

Genus-Wide Pan-Genome Analysis of Oryza Calcium-Dependent Protein Kinase Genes and Their Related Kinases Highlights the Complexity of Protein Domain Architectures and Expression Dynamics




 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
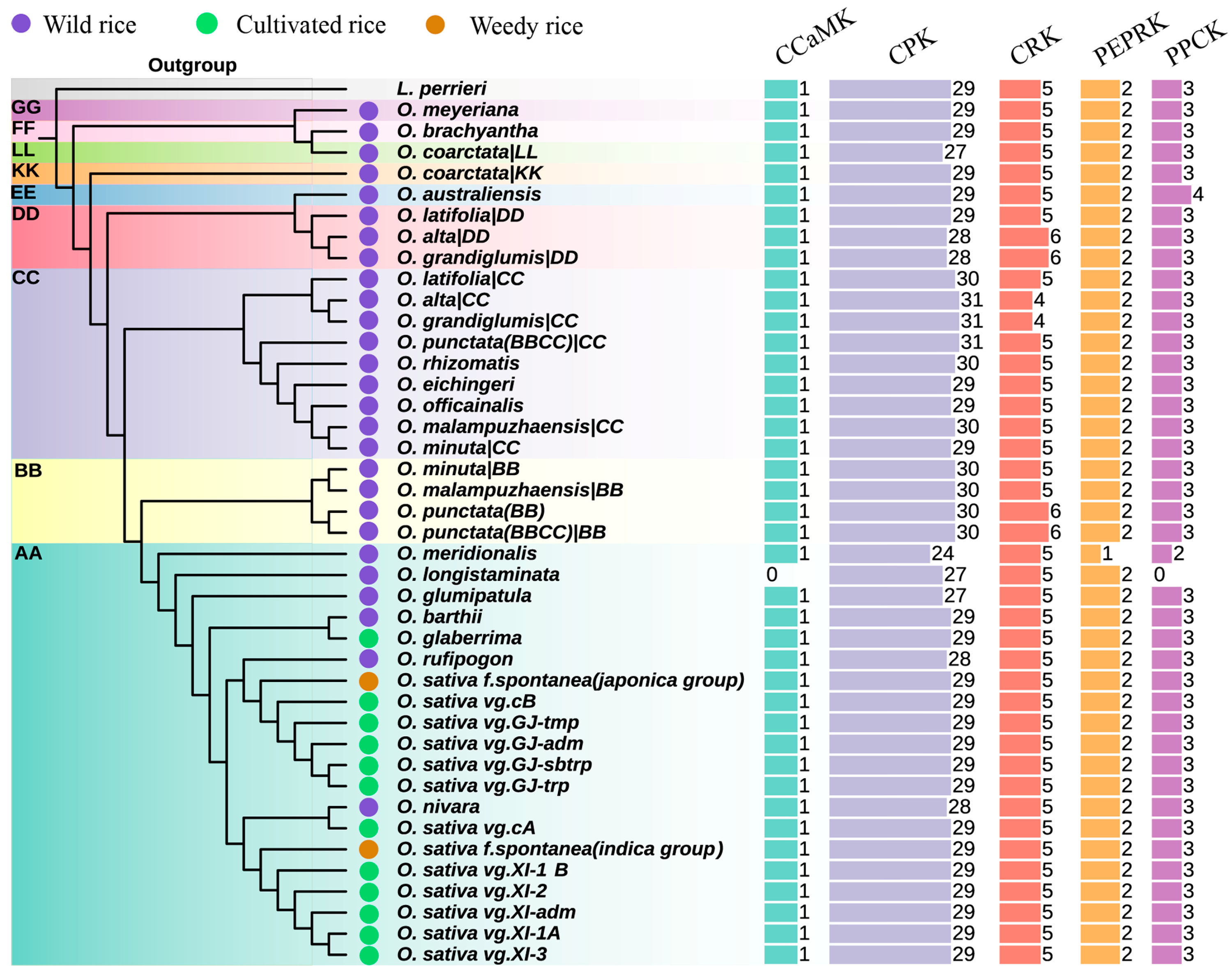
2.1. The Pan-Genome Analyses Enabled Understanding the Repertoire of CPKs and Their Related Kinases in Oryza
2.2. Several Types of Gene Duplication Contribute to the Expansion of CPKs and Their Related Kinases
2.3. Myristoylation and Palmitoylation of CPKs and Their Related Kinases
2.4. Molecular Properties of CPKs and Their Related Kinases
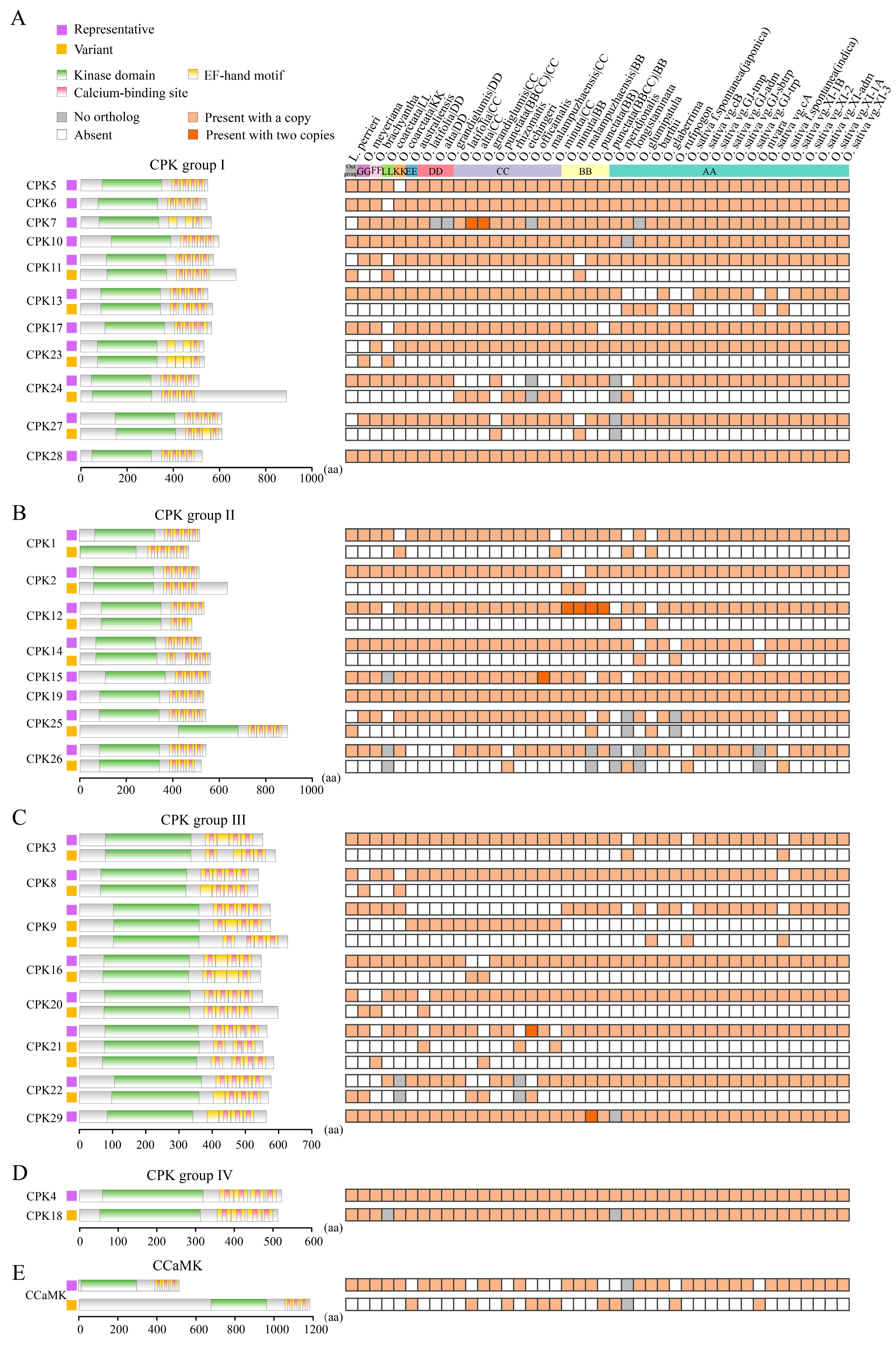
2.5. Landscapes of Domain Architectures Reveal the Diversity of EF-Hand Motifs in CPKs and CCaMKs
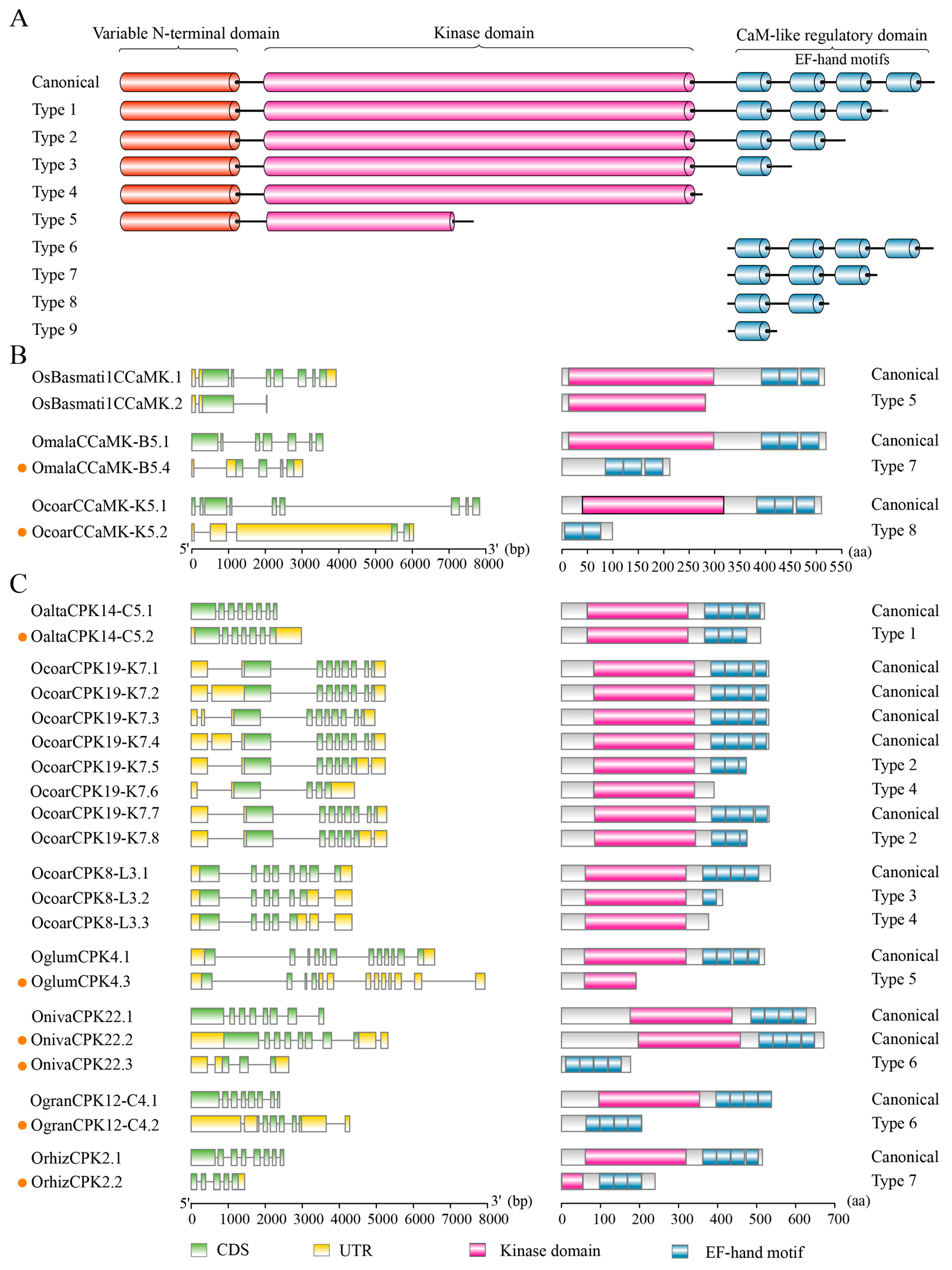
2.6. Alternative Splicing Contributes to the Diversity of Domain Architectures of CPKs and CCaMKs
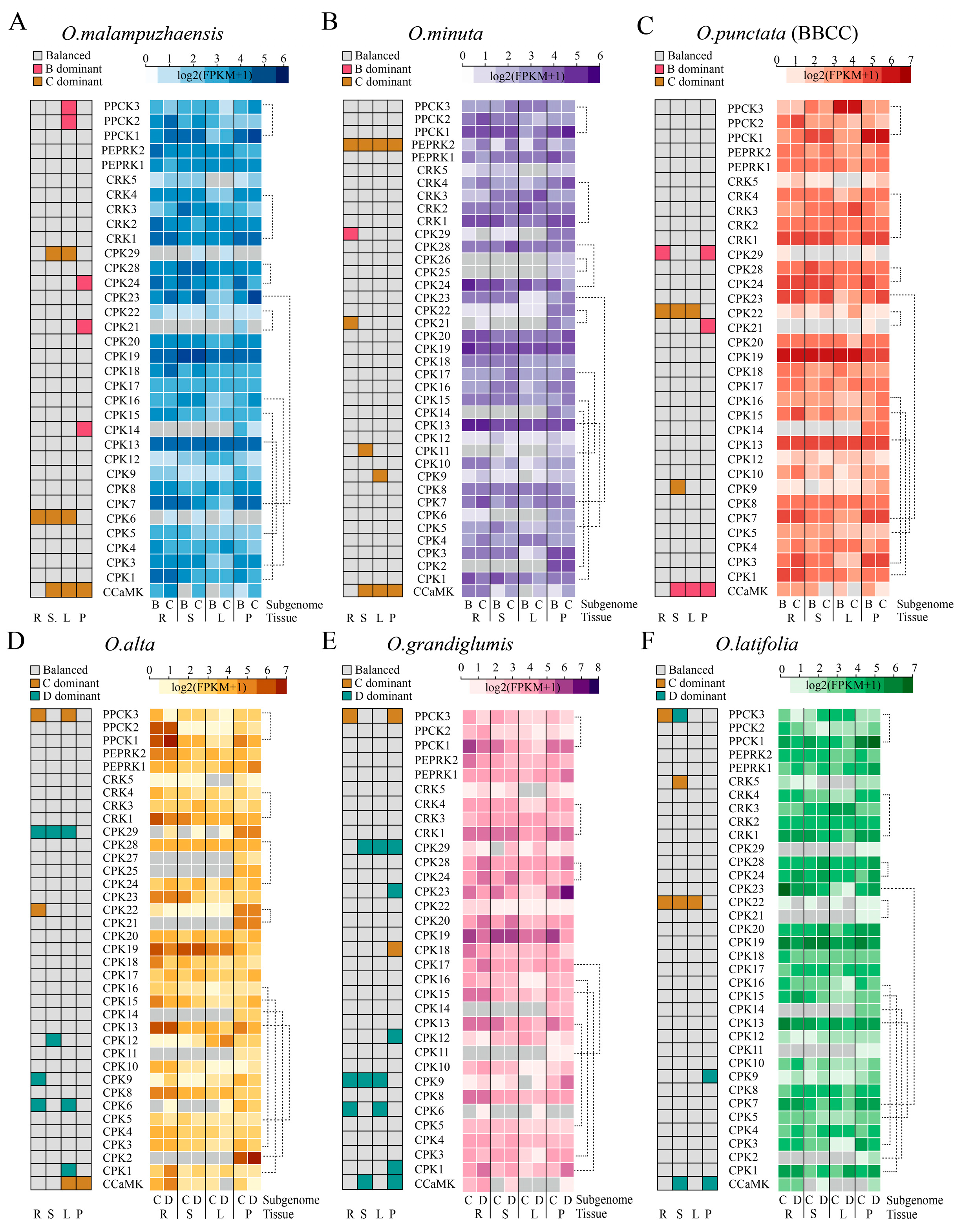
2.7. Homoeolog Expression Patterns of CPKs and Related Kinases Across Tissues and Species
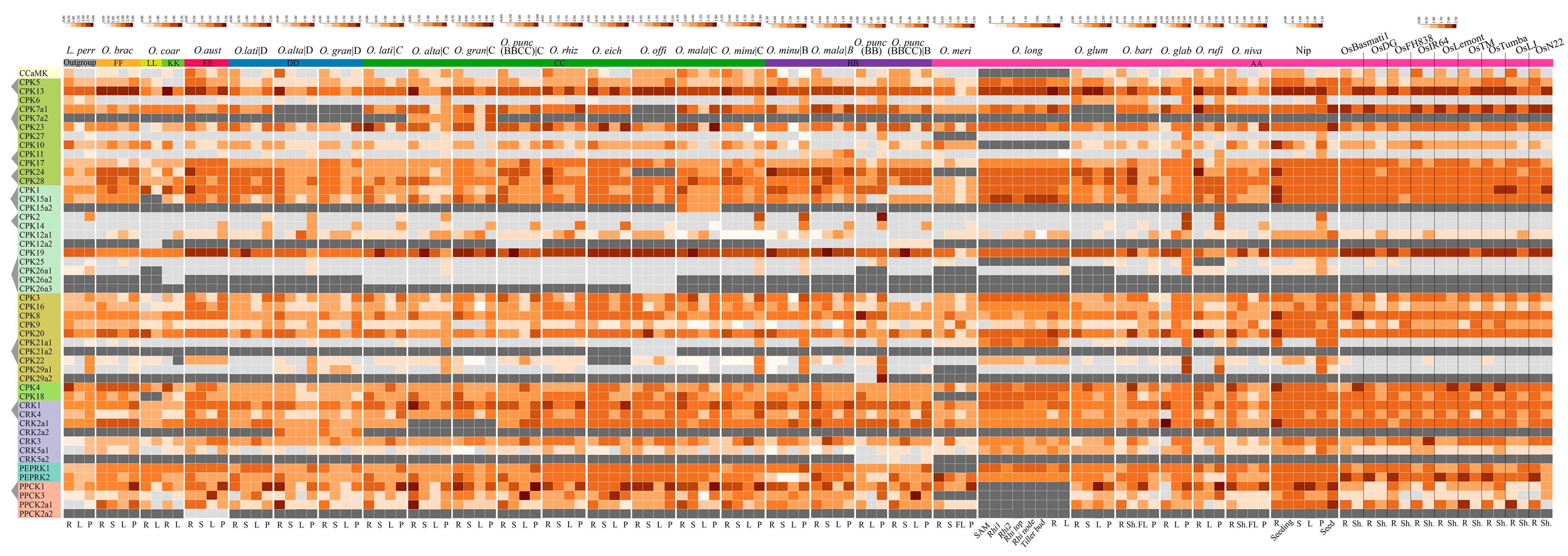
2.8. Transcriptomal Atlas of CPKs and Their Related Kinases Underlines the Expression Dynamics and Indicates Potential Functions
3. Discussion
4. Materials and Methods
4.1. Gene Identification of CPKs and Their Related Kinases
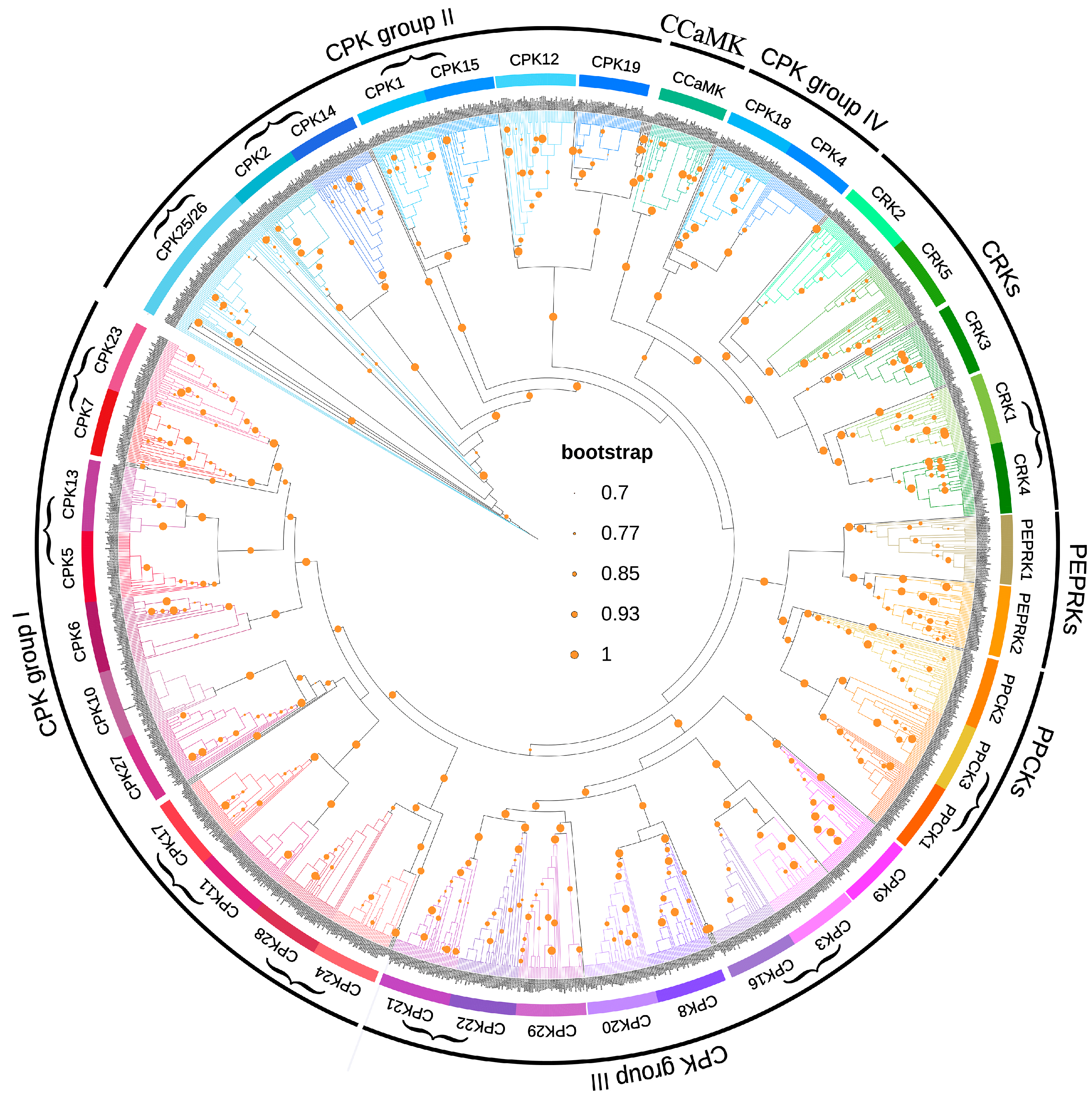
4.2. Construction of Phylogenetic Tree
4.3. Gene Structure and Protein Domain Analysis
4.4. Gene Duplication and Synteny Analysis
4.5. Reference-Based Transcriptome Assembly and Expression Quantification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CPKs | Calcium-dependent protein kinases |
| CRKs | CPK-related kinases |
| PPCKs | Phosphoenolpyruvate carboxylase kinases |
| PEPRKs | PPCK-related kinases |
| CCaMKs | Calcium and calmodulin-dependent kinases |
| VNTD | Variable N-terminal domain |
| JD | Junction domain |
| CaM | Calmodulin |
| CaMLD | Calmodulin (CaM)-like domain |
| CNV | Copy number variation |
| WGD | Whole-genome duplication |
| TD | Tandem duplication |
| PD | Proximal duplication |
| TRD | Transposed duplication |
| DSD | Dispersed duplication |
| AS | Alternative splicing |
| HEB | Homoeolog expression bias |
References
