
Functional Characterization of KNOX and BELL Genes in Temperature-Responsive Floral Morphogenesis of Passion Fruit (Passiflora edulis)

 ,
, 
Abstract

1. Introduction
2. Results
2.1. Identification of the TALE Gene Family in Passion Fruit
2.2. Classification and Phylogenetic Relationships of PeTALEs
2.3. Chromosomal Distribution and Synteny Analysis of PeTALE Genes
2.4. Conserved Motifs and Gene Structure Characteristic Analysis of PeTALE Genes
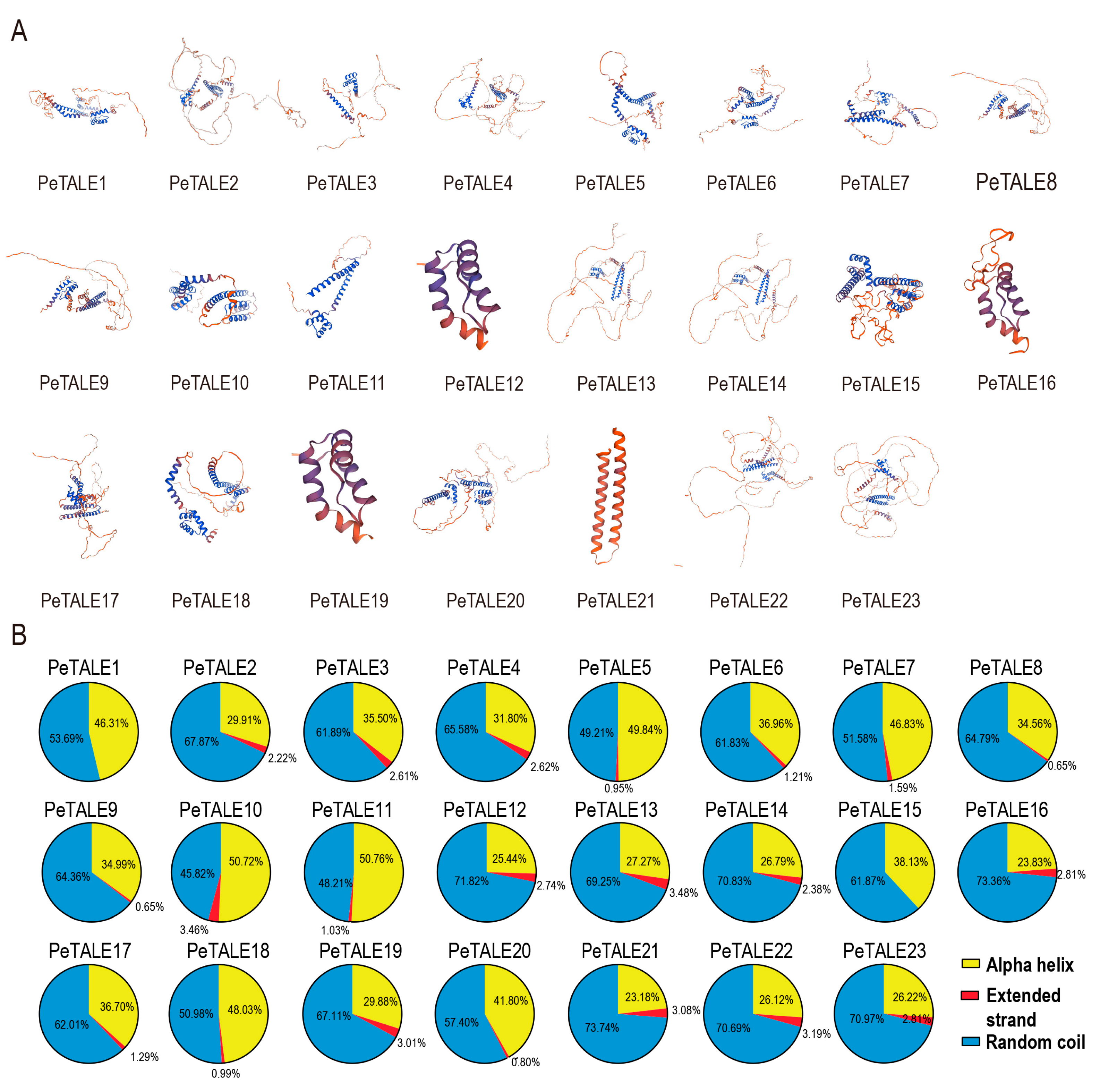
2.5. The Secondary Structure Prediction of PeTALE Proteins
2.6. Expression Profiling of PeTALE Genes For Different Floral Organs and Their Response to Temperature
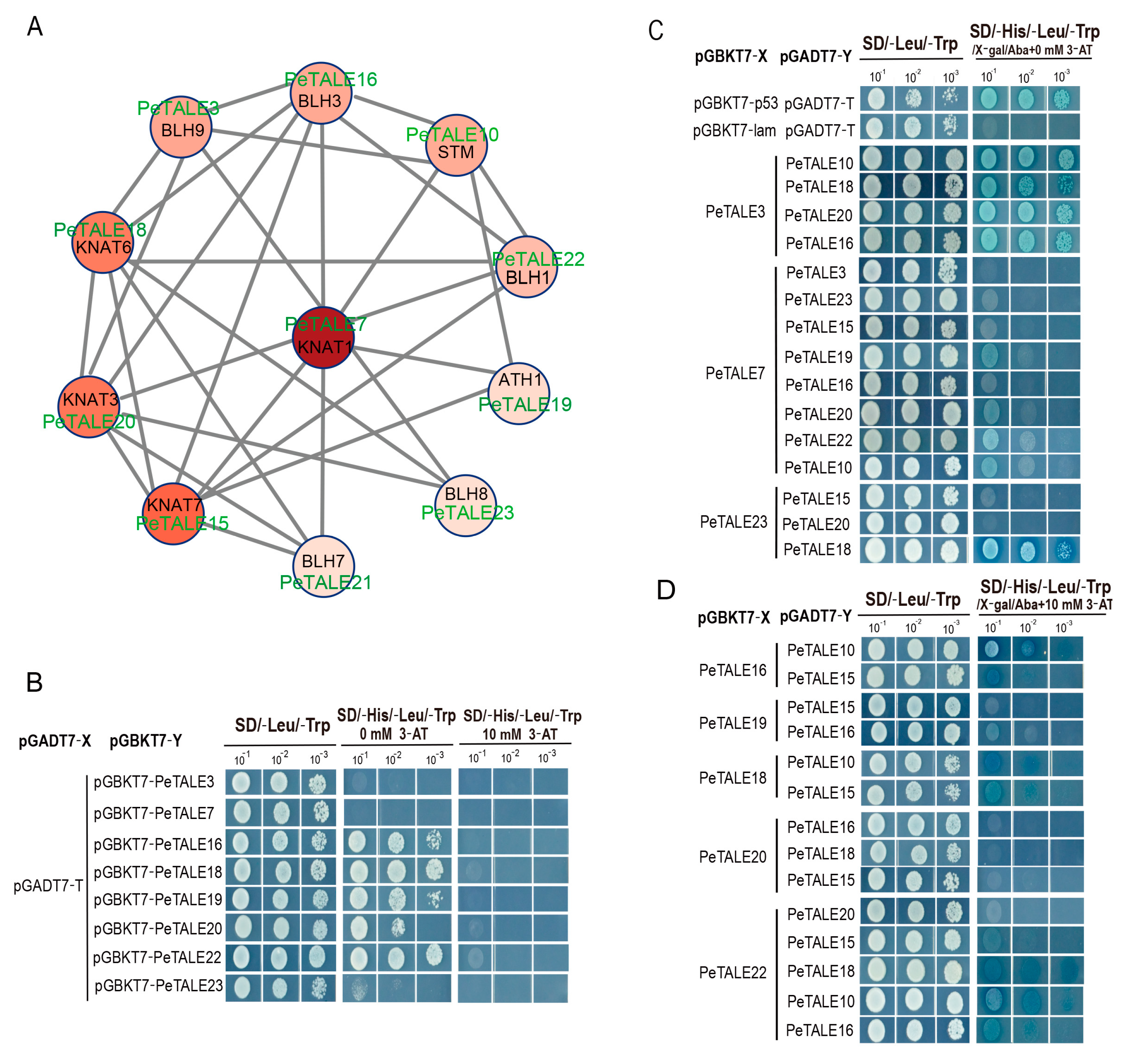
2.7. Protein Interaction of PeTALE Proteins
3. Discussion
3.1. Evolutionary Conservation and Diversification of PeTALE Genes
3.2. PeTALEs in Floral Organogenesis: From Meristem Dynamics to Organ-Specific Regulation
3.3. Temperature-Responsive PeTALEs: Balancing Development and Stress Adaptation
4. Materials and Methods
4.1. Identification of Putative TALE Gene Members in P. edulis
4.2. Sequence Alignment and Phylogenetic Analyses
4.3. Chromosomal Localization and Duplication Analysis of TALE Genes
4.4. Conserved Motif Prediction and Cis-Acting Element Analysis
4.5. Tertiary Structure Analysis of TALE Family Member
4.6. Plant Material and Sample Preparation
4.7. RNA Isolation, qRT-PCR, and Expression Pattern Analysis
4.8. Protein–Protein Interaction (PPI) Network Construction of PeTALEs
4.9. Yeast Two-Hybrid Assay
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sablowski, R. Plant stem cell niches: From signalling to execution. Curr. Opin. Plant Biol. 2011, 14, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, N.; Pautot, V. Ring the BELL and tie the KNOX: Roles for TALEs in gynoeciurn development. Front Plant Sci. 2014, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Hake, S.; Vollbrecht, E.; Freeling, M. Cloning Knotted, the dominant morphological mutant in maize using Ds2 as a transposon tag. EMBO J. 1989, 8, 15–22. [Google Scholar] [CrossRef]
- Aichinger, E.; Kornet, N.; Friedrich, T.; Laux, T. Plant stem cell niches. Annu. Rev. Plant Biol. 2012, 63, 615–636. [Google Scholar] [CrossRef]
- Mukherjee, K.; Brocchieri, L.; Bürglin, T.R. A Comprehensive Classification and Evolutionary Analysis of Plant Homeobox Genes. Mol. Biol. Evol. 2009, 26, 2775–2794. [Google Scholar] [CrossRef]
