
Transcriptomic Profiling Reveals the Involvement of the Phenylpropanoid–Lignin Pathway in the Response of Maize Roots to Zinc Stress

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
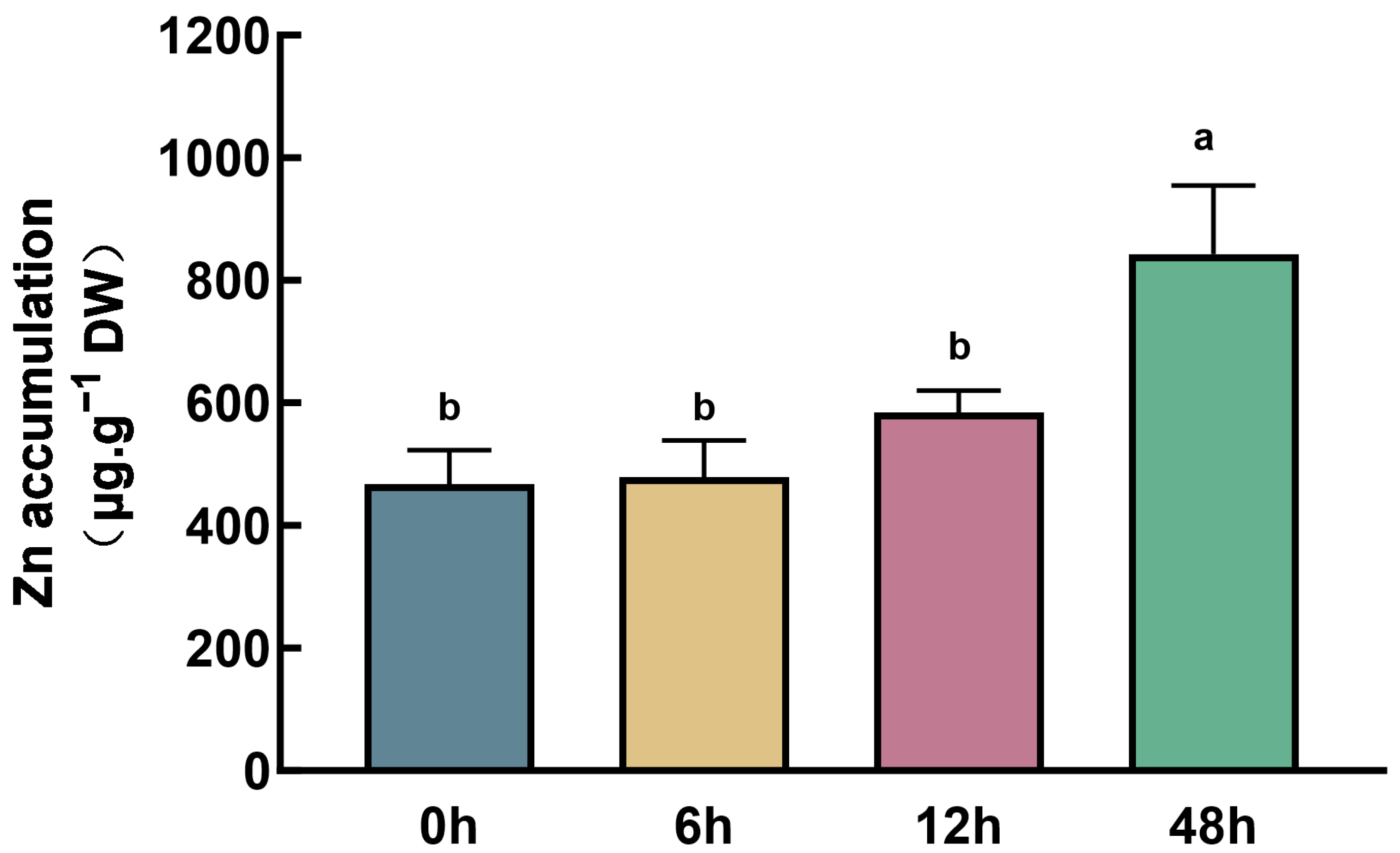
2.1. Zn Treatments and Transcriptome Sequencing
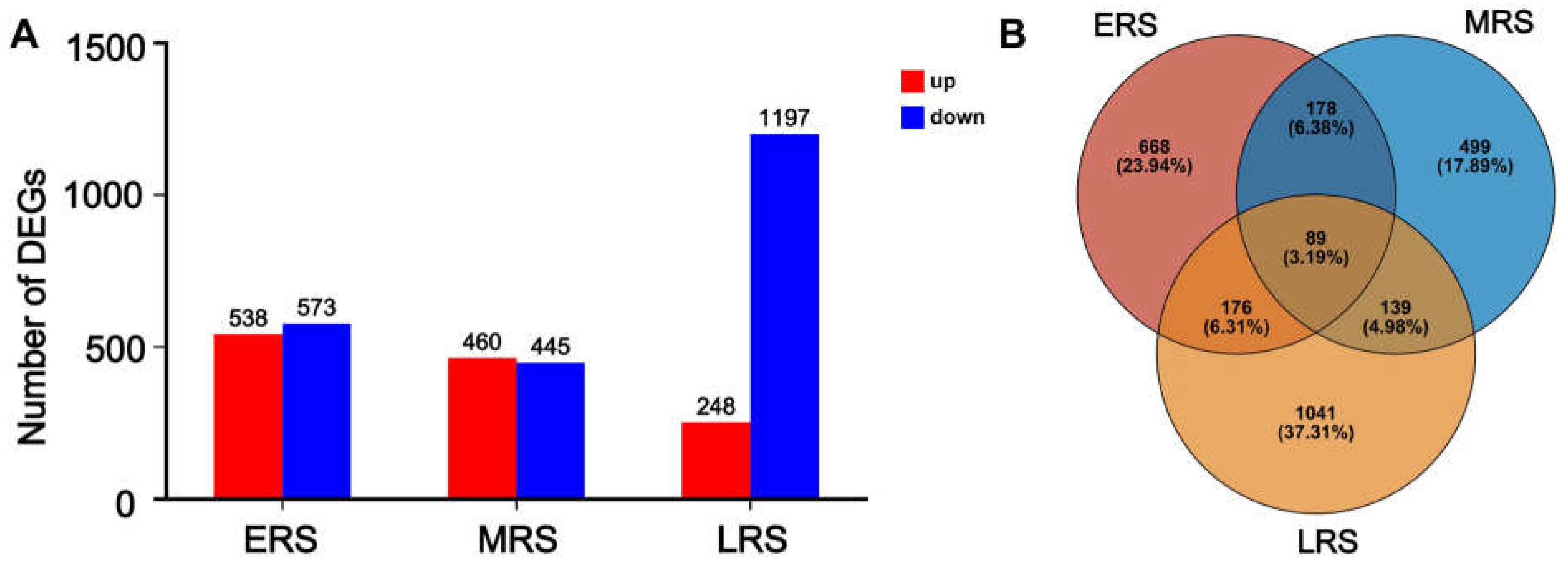
2.2. Identification of Differentially Expressed Genes (DEGs) at Different Treatment Stages
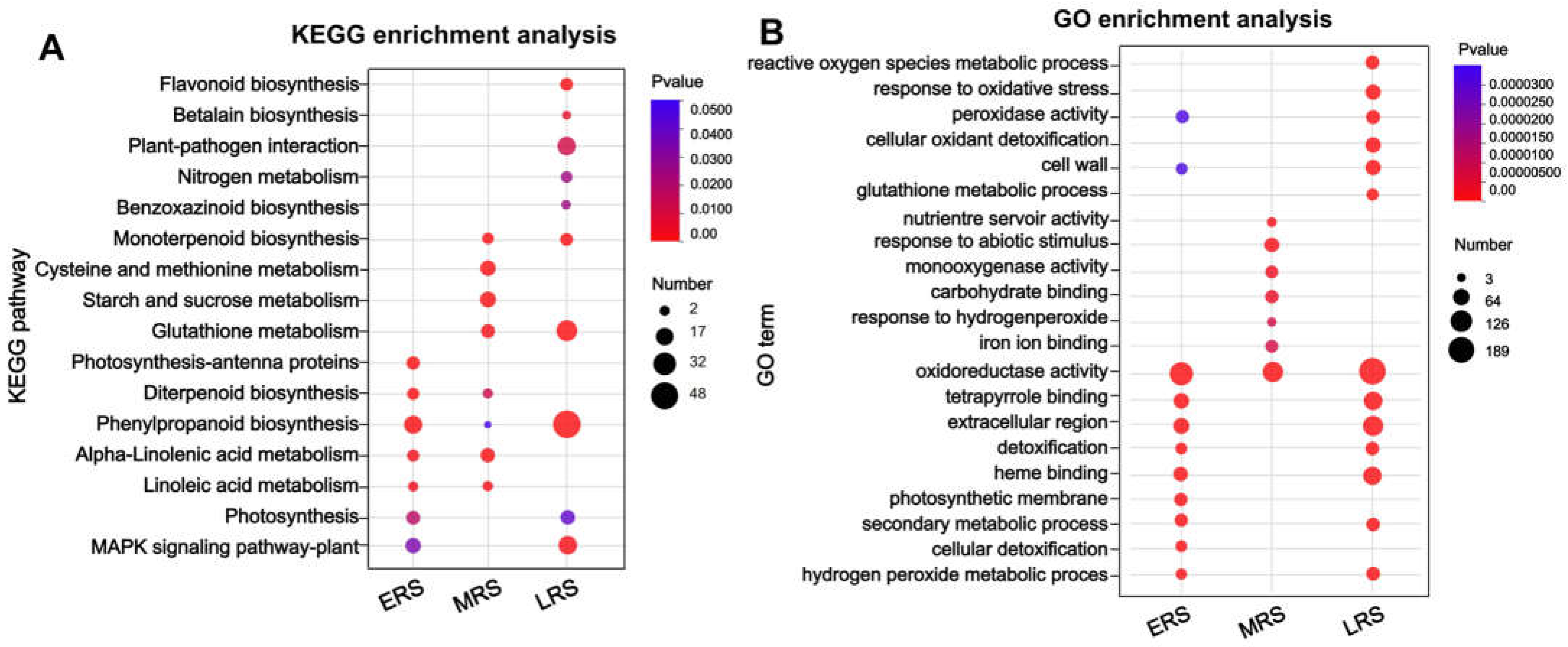
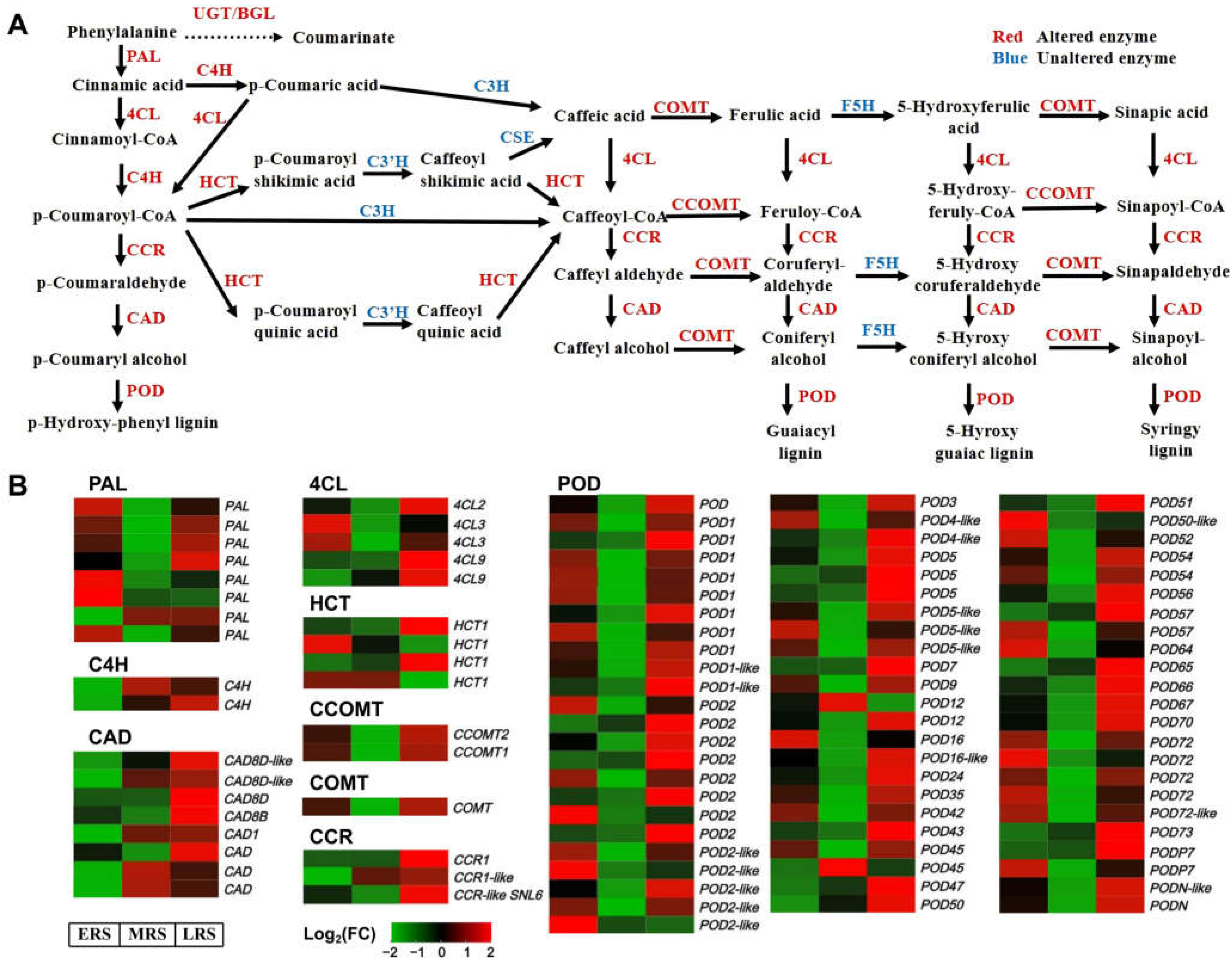
2.3. Zn Stress-Induced Extensive Changes in Phenylpropanoid–Lignin Pathway in Roots
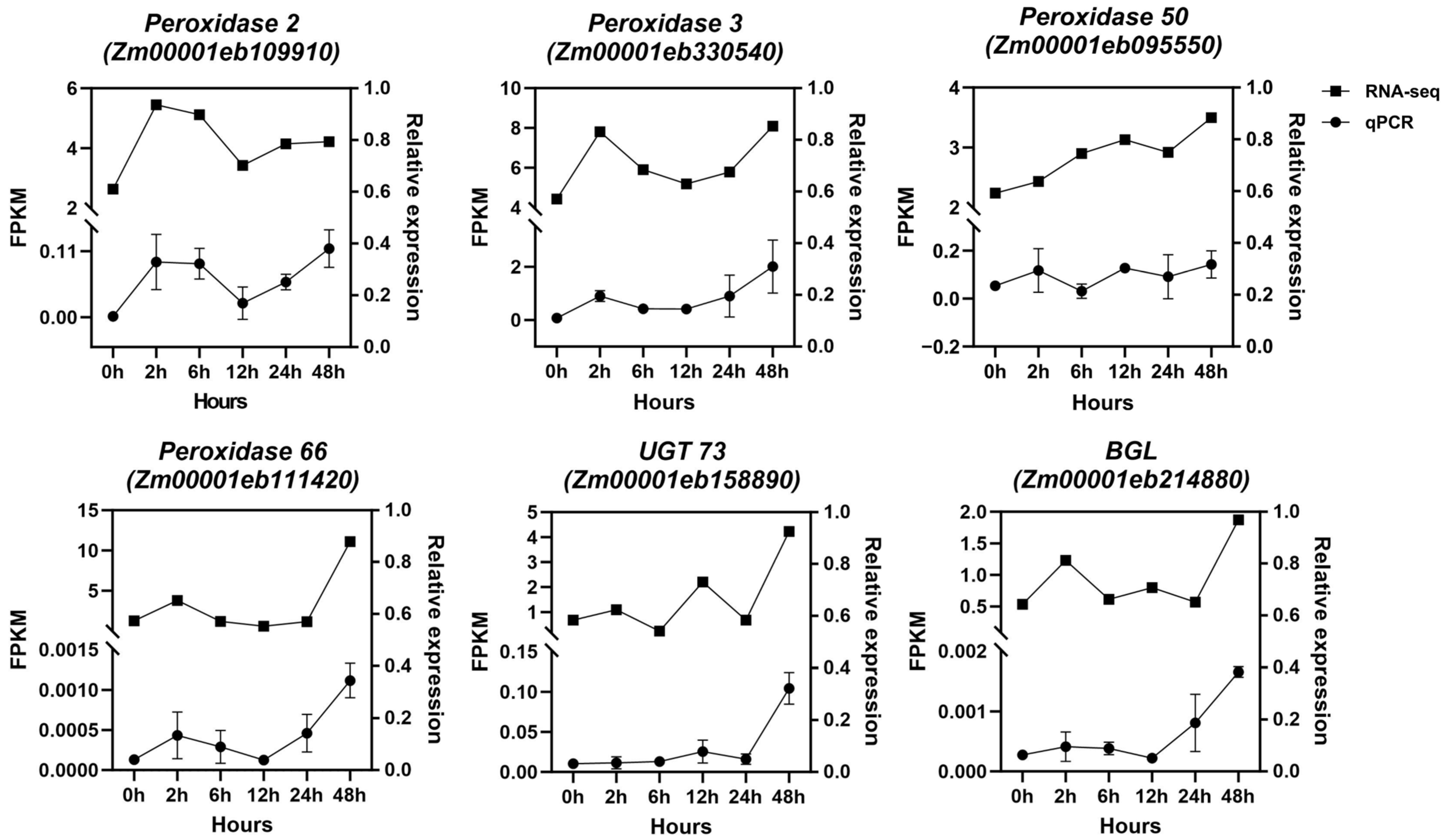
2.4. Validation of Expression Changes of Enzyme Genes in the Phenylpropanoid–Lignin Pathway by qRT-PCR Analysis
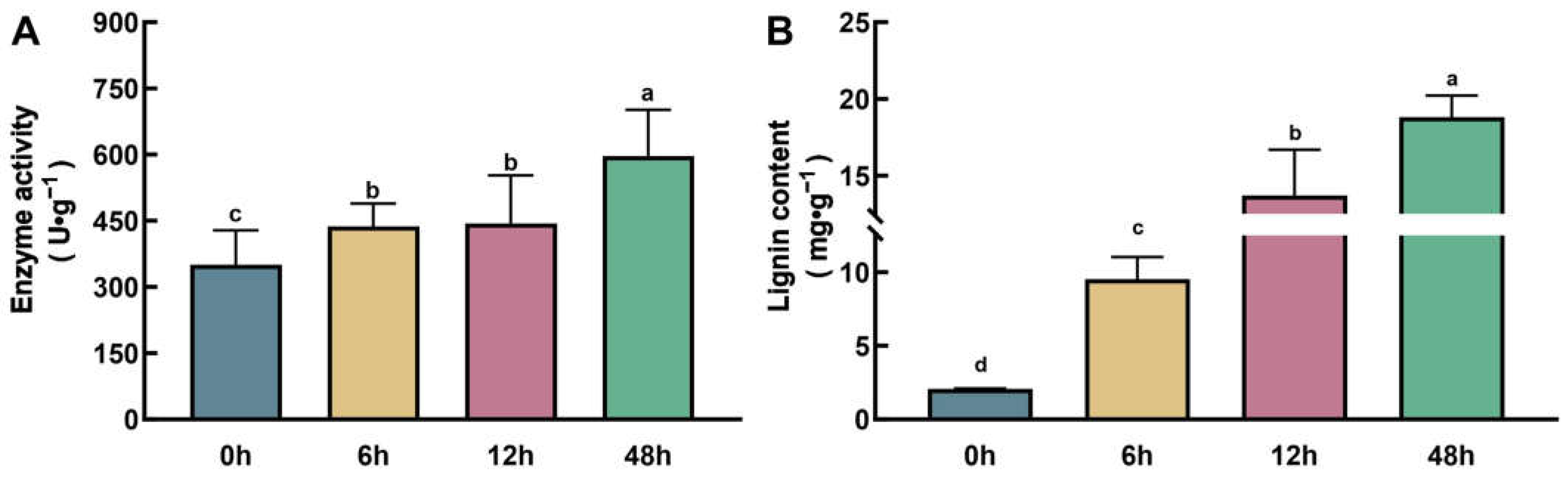
2.5. The Activity of POD Enzymes and the Lignin Content Increased with Zn Exposure
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Zn Treatment
4.2. RNA Extraction, Library Construction, and RNA Sequencing
4.3. Reads Mapping and Quantification of Gene Expression Levels
4.4. Identification of Differentially Expressed Genes
4.5. Mfuzz Analysis
4.6. Functional Enrichment Analysis
4.7. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.8. Determination of Metal Content
4.9. Determination of Antioxidant Enzyme Activity and Lignin Content
4.10. Quantitative RT-PCR (qRT-PCR)