- Wing, R.A.; Purugganan, M.D.; Zhang, Q. The rice genome revolution: From an ancient grain to Green Super Rice. Nat. Rev. Genet. 2018, 19, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Peng, Y.; Qiao, J.; Henry, R.; Qian, Q. Wild rice: Unlocking the future of rice breeding. Plant Biotechnol. J. 2024, 22, 3218–3226. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chen, Z.; Liang, C. Oryza pan-genomics: A new foundation for future rice research and improvement. Crop J. 2021, 9, 622–632. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Liu, H.; Fu, B.; Li, L.; Xie, M.; Song, Y.; Li, X.; Cai, J.; Wan, W.; et al. Genome and comparative transcriptomics of african wild rice Oryza longistaminata provide insights into molecular mechanism of rhizomatousness and self-incompatibility. Mol. Plant 2015, 8, 1683–1686. [Google Scholar] [CrossRef]
- Stein, J.C.; Yu, Y.; Copetti, D.; Zwickl, D.J.; Zhang, L.; Zhang, C.; Chougule, K.; Gao, D.; Iwata, A.; Goicoechea, J.L.; et al. Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza. Nat. Genet. 2018, 50, 285–296. [Google Scholar] [CrossRef]
- Wang, M.; Yu, Y.; Haberer, G.; Marri, P.R.; Fan, C.; Goicoechea, J.L.; Zuccolo, A.; Song, X.; Kudrna, D.; Ammiraju, J.S.; et al. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat. Genet. 2014, 46, 982–988. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, W.; Yang, Y.; Wang, Z.; Sun, J.; Yuan, K.; Rabbi, S.; Khanam, M.; Kabir, M.S.; Seraj, Z.I.; et al. A high-quality chromosome-level wild rice genome of Oryza coarctata. Sci. Data 2023, 10, 701. [Google Scholar] [CrossRef]
- Long, W.; He, Q.; Wang, Y.; Wang, Y.; Wang, J.; Yuan, Z.; Wang, M.; Chen, W.; Luo, L.; Luo, L.; et al. Genome evolution and diversity of wild and cultivated rice species. Nat. Commun. 2024, 15, 9994. [Google Scholar] [CrossRef]
- Xie, X.; Du, H.; Tang, H.; Tang, J.; Tan, X.; Liu, W.; Li, T.; Lin, Z.; Liang, C.; Liu, Y.G. A chromosome-level genome assembly of the wild rice Oryza rufipogon facilitates tracing the origins of Asian cultivated rice. Sci. China Life Sci. 2021, 64, 282–293. [Google Scholar] [CrossRef]
- Cai, X.; He, W.; Qian, Q.; Shang, L. Genetic resource utilization in wild rice species: Genomes and gene bank. New Crops 2025, 2, 100065. [Google Scholar] [CrossRef]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Chen, Y.; Zhou, Y.; Zhang, Y.; Li, M.; Ouyang, Y.; Chebotarov, D.; Mauleon, R.; Zhao, H.; Xie, W.; et al. Rice Gene Index: A comprehensive pan-genome database for comparative and functional genomics of Asian rice. Mol. Plant 2023, 16, 798–801. [Google Scholar] [CrossRef]
- Song, J.M.; Xie, W.Z.; Wang, S.; Guo, Y.X.; Koo, D.H.; Kudrna, D.; Gong, C.; Huang, Y.; Feng, J.W.; Zhang, W.; et al. Two gap-free reference genomes and a global view of the centromere architecture in rice. Mol. Plant 2021, 14, 1757–1767. [Google Scholar] [CrossRef]
