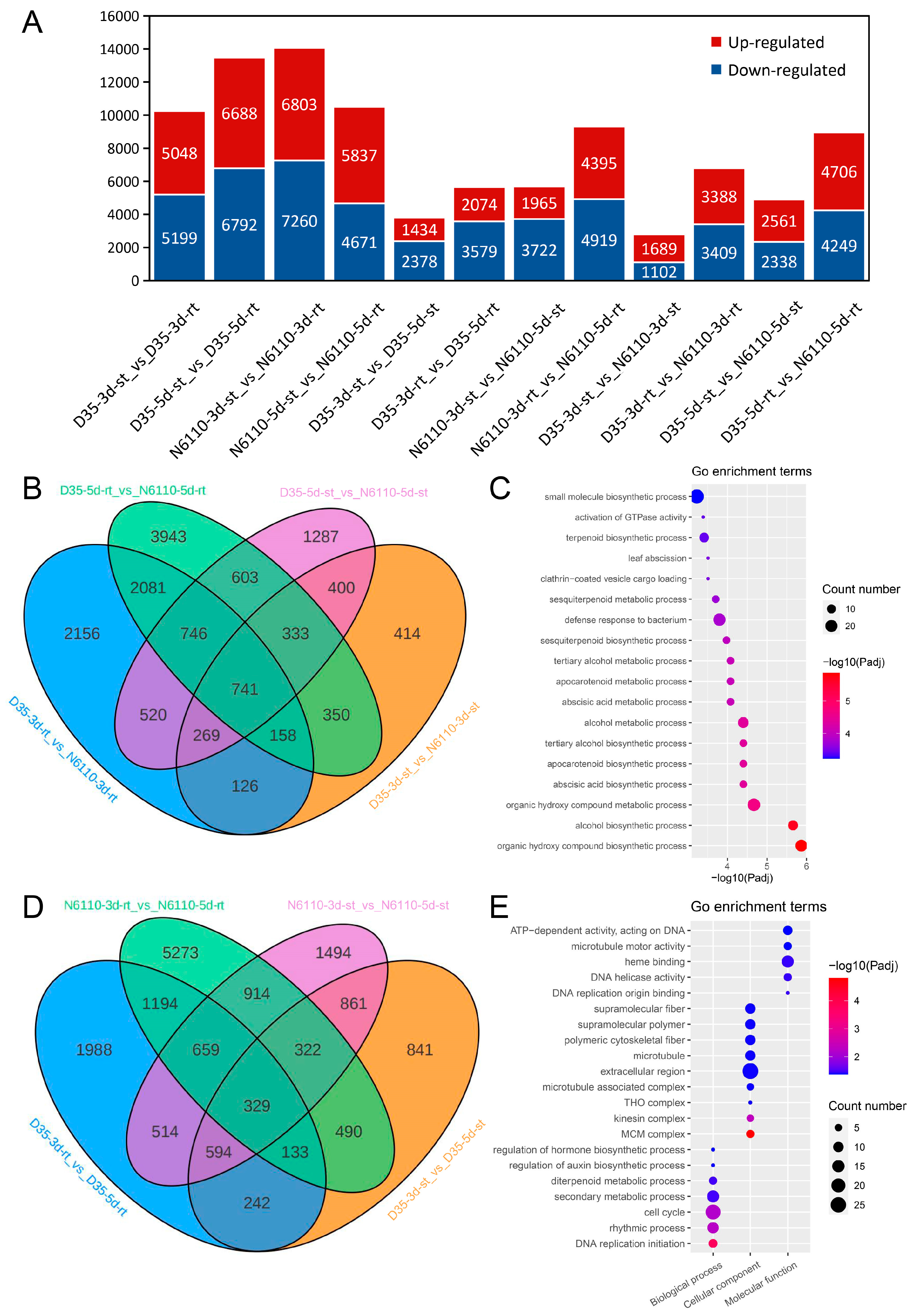
2.3. Transcriptome Analysis for Seedlings
2.4. KEGG Pathway Co-Enrichments of DAMs and DEGs
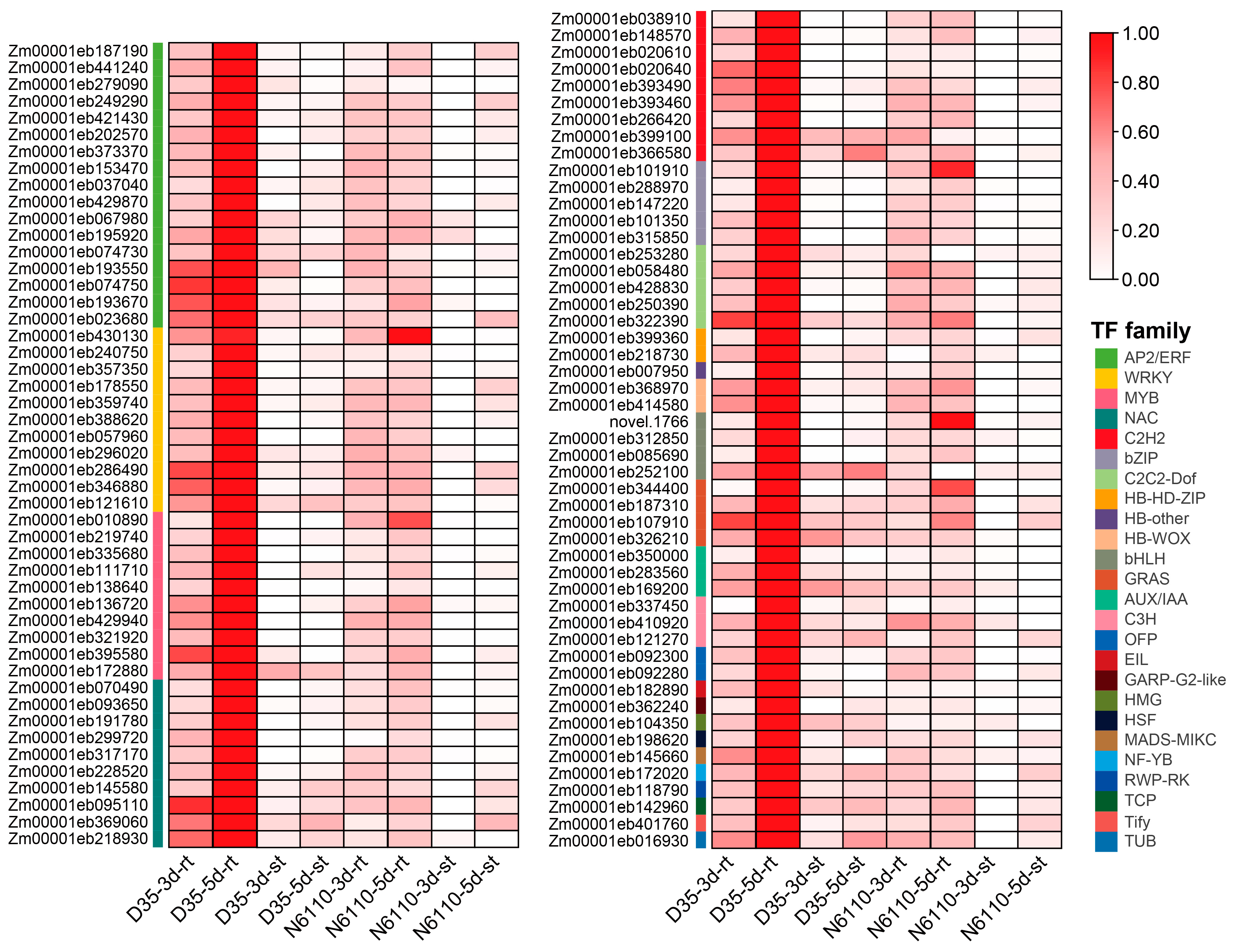
2.5. Prediction of Hormone-Gene Relation Networks
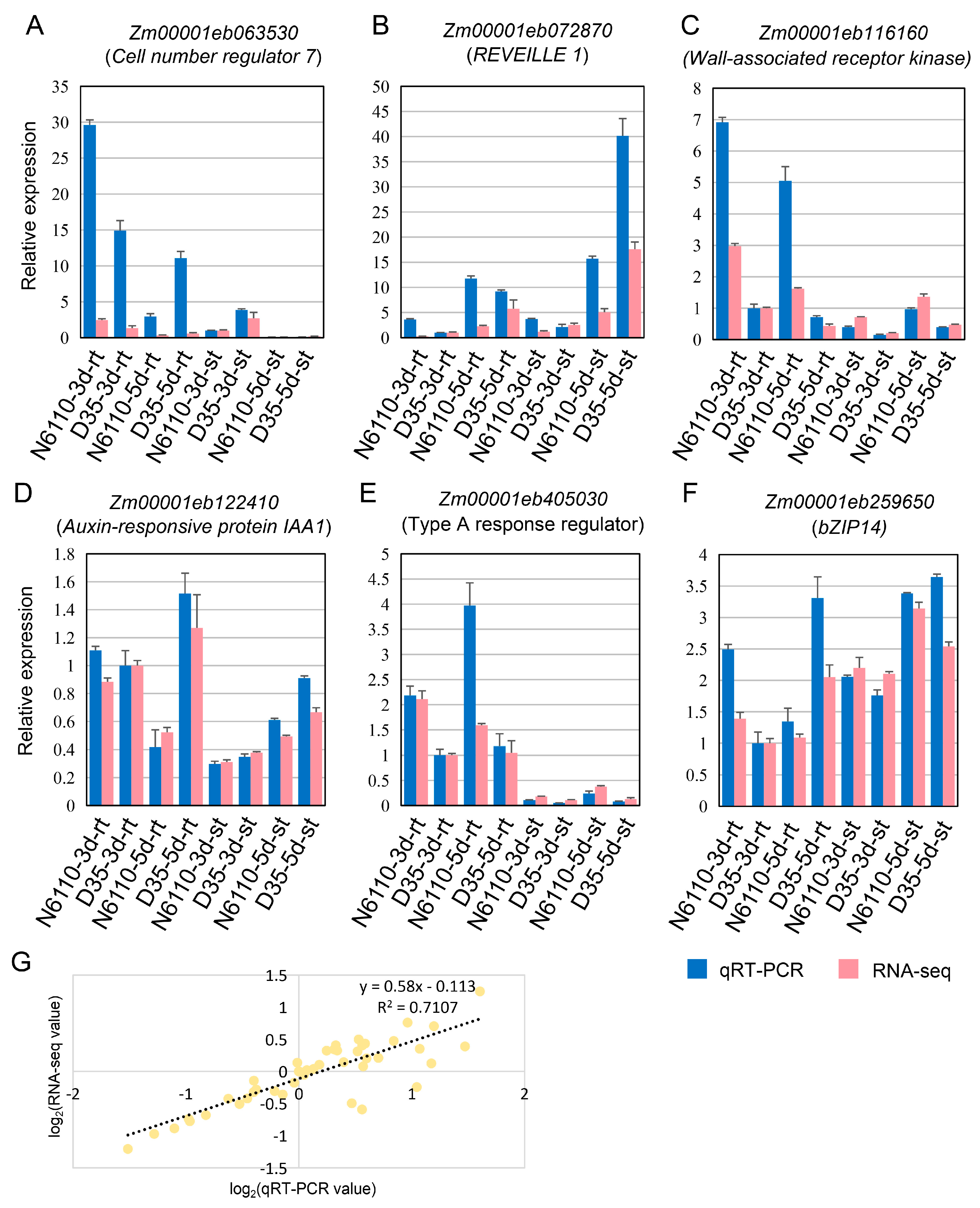
2.6. RNA-Seq Expression Validation via qRT-PCR
3. Discussion
4. Methods and Materials
4.1. Plant Material and Growth
4.2. Determination of Endogenous Phytohormones
4.3. LC-MS/MS Platform and Settings
4.4. Differential Analysis of Phytohormone Abundance
4.5. RNA Isolation and RNA-Seq Analyses
4.6. Network Construction of Hormone-Related DEGs
4.7. Quantitative Real-Time PCR (qRT-PCR) Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Michel, M.R.; Cruz-Requena, M.; Avendaño-Sanchez, M.C.; González-Vazquez, V.M.; Flores-Gallegos, A.C.; Aguilar, C.N.; Espinoza-Velázquez, J.; Rodríguez-Herrera, R. Polyembryony in Maize: A Complex, Elusive, and Potentially Agronomical Useful Trait. In Maize Germplasm—Characterization and Genetic Approaches for Crop Improvement; IntechOpen: London, UK, 2018; pp. 1–20. [Google Scholar]
- González-Vázquez, V.; Espinoza-Velázquez, J.; Mendoza-Villarreal, R.; León-Castillo, H.D.; Torres-Tapia, M. Characterisation of maize germoplasm that combines a high oil content and polyembryony. Univ. Cienc. 2011, 27, 157–167. [Google Scholar]
- Michel-Michel, M.R.; Aguilar-Zárate, P.; Lopez-Badillo, C.M.; Chávez-González, M.L.; Flores-Gallegos, A.C.; Espinoza-Velázquez, J.; Aguilar, C.N.; Rodríguez-Herrera, R. Mineral and fatty acid contents of maize kernels with different levels of polyembryony. Cereal Chem. 2020, 97, 723–732. [Google Scholar] [CrossRef]
- Cruz-Requena, M.; Espinoza-Velázquez, J.; Aguilar, C.N.; Rodríguez-Herrera, R. Polyembryony improvement effect on chemical and physical traits of maize grain. Food Humanit. 2023, 1, 78–84. [Google Scholar] [CrossRef]
- Requena, M.; Herrera, R.R.; González, C.N.A.; Velazquez, J.; Martinez, M.; Cárdenas, J.; Carranza, V.; Antonio, A. Alkaline Cooking Quality of Polyembryonic and Non-Polyembryonic Maize Populations. Adv. J. Food Sci. Technol. 2011, 3, 259–268. [Google Scholar]
- Garcia-Ortiz, J.D.; Flores-Gallegos, A.C.; Espinoza-Velázquez, J.; Ascacio-Valdés, J.A.; Nery-Flores, S.D.; Rodríguez-Herrera, R. Morphological, physicochemical, techno-functional, phytochemical, and antioxidant evaluation of polyembryonic and non-polyembryonic maize sprouts. Biocatal. Agric. Biotechnol. 2023, 47, 102583. [Google Scholar] [CrossRef]
- Miyahira, R.F.; Lopes, J.O.; Antunes, A.E.C. The Use of Sprouts to Improve the Nutritional Value of Food Products: A Brief Review. Plant Foods Hum. Nutr. 2021, 76, 143–152. [Google Scholar] [CrossRef]