- Hamant, O.; Pautot, V. Plant development: A TALE story. Curr. Biol. 2010, 333, 371–381. [Google Scholar] [CrossRef]
- Hay, A.; Tsiantis, M. KNOX genes: Versatile regulators of plant development and diversity. Development 2010, 137, 3153–3165. [Google Scholar] [CrossRef]
- Bolduc, N.; Hake, S. The maize transcription factor KNOTTED1 directly regulates the gibberellin catabolism gene ga2ox1. Plant Cell 2009, 21, 1647–1658. [Google Scholar] [CrossRef]
- Chen, H.; Banerjee, A.K.; Hannapel, D.J. The tandem complex of BEL and KNOX partners is required for transcriptional repression of ga20ox1. Plant J. 2004, 38, 276–284. [Google Scholar] [CrossRef]
- Jasinski, S.; Piazza, P.; Craft, J.; Hay, A.; Woolley, L.; Rieu, I.; Phillips, A.; Hedden, P.; Tsiantis, M. KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr. Biol. 2005, 15, 1560–1565. [Google Scholar] [CrossRef]
- Phelps-Durr, T.L.; Thomas, J.; Vahab, P.; Timmermans, M.C. Maize rough sheath2 and its Arabidopsis orthologue ASYM MERIC LEAVES1 interact with HIRA, a predicted histone chaperone, to maintain KNOX gene silencing and determinacy during organogenesis. Plant Cell 2005, 17, 2886–2898. [Google Scholar] [CrossRef] [PubMed]
- Hake, S.; Smith, H.M.S.; Holtan, H.; Magnani, E.; Mele, G.; Ramirez, J. The role of KNOX genes in plant development. Annu Rev. Cell Dev. Biol. 2004, 20, 125–151. [Google Scholar] [CrossRef]
- Smith, H.M.S.; Hake, S. The interaction of two homeobox genes, BREVIPEDICELLUS and PENNYWISE, regulates internode patterning in the Arabidopsis inflorescence. Plant Cell 2003, 15, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Sharma, P.; Gonzalez, D.H.; Viola, I.L.; Hannapel, D.J. The Impact of the Long-Distance Transport of a BEL1-Like Messenger RNA on Development. Plant Physiol. 2013, 161, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Belles-Boix, E.; Hamant, O.; Witiak, S.M.; Morin, H.; Traas, J.; Pautot, V. KNAT6: An homeobox gene involved in meristem activity and organ separation. Plant Cell 2006, 18, 1900–1907. [Google Scholar] [CrossRef]
- Cnops, G.; Neyt, P.; Raes, J.; Petrarulo, M.; Nelissen, H.; Malenica, N.; Luschnig, C.; Tietz, O.; Ditengou, F.; Palme, K.; et al. The TORNADO1 and TORNADO2 genes function in several patterning processes during early leaf development in Arabidopsis thaliana. Plant Cell 2006, 18, 852–866. [Google Scholar] [CrossRef]
- Jia, P.; Sharif, R.; Li, Y.M.; Sun, T.B.; Li, S.K.; Zhang, X.M.; Dong, Q.L.; Luan, H.A.; Guo, S.P.; Ren, X.L.; et al. The BELL1-like homeobox gene MdBLH14 from apple controls flowering and plant height via repression of MdGA20ox3. Int. J. Biol. Macromol. 2023, 242, 124790. [Google Scholar] [CrossRef]