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Haydon, M.J.; Cobbett, C.S. A novel major facilitator superfamily protein at the tonoplast influences zinc tolerance and accumulation in Arabidopsis. Plant Physiol. 2007, 143, 1705–1719. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A.K.; Pathak, C.; Prasad, G. Scenario of heavy metal contamination in agricultural soil its management. J. Appl. Nat. Sci. 2009, 1, 99. [Google Scholar] [CrossRef]
- Abedi, T.; Gavanji, S.; Mojiri, A. Lead and zinc uptake and toxicity in maize and their management. Plants 2022, 11, 1922. [Google Scholar] [CrossRef]
- Kaur, H.; Garg, N. Zinc toxicity in plants: A review. Planta 2021, 253, 129. [Google Scholar] [CrossRef]
- Tennstedt, P.; Peisker, D.; Böttcher, C.; Trampczynska, A.; Clemens, S. Phytochelatin synthesis is essential for the detoxification of excess zinc and contributes significantly to the accumulation of zinc. Plant Physiol. 2009, 149, 938–948. [Google Scholar] [CrossRef]
- Rout, G.R.; Das, P. Effect of metal toxicity on plant growth and metabolism: I. Zinc. Agronomie 2003, 23, 3–11. [Google Scholar] [CrossRef]
- Ren, Y.; Liu, Y.; Chen, H.; Li, G.; Zhang, X.; Zhao, J. Type 4 metallothionein genes are involved in regulating Zn ion accumulation in late embryo and in controlling early seedling growth in Arabidopsis. Plant Cell Environ. 2012, 35, 770–789. [Google Scholar] [CrossRef]
- Han, T.L.; Tang, T.W.; Zhang, P.H.; Liu, M.; Zhao, J.; Peng, J.S.; Meng, S. Cloning and functional characterization of SpZIP2. Genes 2022, 13, 2395. [Google Scholar] [CrossRef]
- Desbrosses-Fonrouge, A.G.; Voigt, K.; Schröder, A.; Arrivault, S.; Thomine, S.; Krämer, U. Arabidopsis thaliana MTP1 is a Zn transporter in the vacuolar membrane which mediates Zn detoxification and drives leaf Zn accumulation. FEBS Lett. 2005, 579, 4165–4174. [Google Scholar] [CrossRef]