- Casanova-Sáez, R.; Mateo-Bonmatí, E.; Ljung, K. Auxin Metabolism in Plants. Cold Spring Harb. Perspect. Biol. 2021, 13, a039867. [Google Scholar] [CrossRef] [PubMed]
- Kieber, J.J.; Schaller, G.E. Cytokinin signaling in plant development. Development 2018, 145, dev149344. [Google Scholar] [CrossRef]
- Gao, S.; Chu, C. Gibberellin Metabolism and Signaling: Targets for Improving Agronomic Performance of Crops. Plant Cell Physiol. 2020, 61, 1902–1911. [Google Scholar] [CrossRef]
- Chen, K.; Li, G.J.; Bressan, R.A.; Song, C.P.; Zhu, J.K.; Zhao, Y. Abscisic acid dynamics, signaling, and functions in plants. J. Integr. Plant Biol. 2020, 62, 25–54. [Google Scholar] [CrossRef]
- Dubois, M.; Van den Broeck, L.; Inzé, D. The Pivotal Role of Ethylene in Plant Growth. Trends Plant Sci. 2018, 23, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Planas-Riverola, A.; Gupta, A.; Betegón-Putze, I.; Bosch, N.; Ibañes, M.; Caño-Delgado, A.I. Brassinosteroid signaling in plant development and adaptation to stress. Development 2019, 146, dev151894. [Google Scholar] [CrossRef]
- Li, C.; Xu, M.; Cai, X.; Han, Z.; Si, J.; Chen, D. Jasmonate Signaling Pathway Modulates Plant Defense, Growth, and Their Trade-Offs. Int. J. Mol. Sci. 2022, 23, 3945. [Google Scholar] [CrossRef]
- Li, A.; Sun, X.; Liu, L. Action of Salicylic Acid on Plant Growth. Front. Plant Sci. 2022, 13, 878076. [Google Scholar] [CrossRef]
- Alvi, A.F.; Sehar, Z.; Fatma, M.; Masood, A.; Khan, N.A. Strigolactone: An Emerging Growth Regulator for Developing Resilience in Plants. Plants 2022, 11, 2604. [Google Scholar] [CrossRef]
- Weiss, D.; Ori, N. Mechanisms of Cross Talk between Gibberellin and Other Hormones. Plant Physiol. 2007, 144, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, F.; Xie, Q. Balancing growth and adaptation to stress: Crosstalk between brassinosteroid and abscisic acid signaling. Plant Cell Environ. 2020, 43, 2325–2335. [Google Scholar] [CrossRef]
- Song, W.; Liu, L.; Wang, J.; Wu, Z.; Zhang, H.; Tang, J.; Lin, G.; Wang, Y.; Wen, X.; Li, W.; et al. Signature motif-guided identification of receptors for peptide hormones essential for root meristem growth. Cell Res. 2016, 26, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Wei, L.; Xu, J.; Zhai, Q.; Jiang, H.; Chen, R.; Chen, Q.; Sun, J.; Chu, J.; Zhu, L.; et al. Arabidopsis Tyrosylprotein sulfotransferase acts in the auxin/PLETHORA pathway in regulating postembryonic maintenance of the root stem cell niche. Plant Cell 2010, 22, 3692–3709. [Google Scholar] [CrossRef]