- Kondhare, K.R.; Vetal, P.V.; Kalsi, H.S.; Banerjee, A.K. BEL1-like protein (StBEL5) regulates CYCLING DOF FACTOR (StCDF1) through tandem TGAC core motifs in potato. J. Plant Physiol. 2019, 241, 153014. [Google Scholar] [CrossRef]
- Niu, X.L.; Fu, D.Q. The Roles of BLH Transcription Factors in Plant Development and Environmental Response. Int. J. Mol. Sci. 2022, 23, 3731. [Google Scholar] [CrossRef]
- Zhao, K.; Zhang, X.M.; Cheng, Z.H.; Yao, W.J.; Li, R.H.; Jiang, T.B.; Zhou, B.R. Comprehensive analysis of the three-amino-acid-loop-extension gene family and its tissue-differential expression in response to salt stress in poplar. Plant Physiol. Bioch. 2019, 136, 1–12. [Google Scholar] [CrossRef]
- Tsuda, K.; Hake, S. Diverse functions of KNOX transcription factors in the diploid body plan of plants. Curr. Opin. Plant Biol. 2015, 27, 91–96. [Google Scholar] [CrossRef]
- Kim, D.; Cho, Y.H.; Ryu, H.; Kim, Y.; Kim, T.H.; Hwang, I. BLH1 and KNAT3 modulate ABA responses during germination and early seedling development in Arabidopsis. Plant J. 2013, 75, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Jia, P.; Xing, L.B.; Zhang, C.G.; Zhang, D.; Ma, J.J.; Zhao, C.P.; Han, M.Y.; Ren, X.L.; An, N. MdKNOX19, a class II knotted-like transcription factor of apple, plays roles in ABA signaling/sensitivity by targeting ABI5 during organ development. Plant Sci. 2021, 302, 110701. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.X.; Zhang, L.L.; Yan, L.Y.; Xiong, X.X.; Wang, W.J.; Zhang, X.H.; Min, D.H. Genome-wide analysis of TALE superfamily in Triticum aestivum reveals TaKNOX11a is involved in abiotic stress response. BMC Genom. 2022, 23, 89. [Google Scholar] [CrossRef] [PubMed]
- Blümel, M.; Dally, N.; Jung, C. Flowering time regulation in crops—What did we learn from Arabidopsis? Curr. Opin. Biotech. 2015, 32, 121–129. [Google Scholar] [CrossRef]
- Luo, T.; Zhang, J.; Khan, M.N.; Liu, J.; Xu, Z.; Hu, L. Temperature variation caused by sowing dates significantly affects floral initiation and floral bud differentiation processes in rapeseed (Brassica napus L.). Plant Sci. 2018, 271, 40–51. [Google Scholar] [CrossRef]
- Garrity, P.A.; Goodman, M.B.; Samuel, A.D.; Sengupta, P. Running hot and cold: Behavioral strategies, neural circuits, and the molecular machinery for thermotaxis in C. elegans and Drosophila. Genes Dev. 2010, 24, 2365–2382. [Google Scholar] [CrossRef]