- Ammiraju, J.S.; Lu, F.; Sanyal, A.; Yu, Y.; Song, X.; Jiang, N.; Pontaroli, A.C.; Rambo, T.; Currie, J.; Collura, K.; et al. Dynamic evolution of oryza genomes is revealed by comparative genomic analysis of a genus-wide vertical data set. Plant Cell 2008, 20, 3191–3209. [Google Scholar] [CrossRef]
- Chen, E.; Huang, X.; Tian, Z.; Wing, R.A.; Han, B. The genomics of oryza species provides insights into rice domestication and heterosis. Annu. Rev. Plant Biol. 2019, 70, 639–665. [Google Scholar] [CrossRef]
- Wu, D.; Xie, L.; Sun, Y.; Huang, Y.; Jia, L.; Dong, C.; Shen, E.; Ye, C.Y.; Qian, Q.; Fan, L. A syntelog-based pan-genome provides insights into rice domestication and de-domestication. Genome Biol. 2023, 24, 179. [Google Scholar] [CrossRef]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar] [CrossRef]
- Paterson, A.H.; Bowers, J.E.; Chapman, B.A. Ancient polyploidization predating divergence of the cereals, and its consequences for comparative genomics. Proc. Natl. Acad. Sci. USA 2004, 101, 9903–9908. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, J.; Tang, H.; Paterson, A.H. Integrated syntenic and phylogenomic analyses reveal an ancient genome duplication in monocots. Plant Cell 2014, 26, 2792–2802. [Google Scholar] [CrossRef]
- Tang, H.; Bowers, J.E.; Wang, X.; Paterson, A.H. Angiosperm genome comparisons reveal early polyploidy in the monocot lineage. Proc. Natl. Acad. Sci. USA 2010, 107, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, X.; Hu, Y.; Qin, L.; Wang, D.; Jia, J.; Jiao, Y. A recent burst of gene duplications in Triticeae. Plant Commun. 2022, 3, 100268. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Feng, J.; Zhang, Q.; Wang, Y.; Guan, Y.; Wang, R.; Shi, F.; Zeng, F.; Wang, Y.; Chen, M.; et al. Integrative gene duplication and genome-wide analysis as an approach to facilitate wheat reverse genetics: An example in the TaCIPK family. J. Adv. Res. 2024, 61, 19–33. [Google Scholar] [CrossRef]
- Hrabak, E.M.; Chan, C.W.; Gribskov, M.; Harper, J.F.; Choi, J.H.; Halford, N.; Kudla, J.; Luan, S.; Nimmo, H.G.; Sussman, M.R.; et al. The Arabidopsis CDPK-SnRK superfamily of protein kinases. Plant Physiol. 2003, 132, 666–680. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, L.; Cheng, Z.M. The calmodulin fused kinase novel gene family is the major system in plants converting Ca2+ signals to protein phosphorylation responses. Sci. Rep. 2017, 7, 4127. [Google Scholar] [CrossRef]
- Harmon, A.C.; Gribskov, M.; Gubrium, E.; Harper, J.F. The CDPK superfamily of protein kinases. New Phytol. 2001, 151, 175–183. [Google Scholar] [CrossRef]