- Dai, X.; Liu, Z.; Qiao, M.; Li, J.; Li, S.; Xiang, F. ARR12 promotes de novo shoot regeneration in Arabidopsis thaliana via activation of WUSCHEL expression. J. Integr. Plant Biol. 2017, 59, 747–758. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Dong, Y.X.; Hao, Y.J.; Zhang, X.S. DNA METHYLTRANSFERASE1-mediated shoot regeneration is regulated by cytokinin-induced cell cycle in Arabidopsis. New Phytol. 2018, 217, 219–232. [Google Scholar] [CrossRef]
- Wang, J.; Tian, C.; Zhang, C.; Shi, B.; Cao, X.; Zhang, T.Q.; Zhao, Z.; Wang, J.W.; Jiao, Y. Cytokinin Signaling Activates WUSCHEL Expression during Axillary Meristem Initiation. Plant Cell 2017, 29, 1373–1387. [Google Scholar] [CrossRef]
- Zhao, Y.; Cheng, S.; Song, Y.; Huang, Y.; Zhou, S.; Liu, X.; Zhou, D.X. The Interaction between Rice ERF3 and WOX11 Promotes Crown Root Development by Regulating Gene Expression Involved in Cytokinin Signaling. Plant Cell 2015, 27, 2469–2483. [Google Scholar] [CrossRef]
- Zou, X.; Shao, J.; Wang, Q.; Chen, P.; Zhu, Y.; Yin, C. Supraoptimal Cytokinin Content Inhibits Rice Seminal Root Growth by Reducing Root Meristem Size and Cell Length via Increased Ethylene Content. Int. J. Mol. Sci. 2018, 19, 4051. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, J.; Zhai, Q.; Zhou, W.; Qi, L.; Xu, L.; Wang, B.; Chen, R.; Jiang, H.; Qi, J.; et al. The basic helix-loop-helix transcription factor MYC2 directly represses PLETHORA expression during jasmonate-mediated modulation of the root stem cell niche in Arabidopsis. Plant Cell 2011, 23, 3335–3352. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xu, Y.; Ye, S.; Jiang, H.; Chen, Q.; Liu, F.; Zhou, W.; Chen, R.; Li, X.; Tietz, O.; et al. Arabidopsis ASA1 is important for jasmonate-mediated regulation of auxin biosynthesis and transport during lateral root formation. Plant Cell 2009, 21, 1495–1511. [Google Scholar] [CrossRef]
- He, Q.; Yuan, R.; Zhang, T.; An, F.; Wang, N.; Lan, J.; Wang, X.; Zhang, Z.; Pan, Y.; Wang, X.; et al. ArabidopsisTIE1 and TIE2 transcriptional repressors dampen cytokinin response during root development. Sci. Adv. 2022, 8, eabn5075. [Google Scholar] [CrossRef] [PubMed]
- Szemenyei, H.; Hannon, M.; Long, J.A. TOPLESS Mediates Auxin-Dependent Transcriptional Repression During Arabidopsis Embryogenesis. Science 2008, 319, 1384–1386. [Google Scholar] [CrossRef]
- Pauwels, L.; Barbero, G.F.; Geerinck, J.; Tilleman, S.; Grunewald, W.; Pérez, A.C.; Chico, J.M.; Bossche, R.V.; Sewell, J.; Gil, E.; et al. NINJA connects the co-repressor TOPLESS to jasmonate signalling. Nature 2010, 464, 788–791. [Google Scholar] [CrossRef]