- McClung, C.R.; Davis, S.J. Ambient thermometers in plants: From physiological outputs towards mechanisms of thermal sensing. Curr. Biol. 2010, 20, R1086–R1092. [Google Scholar] [CrossRef]
- Ding, Y.; Shi, Y.; Yang, S. Advances and challenges in uncovering cold tolerance regulatory mechanisms in plants. New Phytol. 2019, 222, 1690–1704. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.M.; Lin, H.X.; Chong, K. Crop Improvement Through Temperature Resilience. Annu. Rev. Plant Biol. 2019, 70, 753–780. [Google Scholar] [CrossRef]
- Ocampo, J.; Lopez, W. Genetic Resources and Breeding Prospects in Passiflora Species. In Passiflora: Genetic, Grafting and Biotechnology Approaches; Nova Science Publishers: Hauppauge, NY, USA, 2021; p. 76. [Google Scholar]
- Cerqueira-Silva, C.B.M.; Faleiro, F.G.; de Jesus, O.N.; dos Santos, E.S.L.; de Souza, A.P. Passion fruit (Passiflora spp.) breeding. Adv. Plant Breed. Strateg. Fruits 2018, 3, 929–951. [Google Scholar]
- Utsunomiya, N. Effect of temperature on shoot growth, flowering and fruit growth of purple passion fruit (Passiflora edulis Sims var. edulis). Sci. Hortic. 1992, 52, 63–68. [Google Scholar] [CrossRef]
- Liu, F.-Y.; Peng, Y.-L.; Chang, Y.-S. Effects of Temperature and Ethylene Response Inhibitors on Growth and Flowering of Passion Fruit. Hortic. Sci. Technol. 2015, 33, 356–363. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Yan, M.; Zhao, H.; Zhang, X.; Yuan, Z. Genome-wide Identification and Expression Analysis of TALE Gene Family in Pomegranate (Punica granatum L.). Agronomy 2020, 10, 829. [Google Scholar] [CrossRef]
- Wang, L.; Yang, X.; Gao, Y.; Yang, S. Genome-Wide Identification and Characterization of TALE Superfamily Genes in Soybean (Glycine max L.). Int. J. Mol. Sci. 2021, 22, 4117. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, P.; Cheng, B.; Zhang, Y.; Shen, Y.; Wang, X.; Zhang, Q.; Lou, Q.; Zhang, S.; Wang, B.; et al. Identification of TALE Transcription Factor Family and Expression Patterns Related to Fruit Chloroplast Development in Tomato (Solanum lycopersicum L.). Int. J. Mol. Sci. 2022, 23, 4507. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, K.; Saleh, I.A.; Okla, M.K.; Alaraidh, I.A.; AbdElgawad, H.; Naeem, M.; Ahmad, N.; Fahad, S. TALE gene family: Identification, evolutionary and expression analysis under various exogenous hormones and waterlogging stress in Cucumis sativus L. BMC Plant Biol. 2024, 24, 564. [Google Scholar] [CrossRef]