- Zhang, X.S.; Choi, J.H. Molecular evolution of calmodulin-like domain protein kinases (CDPKs) in plants and protists. J. Mol. Evol. 2001, 53, 214–224. [Google Scholar] [CrossRef]
- Ye, S.; Wang, L.; Xie, W.; Wan, B.; Li, X.; Lin, Y. Expression profile of calcium-dependent protein kinase (CDPKs) genes during the whole lifespan and under phytohormone treatment conditions in rice (Oryza sativa L. ssp. indica). Plant Mol. Biol. 2009, 70, 311–325. [Google Scholar] [CrossRef]
- Asano, T.; Tanaka, N.; Yang, G.; Hayashi, N.; Komatsu, S. Genome-wide identification of the rice calcium-dependent protein kinase and its closely related kinase gene families: Comprehensive analysis of the CDPKs gene family in rice. Plant Cell Physiol. 2005, 46, 356–366. [Google Scholar] [CrossRef]
- Ray, S.; Agarwal, P.; Arora, R.; Kapoor, S.; Tyagi, A.K. Expression analysis of calcium-dependent protein kinase gene family during reproductive development and abiotic stress conditions in rice (Oryza sativa L. ssp. indica). Mol. Genet. Genom. 2007, 278, 493–505. [Google Scholar] [CrossRef]
- Zhang, J.; Lyu, H.; Chen, J.; Cao, X.; Du, R.; Ma, L.; Wang, N.; Zhu, Z.; Rao, J.; Wang, J.; et al. Releasing a sugar brake generates sweeter tomato without yield penalty. Nature 2024, 635, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, Q.; Yan, H.; Cang, X.; Li, W.; He, J.; Zhang, M.; Lou, L.; Wang, R.; Chang, M. Lighting-up wars: Stories of Ca2+ signaling in plant immunity. New Crops 2024, 1, 100027. [Google Scholar] [CrossRef]
- Yip Delormel, T.; Boudsocq, M. Properties and functions of calcium-dependent protein kinases and their relatives in Arabidopsis thaliana. New Phytol. 2019, 224, 585–604. [Google Scholar] [CrossRef]
- Baba, A.I.; Rigo, G.; Ayaydin, F.; Rehman, A.U.; Andrasi, N.; Zsigmond, L.; Valkai, I.; Urbancsok, J.; Vass, I.; Pasternak, T.; et al. Functional analysis of the Arabidopsis thaliana CDPK-related kinase family: AtCRK1 regulates responses to continuous light. Int. J. Mol. Sci. 2018, 19, 1282. [Google Scholar] [CrossRef]
- Rigo, G.; Ayaydin, F.; Tietz, O.; Zsigmond, L.; Kovacs, H.; Pay, A.; Salchert, K.; Darula, Z.; Medzihradszky, K.F.; Szabados, L.; et al. Inactivation of plasma membrane-localized CDPK-RELATED KINASE5 decelerates PIN2 exocytosis and root gravitropic response in Arabidopsis. Plant Cell 2013, 25, 1592–1608. [Google Scholar] [CrossRef]