- Wang, L.; Wang, B.; Yu, H.; Guo, H.; Lin, T.; Kou, L.; Wang, A.; Shao, N.; Ma, H.; Xiong, G.; et al. Transcriptional regulation of strigolactone signalling in Arabidopsis. Nature 2020, 583, 277–281. [Google Scholar] [CrossRef]
- Geng, L.; Li, Q.; Jiao, L.; Xiang, Y.; Deng, Q.; Zhou, D.X.; Zhao, Y. WOX11 and CRL1 act synergistically to promote crown root development by maintaining cytokinin homeostasis in rice. New Phytol. 2022, 237, 204–216. [Google Scholar] [CrossRef]
- Márquez, G.; Alarcón, M.V.; Salguero, J. Cytokinin Inhibits Lateral Root Development at the Earliest Stages of Lateral Root Primordium Initiation in Maize Primary Root. J. Plant Growth Regul. 2018, 38, 83–92. [Google Scholar] [CrossRef]
- Gao, S.; Fang, J.; Xu, F.; Wang, W.; Sun, X.; Chu, J.; Cai, B.; Feng, Y.; Chu, C. CYTOKININ OXIDASE/DEHYDROGENASE4 Integrates Cytokinin and Auxin Signaling to Control Rice Crown Root Formation. Plant Physiol. 2014, 165, 1035–1046. [Google Scholar] [CrossRef]
- Hayashi, K.-i.; Arai, K.; Aoi, Y.; Tanaka, Y.; Hira, H.; Guo, R.; Hu, Y.; Ge, C.; Zhao, Y.; Kasahara, H.; et al. The main oxidative inactivation pathway of the plant hormone auxin. Nat. Commun. 2021, 12, 6752. [Google Scholar] [CrossRef]
- Bartel, B.; Fink, G.R. ILR1, an Amidohydrolase That Releases Active Indole-3-Acetic Acid from Conjugates. Science 1995, 268, 1745–1748. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Liu, L.; Pei, Y.; Liu, Z.; Feng, X.; Li, X. Transcriptomic and metabolomic profiling provide insight into the role of sugars and hormones in leaf senescence of Pinellia ternata. Plant Cell Rep. 2024, 43, 125. [Google Scholar] [CrossRef] [PubMed]
- Quirino, B.F.; Normanly, J.; Amasino, R.M. Diverse range of gene activity during Arabidopsis thaliana leaf senescence includes pathogen-independent induction of defense-related genes. Plant Mol. Biol. 1999, 40, 267–278. [Google Scholar] [CrossRef]
- Major, I.T.; Guo, Q.; Zhai, J.; Kapali, G.; Kramer, D.M.; Howe, G.A. A Phytochrome B-Independent Pathway Restricts Growth at High Levels of Jasmonate Defense. Plant Physiol. 2020, 183, 733–749. [Google Scholar] [CrossRef]
- Xiong, Q.; Ma, B.; Lu, X.; Huang, Y.-H.; He, S.-J.; Yang, C.; Yin, C.-C.; Zhao, H.; Zhou, Y.; Zhang, W.-K.; et al. Ethylene-Inhibited Jasmonic Acid Biosynthesis Promotes Mesocotyl/Coleoptile Elongation of Etiolated Rice Seedlings. Plant Cell 2017, 29, 1053–1072. [Google Scholar] [CrossRef]
- Wasternack, C. Jasmonates: An Update on Biosynthesis, Signal Transduction and Action in Plant Stress Response, Growth and Development. Ann. Bot. 2007, 100, 681–697. [Google Scholar] [CrossRef]