- Scofield, S.; Murray, J.A. KNOX gene function in plant stem cell niches. Plant Mol. Biol. 2006, 60, 929–946. [Google Scholar] [CrossRef]
- Ray, A.; Robinson-Beers, K.; Ray, S.; Baker, S.C.; Lang, J.D.; Preuss, D.; Milligan, S.B.; Gasser, C.S. Arabidopsis floral homeotic gene BELL (BEL1) controls ovule development through negative regulation of AGAMOUS gene (AG). Proc. Natl. Acad. Sci. USA 1994, 91, 5761–5765. [Google Scholar] [CrossRef]
- Que, F.; Liu, Q.; Zha, R.; Xiong, A.; Wei, Q. Genome-Wide Identification, Expansion, and Evolution Analysis of Homeobox Gene Family Reveals TALE Genes Important for Secondary Cell Wall Biosynthesis in Moso Bamboo (Phyllostachys edulis). Int. J. Mol. Sci. 2022, 23, 4112. [Google Scholar] [CrossRef]
- Bencivenga, S.; Simonini, S.; Benková, E.; Colombo, L. The transcription factors BEL1 and SPL are required for cytokinin and auxin signaling during ovule development in Arabidopsis. Plant Cell 2012, 24, 2886–2897. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Quan, S.; Zhang, Z.; Kang, C.; Liu, J.; Niu, J. Genome-wide Identification, Characterization and Expression profile of TALE gene family in (Juglans regia L.). Sci. Hortic. 2022, 297, 110945. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, J.; Wu, X.; Hu, G.; Fan, S.; Ma, Q. Evolutionary Relationships and Divergence of KNOTTED1-Like Family Genes Involved in Salt Tolerance and Development in Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2021, 12, 774161. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Han, J.; Fang, X.; Xu, H.; Liang, C.; Li, D.; Yang, Y.; Cui, Z.; Wang, R.; et al. Genome-Wide Analysis of KNOX Transcription Factors and Expression Pattern of Dwarf-Related KNOX Genes in Pear. Front. Plant Sci. 2022, 13, 806765. [Google Scholar] [CrossRef]
- Yang, Q.; Yuan, C.; Cong, T.; Wang, J.; Zhang, Q. Genome-wide identification of three-amino-acid-loop-extension gene family and their expression profile under hormone and abiotic stress treatments during stem development of Prunus mume. Front. Plant Sci. 2022, 13, 1006360. [Google Scholar] [CrossRef]