- Remy, E.; Cabrito, T.R.; Batista, R.A.; Hussein, M.A.M.; Teixeira, M.C. Intron retention in the 5′ UTR of the novel ZIF2 transporter enhances translation to promote zinc tolerance in Arabidopsis. PLoS Genet. 2014, 10, e1004375. [Google Scholar] [CrossRef] [PubMed]
- Barrameda-Medina, Y.; Montesinos-Pereira, D.; Romero, L.; Blasco, B.; Ruiz, J.M. Role of GSH homeostasis under Zn toxicity in plants with different Zn tolerance. Plant Sci. 2014, 227, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Feng, K.; Xie, M.; Barros, J.; Tschaplinski, T.J.; Tuskan, G.A.; Muchero, W.; Chen, J.G. Phylogenetic occurrence of the phenylpropanoid pathway and lignin biosynthesis in plants. Front Plant Sci. 2021, 12, 704697. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, L.; Zheng, L. Lignins: Biosynthesis and biological functions in plants. Int. J. Mol. Sci. 2018, 19, 335. [Google Scholar] [CrossRef]
- Peng, J.S.; Guan, Y.H.; Lin, X.J.; Xu, X.J.; Xiao, L.; Wang, H.H.; Meng, S. Comparative understanding of metal hyperaccumulation in plants: A mini-review. Environ. Geochem. Health 2021, 43, 1599–1607. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, Q.; Liu, W.; Zhang, J. Exogenous Brassinosteroid Enhances Zinc tolerance by activating the Phenylpropanoid Biosynthesis pathway in Citrullus lanatus L. Plant Signal Behav. 2023, 18, 2186640. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, X.; Huang, Y.; Zhu, L.; Zhang, S.; Zhao, Y.; Guo, J.; Chen, J.; Chen, R. Identification and characterization of the zinc-regulated transporters, iron-regulated transporter-like protein (ZIP) gene family in maize. BMC Plant Biol. 2013, 13, 114. [Google Scholar] [CrossRef]