- Huang, H.; Liu, B.; Liu, L.; Song, S. Jasmonate action in plant growth and development. J. Exp. Bot. 2017, 68, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, H.H.A.; Wang, H.; Song, J.; Li, X. Effects of genotypes and explants on garlic callus production and endogenous hormones. Sci. Rep. 2020, 10, 4867. [Google Scholar] [CrossRef]
- Attaran, E.; Zeier, T.E.; Griebel, T.; Zeier, J. Methyl Salicylate Production and Jasmonate Signaling Are Not Essential for Systemic Acquired Resistance in Arabidopsis. Plant Cell 2009, 21, 954–971. [Google Scholar] [CrossRef]
- Janda, M.; Ruelland, E. Magical mystery tour: Salicylic acid signalling. Environ. Exp. Bot. 2015, 114, 117–128. [Google Scholar] [CrossRef]
- Sasek, V.; Janda, M.; Delage, E.; Puyaubert, J.; Guivarc’h, A.; Lopez Maseda, E.; Dobrev, P.I.; Caius, J.; Boka, K.; Valentova, O.; et al. Constitutive salicylic acid accumulation in pi4kIIIbeta1beta2 Arabidopsis plants stunts rosette but not root growth. New Phytol. 2014, 203, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Meguro, A.; Sato, Y. Salicylic acid antagonizes abscisic acid inhibition of shoot growth and cell cycle progression in rice. Sci. Rep. 2014, 4, 4555. [Google Scholar] [CrossRef]
- Tan, S.; Abas, M.; Verstraeten, I.; Glanc, M.; Molnár, G.; Hajný, J.; Lasák, P.; Petřík, I.; Russinova, E.; Petrášek, J.; et al. Salicylic Acid Targets Protein Phosphatase 2A to Attenuate Growth in Plants. Curr. Biol. 2020, 30, 381–395.e388. [Google Scholar] [CrossRef]
- Pasternak, T.; Groot, E.P.; Kazantsev, F.V.; Teale, W.; Omelyanchuk, N.; Kovrizhnykh, V.; Palme, K.; Mironova, V.V. Salicylic Acid Affects Root Meristem Patterning via Auxin Distribution in a Concentration-Dependent Manner. Plant Physiol. 2019, 180, 1725–1739. [Google Scholar] [CrossRef]
- Mazzoni-Putman, S.M.; Brumos, J.; Zhao, C.; Alonso, J.M.; Stepanova, A.N. Auxin Interactions with Other Hormones in Plant Development. Cold Spring Harb. Perspect. Biol. 2021, 13, a039990. [Google Scholar] [CrossRef]
- Cheng, X.; Jiang, H.; Zhang, J.; Qian, Y.; Zhu, S.; Cheng, B. Overexpression of type-A rice response regulators, OsRR3 and OsRR5, results in lower sensitivity to cytokinins. Gen. Mol. Res. 2010, 9, 348–359. [Google Scholar] [CrossRef]
- Zhang, W.; To, J.P.C.; Cheng, C.Y.; Eric Schaller, G.; Kieber, J.J. Type-A response regulators are required for proper root apical meristem function through post-transcriptional regulation of PIN auxin efflux carriers. Plant J. 2011, 68, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-F.; Zhou, Q.; Ma, Z.-H.; Zuo, C.-W.; Chu, M.-Y.; Mao, J.; Chen, B.-H. Regulatory mechanism of GA3 application on grape (Vitis vinifera L.) berry size. Plant Physiol. Biochem. 2024, 210, 108543. [Google Scholar] [CrossRef]