- An, C.; Liao, J.; Lu, L.; Cai, X.; Liu, R.; Chen, S.; Shen, M.; Wang, X.; Qin, Y.; Zheng, P. From gene expression to flower patterns: Genome-wide characterization of the MADS-box gene family in passion fruit (Passiflora edulis). Trop. Plants 2024, 3, e004. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Lu, S.N.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Wang, Y.P.; Tang, H.B.; DeBarry, J.D.; Tan, X.; Li, J.P.; Wang, X.Y.; Lee, T.H.; Jin, H.Z.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Janson, G.; Zhang, C.X.; Prado, M.G.; Paiardini, A. PyMod 2.0: Improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics 2017, 33, 444–446. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Chromosome | Size (aa) | MW (kDa) | pI | Instability Index | A.I. | GRAVY | Predicted Location |
|---|---|---|---|---|---|---|---|---|---|
| PeTALE1 | P_edulia010000512.g | LG01 | 393 | 44.913 | 5.45 | 39.81 | 68.78 | −0.524 | Nucleus |
| PeTALE2 | P_edulia010000849.g | LG01 | 632 | 68.668 | 6.49 | 42.76 | 67.45 | −0.609 | Nucleus |
| PeTALE3 | P_edulia010001174.g | LG01 | 307 | 34.041 | 6.84 | 41.00 | 69.90 | −0.579 | Nucleus |
| PeTALE4 | P_edulia010001451.g | LG01 | 610 | 67.089 | 8.70 | 42.84 | 63.33 | −0.649 | Nucleus |
| PeTALE5 | P_edulia010003102.g | LG01 | 315 | 35.834 | 4.70 | 45.00 | 74.73 | −0.723 | Nucleus |
| PeTALE6 | P_edulia010004158.g | LG01 | 414 | 47.280 | 6.04 | 53.44 | 60.12 | −0.858 | Nucleus |
| PeTALE7 | P_edulia010004162.g | LG01 | 378 | 43.216 | 6.21 | 52.59 | 60.95 | −0.865 | Nucleus |
| PeTALE8 | P_edulia010004665.g | LG01 | 463 | 51.731 | 5.85 | 45.85 | 82.59 | −0.419 | Nucleus |
| PeTALE9 | P_edulia010004708.g | LG01 | 463 | 51.796 | 5.90 | 44.65 | 79.85 | −0.455 | Nucleus |
| PeTALE10 | P_edulia010004901.g | LG01 | 347 | 38.850 | 6.02 | 44.83 | 66.40 | −0.505 | Nucleus |
| PeTALE11 | P_edulia020006626.g | LG02 | 195 | 22.506 | 6.66 | 56.48 | 64.00 | −0.923 | Nucleus |
| PeTALE12 | P_edulia020006883.g | LG02 | 731 | 80.057 | 7.02 | 48.53 | 71.70 | −0.616 | Nucleus |
| PeTALE13 | P_edulia020007397.g | LG02 | 660 | 73.090 | 5.50 | 51.09 | 65.94 | −0.622 | Nucleus |
| PeTALE14 | P_edulia020007434.g | LG02 | 672 | 74.178 | 5.59 | 52.11 | 65.06 | −0.618 | Nucleus |
| PeTALE15 | P_edulia050012005.g | LG05 | 396 | 44.497 | 7.35 | 55.42 | 83.99 | −0.484 | Nucleus |
| PeTALE16 | P_edulia060015581.g | LG06 | 747 | 82.694 | 6.03 | 46.17 | 70.12 | −0.622 | Nucleus |
| PeTALE17 | P_edulia060015582.g | LG06 | 466 | 52.262 | 6.28 | 53.85 | 81.65 | −0.449 | Nucleus |
| PeTALE18 | P_edulia070016949.g | LG07 | 304 | 34.545 | 4.95 | 45.36 | 60.33 | −0.718 | Nucleus |
| PeTALE19 | P_edulia070018156.g | LG07 | 599 | 65.746 | 5.76 | 45.63 | 78.85 | −0.385 | Nucleus |
| PeTALE20 | P_edulia070018309.g | LG07 | 500 | 55.357 | 6.18 | 43.87 | 73.40 | −0.549 | Nucleus |