- Xu, Y.G.; Wang, B.S.; Yu, J.J.; Ao, G.M.; Zhao, Q. Cloning and characterisation of ZmZLP1, a gene encoding an endoplasmic reticulum-localised zinc transporter in Zea mays. Funct. Plant Biol. 2010, 37, 194. [Google Scholar] [CrossRef]
- Mallikarjuna, M.G.; Thirunavukkarasu, N.; Sharma, R.; Shiriga, K.; Hossain, F.; Bhat, J.S.; Mithra, A.C.; Marla, S.S.; Manjaiah, K.M.; Rao, A.; et al. Comparative transcriptome analysis of iron and zinc deficiency in maize (Zea mays L.). Plants 2020, 9, 1812. [Google Scholar] [CrossRef]
- Chao, Z.; Chen, Y.; Ji, C.; Wang, Y.; Huang, X.; Zhang, C.; Yang, J.; Song, T.; Wu, J.; Guo, L.; et al. A genome-wide association study identifies a transporter for zinc uploading to maize kernels. EMBO Rep. 2023, 24, e55542. [Google Scholar] [CrossRef]
- Liu, B.; Yu, H.; Yang, Q.; Ding, L.; Sun, F.; Qu, J.; Feng, W.; Yang, Q.; Li, W.; Fu, F. Zinc Transporter ZmLAZ1-4 modulates zinc homeostasis on plasma and vacuolar membrane in maize. Front. Plant Sci. 2022, 13, 881055. [Google Scholar] [CrossRef]
- Dong, N.Q.; Lin, H.X. Contribution of phenylpropanoid metabolism to plant development and plant-environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Dai, J.; Liu, Y.; Wang, Y.; Zhang, Q.; Lou, Y.; Qiu, Y. NAE1 protein: A prognostic, immunomodulatory, and therapeutic biomarker associated with neddylation in hepatocellular carcinoma. Int. J. Biol. Macromol. 2025, 310, 143539. [Google Scholar] [CrossRef] [PubMed]