- Wang, T.; Guo, J.; Peng, Y.; Lyu, X.; Liu, B.; Sun, S.; Wang, X. Light-induced mobile factors from shoots regulate rhizobium-triggered soybean root nodulation. Science 2021, 374, 65–71. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, T.; Ni, L.; Chen, C.; Jiang, J.; Cui, Z.; Wang, S.; Xu, F.; Yan, R.; Jiang, M. Phosphorylation of OsRbohB by the protein kinase OsDMI3 promotes H2O2 production to potentiate ABA responses in rice. Mol. Plant 2023, 16, 882–902. [Google Scholar] [CrossRef]
- Feria, A.B.; Bosch, N.; Sanchez, A.; Nieto-Ingelmo, A.I.; de la Osa, C.; Echevarria, C.; Garcia-Maurino, S.; Monreal, J.A. Phosphoenolpyruvate carboxylase (PEPC) and PEPC-kinase (PEPC-k) isoenzymes in Arabidopsis thaliana: Role in control and abiotic stress conditions. Planta 2016, 244, 901–913. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Li, C.; Buell, C.R. The rice genome annotation project: An updated database for mining the rice genome. Nucleic Acids Res. 2025, 53, D1614–D1622. [Google Scholar] [CrossRef]
- Shang, L.; He, W.; Wang, T.; Yang, Y.; Xu, Q.; Zhao, X.; Yang, L.; Zhang, H.; Li, X.; Lv, Y.; et al. A complete assembly of the rice Nipponbare reference genome. Mol. Plant 2023, 16, 1232–1236. [Google Scholar] [CrossRef]
- Sakai, H.; Lee, S.S.; Tanaka, T.; Numa, H.; Kim, J.; Kawahara, Y.; Wakimoto, H.; Yang, C.C.; Iwamoto, M.; Abe, T.; et al. Rice Annotation Project Database (RAP-DB): An Integrative and Interactive Database for Rice Genomics. Plant Cell Physiol. 2013, 54, e6. [Google Scholar] [CrossRef] [PubMed]
- Vizueta, J.; Sanchez-Gracia, A.; Rozas, J. Bitacora: A comprehensive tool for the identification and annotation of gene families in genome assemblies. Mol. Ecol. Resour. 2020, 20, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Keilwagen, J.; Wenk, M.; Erickson, J.L.; Schattat, M.H.; Grau, J.; Hartung, F. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 2016, 44, e89. [Google Scholar] [CrossRef]
- Saito, S.; Hamamoto, S.; Moriya, K.; Matsuura, A.; Sato, Y.; Muto, J.; Noguchi, H.; Yamauchi, S.; Tozawa, Y.; Ueda, M.; et al. N-myristoylation and S-acylation are common modifications of Ca2+-regulated Arabidopsis kinases and are required for activation of the SLAC1 anion channel. New Phytol. 2018, 218, 1504–1521. [Google Scholar] [CrossRef]
- Martin, M.L.; Busconi, L. Membrane localization of a rice calcium-dependent protein kinase (CDPK) is mediated by myristoylation and palmitoylation. Plant J. 2000, 24, 429–435. [Google Scholar] [CrossRef]
- Du, W.; Wang, Y.; Liang, S.; Lu, Y. Biochemical and expression analysis of an Arabidopsis calcium-dependent protein kinase-related kinase. Plant Sci. 2005, 168, 1181–1192. [Google Scholar] [CrossRef]