| PeTALE21 | P_edulia080019381.g | LG08 | 811 | 89.581 | 6.16 | 47.89 | 67.36 | −0.618 | Nucleus |
| PeTALE22 | P_edulia080019551.g | LG08 | 689 | 75.049 | 8.75 | 47.31 | 69.38 | −0.603 | Nucleus |
| PeTALE23 | P_edulia080019849.g | LG08 | 675 | 74.569 | 5.68 | 47.60 | 68.24 | −0.631 | Nucleus |
| Duplicated Gene Pairs | Ka | Ks | Ka/Ks | Group | Duplicated Type |
|---|---|---|---|---|---|
| PeTALE1/PeTALE5 | 0.31 | 2.29 | 0.14 | KNOX-III/KNOX-III | Segmental |
| PeTALE2/PeTALE4 | 0.13 | 0.65 | 0.21 | BELL-IV/BELL-IV | Segmental |
| PeTALE8/PeTALE9 | 0.01 | 0.02 | 0.48 | BELL-V/BELL-V | tandem |
| PeTALE8/PeTALE17 | 0.13 | 0.53 | 0.25 | BELL-V/BELL-V | Segmental |
| PeTALE13/PeTALE14 | 0.01 | 0.02 | 0.39 | BELL-V/BELL-V | tandem |
| PeTALE13/PeTALE16 | 0.51 | 2.81 | 0.18 | BELL-V/BELL-V | Segmental |
| PeTALE13/PeTALE23 | 0.12 | 0.54 | 0.23 | BELL-V/BELL-V | Segmental |
| PeTALE14/PeTALE23 | 0.12 | 0.51 | 0.24 | BELL-V/BELL-V | Segmental |
| PeTALE12/PeTALE22 | 0.13 | 0.6 | 0.21 | BELL-IV/BELL-IV | Segmental |
| PeTALE16/PeTALE23 | 0.48 | 1.92 | 0.25 | BELL-V/BELL-V | Segmental |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, X.; Miao, J.; Zu, W.; Zhou, R.; Zheng, L.; Wei, Y.; Lai, C.; Qin, R.; Zheng, P.; Wei, X.; et al. Functional Characterization of KNOX and BELL Genes in Temperature-Responsive Floral Morphogenesis of Passion Fruit (Passiflora edulis). Plants 2025, 14, 1440. https://doi.org/10.3390/plants14101440
Jiang X, Miao J, Zu W, Zhou R, Zheng L, Wei Y, Lai C, Qin R, Zheng P, Wei X, et al. Functional Characterization of KNOX and BELL Genes in Temperature-Responsive Floral Morphogenesis of Passion Fruit (Passiflora edulis). Plants. 2025; 14(10):1440. https://doi.org/10.3390/plants14101440
Chicago/Turabian StyleJiang, Xinni, Jie Miao, Weifan Zu, Ruohan Zhou, Lexin Zheng, Ying Wei, Chunmei Lai, Rongjuan Qin, Ping Zheng, Xiuqing Wei, and et al. 2025. "Functional Characterization of KNOX and BELL Genes in Temperature-Responsive Floral Morphogenesis of Passion Fruit (Passiflora edulis)" Plants 14, no. 10: 1440. https://doi.org/10.3390/plants14101440
APA StyleJiang, X., Miao, J., Zu, W., Zhou, R., Zheng, L., Wei, Y., Lai, C., Qin, R., Zheng, P., Wei, X., Xu, J., Qin, Y., & Niu, X. (2025). Functional Characterization of KNOX and BELL Genes in Temperature-Responsive Floral Morphogenesis of Passion Fruit (Passiflora edulis). Plants, 14(10), 1440. https://doi.org/10.3390/plants14101440