- Mager, S.; Schönberger, B.; Ludewig, U. The transcriptome of zinc deficient maize roots and its relationship to DNA methylation loss. BMC Plant Biol. 2018, 18, 372. [Google Scholar] [CrossRef]
- Ganie, S.A.; Mazumder, A.; Kiran, K.; Hossain, F.; Sharma, R.; Mondal, T.K. Transcriptional dynamics of Zn-accumulation in developing kernels of maize reveals important Zn-uptake mechanisms. Genomics 2020, 112, 3435–3447. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Song, S.; Xu, F.; Pan, Y.; Wang, H. Transcriptomic and proteomic analyses reveal new insight into chlorophyll synthesis and chloroplast structure of maize leaves under zinc deficiency stress. J. Proteom. 2019, 199, 123–134. [Google Scholar] [CrossRef]
- Van, H.T.; Hoang, V.H.; Nga, L.T.Q.; Nguyen, V.Q. Effects of Zn pollution on soil: Pollution sources, impacts and solutions. Surf. Interfaces 2024, 17, 100360. [Google Scholar] [CrossRef]
- Zahra, H.; Latifeh, P. Zinc toxicity on antioxidative response in (Zea mays L.) at two different pH. J. Stress Physiol. Biochem. 2013, 9, 66–73. [Google Scholar]
- Liu, Q.; Zheng, L.; He, F.; Zhao, F.J.; Shen, Z.; Zheng, L. Transcriptional and physiological analyses identify a regulatory role for hydrogen peroxide in the lignin biosynthesis of copper-stressed rice roots. Plant Soil 2015, 387, 323–336. [Google Scholar] [CrossRef]