- Gomi, K.; Sasaki, A.; Itoh, H.; Ueguchi-Tanaka, M.; Ashikari, M.; Kitano, H.; Matsuoka, M. GID2, an F-box subunit of the SCF E3 complex, specifically interacts with phosphorylated SLR1 protein and regulates the gibberellin-dependent degradation of SLR1 in rice. Plant J. 2004, 37, 626–634. [Google Scholar] [CrossRef]
- Wong, S.-M.; Tao, T.; Zhou, C.-J.; Wang, Q.; Chen, X.-R.; Sun, Q.; Zhao, T.-Y.; Ye, J.-C.; Wang, Y.; Zhang, Z.-Y.; et al. Rice black streaked dwarf virus P7-2 forms a SCF complex through binding to Oryza sativa SKP1-like proteins, and interacts with GID2 involved in the gibberellin pathway. PLoS ONE 2017, 12, e0177518. [Google Scholar] [CrossRef]
- Yin, L.; Zander, M.; Huang, S.-S.C.; Xie, M.; Song, L.; Guzmán, J.P.S.; Hann, E.; Shanbhag, B.K.; Ng, S.; Jain, S.; et al. Transcription Factor Dynamics in Cross-Regulation of Plant Hormone Signaling Pathways. bioRxiv 2023. [Google Scholar] [CrossRef]
- Xie, Z.; Nolan, T.M.; Jiang, H.; Yin, Y. AP2/ERF Transcription Factor Regulatory Networks in Hormone and Abiotic Stress Responses in Arabidopsis. Front. Plant Sci. 2019, 10, 228. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.; Wang, Y.; Liu, Z.; Gao, C. Comprehensive Analysis of MYB Gene Family and Their Expressions Under Abiotic Stresses and Hormone Treatments in Tamarix hispida. Front. Plant Sci. 2018, 9, 1303. [Google Scholar] [CrossRef]
- Gupta, S.; Malviya, N.; Kushwaha, H.; Nasim, J.; Bisht, N.C.; Singh, V.K.; Yadav, D. Insights into structural and functional diversity of Dof (DNA binding with one finger) transcription factor. Planta 2015, 241, 549–562. [Google Scholar] [CrossRef]
- Manzoor, M.A.; Sabir, I.A.; Shah, I.H.; Wang, H.; Yu, Z.; Rasool, F.; Mazhar, M.Z.; Younas, S.; Abdullah, M.; Cai, Y. Comprehensive Comparative Analysis of the GATA Transcription Factors in Four Rosaceae Species and Phytohormonal Response in Chinese Pear (Pyrus bretschneideri) Fruit. Int. J. Mol. Sci. 2021, 22, 12492. [Google Scholar] [CrossRef]
- Wang, H.; Jones, B.; Li, Z.; Frasse, P.; Delalande, C.; Regad, F.; Chaabouni, S.; Latché, A.; Pech, J.-C.; Bouzayen, M. The Tomato Aux/IAA Transcription Factor IAA9Is Involved in Fruit Development and Leaf Morphogenesis. Plant Cell 2005, 17, 2676–2692. [Google Scholar] [CrossRef]
- Rong, M.; Gao, S.-X.; Wen, D.; Xu, Y.-H.; Wei, J.-H. The LOB domain protein, a novel transcription factor with multiple functions: A review. Plant Physiol. Biochem. 2024, 214, 108922. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Duan, Y.; Zheng, H.; Tang, H.; Zheng, L.; Yu, X. Genome-Wide Identification and Characterization of the RWP-RK Proteins in Zanthoxylum armatum. Genes 2024, 15, 665. [Google Scholar] [CrossRef] [PubMed]