- Ramirez-Gonzalez, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef]
- Tan, L.; Li, X.; Liu, F.; Sun, X.; Li, C.; Zhu, Z.; Fu, Y.; Cai, H.; Wang, X.; Xie, D.; et al. Control of a key transition from prostrate to erect growth in rice domestication. Nat. Genet. 2008, 40, 1360–1364. [Google Scholar] [CrossRef]
- Hu, L.; Wu, Y.; Wu, D.; Rao, W.; Guo, J.; Ma, Y.; Wang, Z.; Shangguan, X.; Wang, H.; Xu, C.; et al. The coiled-coil and nucleotide binding domains of BROWN PLANTHOPPER RESISTANCE14 function in signaling and resistance against planthopper in rice. Plant Cell 2017, 29, 3157–3185. [Google Scholar] [CrossRef]
- Lin, C.C.; Lee, W.J.; Zeng, C.Y.; Chou, M.Y.; Lin, T.J.; Lin, C.S.; Ho, M.C.; Shih, M.C. SUB1A-1 anchors a regulatory cascade for epigenetic and transcriptional controls of submergence tolerance in rice. PNAS Nexus 2023, 2, pgad229. [Google Scholar] [CrossRef]
- Shang, L.; Li, X.; He, H.; Yuan, Q.; Song, Y.; Wei, Z.; Lin, H.; Hu, M.; Zhao, F.; Zhang, C.; et al. A super pan-genomic landscape of rice. Cell Res. 2022, 32, 878–896. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef]
- Dressano, K.; Weckwerth, P.R.; Poretsky, E.; Takahashi, Y.; Villarreal, C.; Shen, Z.; Schroeder, J.I.; Briggs, S.P.; Huffaker, A. Dynamic regulation of Pep-induced immunity through post-translational control of defence transcript splicing. Nat. Plants 2020, 6, 1008–1019. [Google Scholar] [CrossRef]
- Almadanim, M.C.; Goncalves, N.M.; Rosa, M.T.G.; Alexandre, B.M.; Cordeiro, A.M.; Rodrigues, M.; Saibo, N.J.M.; Soares, C.M.; Romao, C.V.; Oliveira, M.M.; et al. The rice cold-responsive calcium-dependent protein kinase OsCPK17 is regulated by alternative splicing and post-translational modifications. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 231–246. [Google Scholar] [CrossRef]
- Boudsocq, M.; Droillard, M.J.; Regad, L.; Lauriere, C. Characterization of Arabidopsis calcium-dependent protein kinases: Activated or not by calcium? Biochem. J. 2012, 447, 291–299. [Google Scholar] [CrossRef]
- Loranger, M.E.W.; Huffaker, A.; Monaghan, J. Truncated variants of Ca2+-dependent protein kinases: A conserved regulatory mechanism? Trends Plant Sci. 2021, 26, 1002–1005. [Google Scholar] [CrossRef]
- Wan, B.; Lin, Y.; Mou, T. Expression of rice Ca2+-dependent protein kinases (CDPKs) genes under different environmental stresses. FEBS Lett. 2007, 581, 1179–1189. [Google Scholar] [CrossRef]