- Gao, L.; Peng, K.; Chen, Y.; Wang, G.; Shen, Z. Roles of apoplastic peroxidases, laccases, and lignification in the manganese tolerance of hyperaccumulator Phytolacca americana. Acta Physiol. Plant. 2012, 34, 151–159. [Google Scholar] [CrossRef]
- Lin, C.C.; Chen, L.M.; Liu, Z.H. Rapid effect of copper on lignin biosynthesis in soybean roots. Plant Sci. 2005, 168, 855–861. [Google Scholar] [CrossRef]
- Van de Mortel, J.E.; Villanueva, L.A.; Schat, H.; Kwekkeboom, J.; Coughlan, S.; Moerland, P.D.; Van Themaat, E.V.L.; Koornneef, M.; Aarts, M.G.M. Large expression differences in genes for iron and zinc homeostasis, stress response, and lignin biosynthesis distinguish roots of Arabidopsis thaliana and the related metal hyperaccumulator Thlaspi caerulescens. Plant Physiol. 2006, 142, 1127–1147. [Google Scholar] [CrossRef]
- Yang, Y.J.; Cheng, L.M.; Liu, Z.H. Rapid effect of cadmium on lignin biosynthesis in soybean roots. Plant Sci. 2007, 172, 632–639. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, S.; Shan, X.Q. Adsorption of metal ions on lignin. J. Hazard Mater. 2008, 151, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.S.; Zhang, X.J.; Xiong, J.N.; Zhou, Y.; Wang, W.L.; Chen, S.Y.; Zhang, D.W.; Gu, T.Y. Characterization of genes involved in micronutrients and toxic metals detoxification in Brassica napus by genome-wide cDNA library screening. Metallomics 2023, 15, mfad068. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Meng, Z.; Chen, C. Screening of potential biomarkers in peripheral blood of patients with depression based on weighted gene co-expression network analysis and machine learning algorithms. Front. Psychiatry 2022, 13, 1009911. [Google Scholar] [CrossRef]