- Mahendrawada, L.; Warfield, L.; Donczew, R.; Hahn, S. Low overlap of transcription factor DNA binding and regulatory targets. Nature 2025, 642, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.C.; Liao, P.M.; Kuo, W.W.; Lin, T.P. The Arabidopsis Ethylene Response Factor1 Regulates Abiotic Stress-Responsive Gene Expression by Binding to Different cis-Acting Elements in Response to Different Stress Signals. Plant Physiol. 2013, 162, 1566–1582. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, J.; Chen, S.; Zhang, Z.; Wan, G.; Mao, J.; Wang, Z.; Tan, S.; Xiang, C. ERF1 inhibits lateral root emergence by promoting local auxin accumulation and repressing ARF7 expression. Cell Rep. 2023, 42, 112565. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, P.; Chen, S.; Sun, L.; Mao, J.; Tan, S.; Xiang, C. The ABI3-ERF1 module mediates ABA-auxin crosstalk to regulate lateral root emergence. Cell Rep. 2023, 42, 112809. [Google Scholar] [CrossRef]
- Zhao, X.; Niu, Y.; Hossain, Z.; Zhao, B.; Bai, X.; Mao, T. New insights into light spectral quality inhibits the plasticity elongation of maize mesocotyl and coleoptile during seed germination. Front. Plant Sci. 2023, 14, 11523999. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, C.; Yan, X.; Zhang, J.; Xu, J. Simultaneous analysis of ten phytohormones in Sargassum horneri by high-performance liquid chromatography with electrospray ionization tandem mass spectrometry. J. Sep. Sci. 2016, 39, 1804–1813. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Hufford, M.B.; Seetharam, A.S.; Woodhouse, M.R.; Chougule, K.M.; Ou, S.; Liu, J.; Ricci, W.A.; Guo, T.; Olson, A.; Qiu, Y.; et al. De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 2021, 373, 655–662. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, Z.; Wang, J.; Liu, H.; Zhou, L.; Zhong, S.; Li, Y.; Zhu, C.; Liu, J.; Lin, Z. The tin1 gene retains the function of promoting tillering in maize. Nat. Commun. 2019, 10, 5608. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Chr | Related DAMs | TF Family | Gene Annotation |
|---|---|---|---|---|
| Zm00001eb023680 | chr1 | DHZROG, cZROG | AP2/ERF | Ethylene-responsive transcription factor 5 |
| Zm00001eb067800 | chr2 | MeSAG | AP2/ERF | EREBP-like factor |
| Zm00001eb074900 | chr2 | DHZROG | AP2/ERF | Ethylene-responsive transcription factor 6 |
| Zm00001eb074910 | chr2 | MeSAG | AP2/ERF | EREBP-like factor |
| Zm00001eb168040 | chr4 | DHZROG | AP2/ERF | Ethylene-responsive transcription factor TINY |