- Valmonte, G.R.; Arthur, K.; Higgins, C.M.; MacDiarmid, R.M. Calcium-dependent protein kinases in plants: Evolution, expression and function. Plant Cell Physiol. 2014, 55, 551–569. [Google Scholar] [CrossRef]
- Yadav, A.; Garg, T.; Singh, H.; Yadav, S.R. Tissue-specific expression pattern of calcium-dependent protein kinases-related kinases (CRKs) in rice. Plant Signal. Behav. 2020, 15, 1809846. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Chen, Y.; Song, W.; Xie, X.; Wang, Z.; Guan, P.; Peng, H.; Jiao, Y.; Ni, Z.; Sun, Q.; Guo, W. A collinearity-incorporating homology inference strategy for connecting emerging assemblies in the triticeae tribe as a pilot practice in the plant pangenomic era. Mol. Plant 2020, 13, 1694–1708. [Google Scholar] [CrossRef] [PubMed]
- Blum, M.; Andreeva, A.; Florentino, L.C.; Chuguransky, S.R.; Grego, T.; Hobbs, E.; Pinto, B.L.; Orr, A.; Paysan-Lafosse, T.; Ponamareva, I.; et al. InterPro: The protein sequence classification resource in 2025. Nucleic Acids Res. 2025, 53, D444–D456. [Google Scholar] [CrossRef]
- de Castro, E.; Sigrist, C.J.; Gattiker, A.; Bulliard, V.; Langendijk-Genevaux, P.S.; Gasteiger, E.; Bairoch, A.; Hulo, N. ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006, 34, W362–W365. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Marcais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Jia, K.H.; Wang, Z.X.; Wang, L.; Li, G.Y.; Zhang, W.; Wang, X.L.; Xu, F.J.; Jiao, S.Q.; Zhou, S.S.; Liu, H.; et al. SubPhaser: A robust allopolyploid subgenome phasing method based on subgenome-specific k-mers. New Phytol. 2022, 235, 801–809. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Zhang, C.; Rabiee, M.; Sayyari, E.; Mirarab, S. ASTRAL-III: Polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018, 19, 153. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zheng, Y.; Li, H.; Luo, X.; He, Z.; Cao, S.; Shi, Y.; Zhao, Q.; Xue, Y.; Zuo, Z.; et al. GPS-Lipid: A robust tool for the prediction of multiple lipid modification sites. Sci. Rep. 2016, 6, 28249. [Google Scholar] [CrossRef]
- Thumuluri, V.; Almagro Armenteros, J.J.; Johansen, A.R.; Nielsen, H.; Winther, O. DeepLoc 2.0: Multi-label subcellular localization prediction using protein language models. Nucleic Acids Res. 2022, 50, W228–W234. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. Imeta 2023, 2, e107. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Shao, M.; Kingsford, C. Accurate assembly of transcripts through phase-preserving graph decomposition. Nat. Biotechnol. 2017, 35, 1167–1169. [Google Scholar] [CrossRef]