- Hu, H.; He, Y.; Gao, Y.; Chen, S.; Gu, T.; Peng, J. NnMTP10 from Nelumbo nucifera acts as a transporter mediating manganese and iron efflux. Plant Mol. Biol. 2025, 115, 26. [Google Scholar] [CrossRef]
- Peng, J.S.; Zhang, B.C.; Chen, H.; Wang, M.Q.; Wang, Y.T.; Li, H.M.; Cao, S.X.; Yi, H.Y.; Wang, H.; Zhou, Y.H.; et al. Galactosylation of rhamnogalacturonan-II for cell wall pectin biosynthesis is critical for root apoplastic iron reallocation in Arabidopsis. Mol. Plant 2021, 14, 1640–1651. [Google Scholar] [CrossRef]
- Zhao, M.; Yang, Y.; Han, T.L.; Peng, J.S.; Gong, J.M.; Meng, S. Targeted expression of OsHMA3 driven by the OsYSL16 promoter decreases Cd accumulation in grains. Environ. Exp. Bot. 2025, 233, 106138. [Google Scholar] [CrossRef]
- Zhang, P.H.; Zhang, X.J.; Tang, T.W.; Hu, H.L.; Bai, N.N.; Zhang, D.W.; Meng, S.; Peng, J.S. Isolation of three metallothionein genes and their roles in mediating cadmium resistance. Agronomy 2022, 12, 2971. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, C.; Meng, Z.; Li, Y.; Abid, M.A.; Askari, M.; Wang, P.; Wang, Y.; Sun, G.; Cai, Y.; et al. Leveraging Atriplex hortensis choline monooxygenase to improve chilling tolerance in cotton. Environ. Exp. Bot. 2019, 162, 364–373. [Google Scholar] [CrossRef]