| Zm00001eb193550 | chr4 | DHZROG, cZROG | AP2/ERF | Ethylene-responsive transcription factor RAP2-1 |
| Zm00001eb232560 | chr5 | DHZROG | AP2/ERF | EREBP-like factor |
| Zm00001eb307550 | chr7 | DHZROG | AP2/ERF | Ethylene-responsive transcription factor 1 |
| Zm00001eb338940 | chr8 | DHZROG | AP2/ERF | Ethylene-responsive transcription factor 4-like |
| Zm00001eb343790 | chr8 | H2JA | AP2/ERF | EREBP-like factor |
| Zm00001eb373370 | chr9 | H2JA, DHZROG | AP2/ERF | EREBP-like factor |
| Zm00001eb014590 | chr1 | IAA-Val-Me | bZIP | bZIP transcription factor 1/2/11/44/53 |
| Zm00001eb028160 | chr1 | H2JA | bZIP | bZIP transcription factor 1/2/11/44/53 |
| Zm00001eb048370 | chr1 | MeSAG | C2C2-Dof | -- |
| Zm00001eb066100 | chr2 | DHZROG | C2C2-Dof | -- |
| Zm00001eb253280 | chr5 | DHZROG, cZROG | C2C2-Dof | -- |
| Zm00001eb322390 | chr7 | H2JA, DHZROG | C2C2-Dof | -- |
| Zm00001eb215440 | chr5 | MeSAG | C2C2-GATA | FAR1 transcription factor |
| Zm00001eb117380 | chr2 | DHZROG | LOB | LOB domain-containing protein 16 |
| Zm00001eb001670 | chr1 | H2JA | MADS-MIKC | MADS-box transcription factor |
| Zm00001eb393670 | chr9 | H2JA | MADS-MIKC | MADS-box transcription factor |
| Zm00001eb221780 | chr5 | DHZROG | MYB | Transcription factor MYB30 |
| Zm00001eb247570 | chr5 | H2JA | MYB | Myb-related protein MYB4 |
| Zm00001eb320950 | chr7 | IAA-Val-Me | RWP-RK | -- |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, M.; Li, G.; Li, K.; Gao, J.; Li, M.; Liu, H.; Wang, Y.; Kang, K.; Zhang, D.; Li, W. Comparative Analysis of Differentially Expressed Genes and Metabolites in Waxy Maize Inbred Lines with Distinct Twin-Shoot Phenotypes. Plants 2025, 14, 1951. https://doi.org/10.3390/plants14131951
Qin M, Li G, Li K, Gao J, Li M, Liu H, Wang Y, Kang K, Zhang D, Li W. Comparative Analysis of Differentially Expressed Genes and Metabolites in Waxy Maize Inbred Lines with Distinct Twin-Shoot Phenotypes. Plants. 2025; 14(13):1951. https://doi.org/10.3390/plants14131951
Chicago/Turabian StyleQin, Mengfan, Guangyu Li, Kun Li, Jing Gao, Meng Li, Hao Liu, Yifeng Wang, Keke Kang, Da Zhang, and Wu Li. 2025. "Comparative Analysis of Differentially Expressed Genes and Metabolites in Waxy Maize Inbred Lines with Distinct Twin-Shoot Phenotypes" Plants 14, no. 13: 1951. https://doi.org/10.3390/plants14131951
APA StyleQin, M., Li, G., Li, K., Gao, J., Li, M., Liu, H., Wang, Y., Kang, K., Zhang, D., & Li, W. (2025). Comparative Analysis of Differentially Expressed Genes and Metabolites in Waxy Maize Inbred Lines with Distinct Twin-Shoot Phenotypes. Plants, 14(13), 1951. https://doi.org/10.3390/plants14131951