- Pertea, G.; Pertea, M. GFF Utilities: GffRead and GffCompare. F1000Research 2020, 9, ISCB. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [PubMed]
- Lian, Q.; Huettel, B.; Walkemeier, B.; Mayjonade, B.; Lopez-Roques, C.; Gil, L.; Roux, F.; Schneeberger, K.; Mercier, R. A pan-genome of 69 Arabidopsis thaliana accessions reveals a conserved genome structure throughout the global species range. Nat. Genet. 2024, 56, 982–991. [Google Scholar] [CrossRef]
- Jayakodi, M.; Lu, Q.; Pidon, H.; Rabanus-Wallace, M.T.; Bayer, M.; Lux, T.; Guo, Y.; Jaegle, B.; Badea, A.; Bekele, W.; et al. Structural variation in the pangenome of wild and domesticated barley. Nature 2024, 636, 654–662. [Google Scholar] [CrossRef]
- Jiao, C.; Xie, X.; Hao, C.; Chen, L.; Xie, Y.; Garg, V.; Zhao, L.; Wang, Z.; Zhang, Y.; Li, T.; et al. Pan-genome bridges wheat structural variations with habitat and breeding. Nature 2025, 637, 384–393. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, H.; Long, Y.; Shu, Y.; Zhai, J. Plant Public RNA-seq Database: A comprehensive online database for expression analysis of ~45,000 plant public RNA-Seq libraries. Plant Biotechnol. J. 2022, 20, 806–808. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, F.; Li, L.; Chen, M.; Chang, J.; Tu, M.; He, G.; Li, Y.; Yang, G. Genus-Wide Pan-Genome Analysis of Oryza Calcium-Dependent Protein Kinase Genes and Their Related Kinases Highlights the Complexity of Protein Domain Architectures and Expression Dynamics. Plants 2025, 14, 1542. https://doi.org/10.3390/plants14101542
Shi F, Li L, Chen M, Chang J, Tu M, He G, Li Y, Yang G. Genus-Wide Pan-Genome Analysis of Oryza Calcium-Dependent Protein Kinase Genes and Their Related Kinases Highlights the Complexity of Protein Domain Architectures and Expression Dynamics. Plants. 2025; 14(10):1542. https://doi.org/10.3390/plants14101542
Chicago/Turabian StyleShi, Fu, Li Li, Mingjie Chen, Junli Chang, Min Tu, Guangyuan He, Yin Li, and Guangxiao Yang. 2025. "Genus-Wide Pan-Genome Analysis of Oryza Calcium-Dependent Protein Kinase Genes and Their Related Kinases Highlights the Complexity of Protein Domain Architectures and Expression Dynamics" Plants 14, no. 10: 1542. https://doi.org/10.3390/plants14101542
APA StyleShi, F., Li, L., Chen, M., Chang, J., Tu, M., He, G., Li, Y., & Yang, G. (2025). Genus-Wide Pan-Genome Analysis of Oryza Calcium-Dependent Protein Kinase Genes and Their Related Kinases Highlights the Complexity of Protein Domain Architectures and Expression Dynamics. Plants, 14(10), 1542. https://doi.org/10.3390/plants14101542