- Li, F.; Zhang, Y.; Tian, C.; Wang, X.; Zhou, L.; Jiang, J.; Wang, L.; Chen, F.; Chen, S. Molecular module of CmMYB15-like-Cm4CL2 regulating lignin biosynthesis of chrysanthemum (Chrysanthemum morifolium) in response to aphid (Macrosiphoniella sanborni) feeding. New Phytol. 2023, 237, 1776–1793. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Xu, Z.J.; Peng, J.S.; Zhao, J.; Zhang, G.B.; Xie, J.; Yi, Z.X.; Zhang, J.H.; Gong, J.M.; Ye, N.H.; et al. OsProT1 and OsProT3 Function to Mediate Proline-and γ-aminobutyric acid-specific Transport in Yeast and are Differentially Expressed in Rice (Oryza sativa L.). Rice 2019, 12, 79. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Gu, T.; Gao, Y.; Qu, J.; Zheng, H.; Guan, Y.; Peng, J. Transcriptomic Profiling Reveals the Involvement of the Phenylpropanoid–Lignin Pathway in the Response of Maize Roots to Zinc Stress. Plants 2025, 14, 1657. https://doi.org/10.3390/plants14111657
Zhou Y, Gu T, Gao Y, Qu J, Zheng H, Guan Y, Peng J. Transcriptomic Profiling Reveals the Involvement of the Phenylpropanoid–Lignin Pathway in the Response of Maize Roots to Zinc Stress. Plants. 2025; 14(11):1657. https://doi.org/10.3390/plants14111657
Chicago/Turabian StyleZhou, Ying, Tianyu Gu, Yan Gao, Jingtao Qu, Hongjian Zheng, Yuan Guan, and Jiashi Peng. 2025. "Transcriptomic Profiling Reveals the Involvement of the Phenylpropanoid–Lignin Pathway in the Response of Maize Roots to Zinc Stress" Plants 14, no. 11: 1657. https://doi.org/10.3390/plants14111657
APA StyleZhou, Y., Gu, T., Gao, Y., Qu, J., Zheng, H., Guan, Y., & Peng, J. (2025). Transcriptomic Profiling Reveals the Involvement of the Phenylpropanoid–Lignin Pathway in the Response of Maize Roots to Zinc Stress. Plants, 14(11), 1657. https://doi.org/10.3390/plants14111657