
A Distinct Arabidopsis Latent Virus 1 Isolate Was Found in Wild Brassica hirta Plants and Bees, Suggesting the Potential Involvement of Pollinators in Virus Spread
 , , , and
, , , and 
Abstract

1. Introduction
2. Materials and Methods
2.1. Wild Plant Collections
2.2. Viral Particle Purification and Transmission Electron Microscopy Visualization
2.3. Viral RNA Extractions from Purified Virions
2.4. Double-Stranded (ds) cDNA Synthesis
2.5. Viral RNA Extractions from Plants
2.6. RT-PCR Amplifications
2.7. Virus Host Range Analyses
2.8. Viral Genome Assembly
2.9. Characterization of the Viral Coat Protein
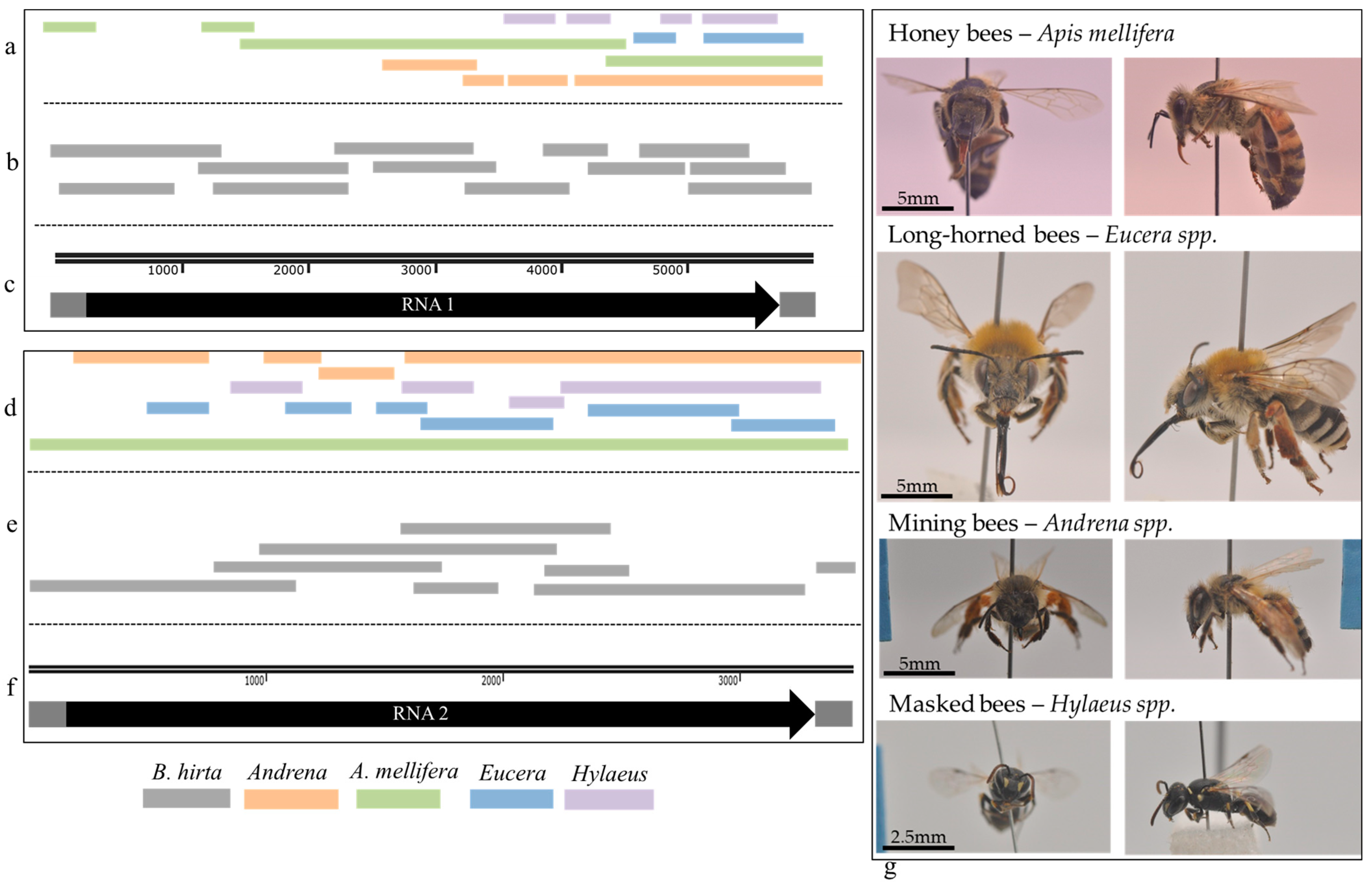
2.10. Bee Sampling
2.11. Local Flora Characterization
2.12. Viral RNA Extractions from Bees
2.13. High Throughput Sequencing (HTS) Analyses
2.14. Statistical Analysis
2.15. A Phylogenetic Tree Analysis
3. Results
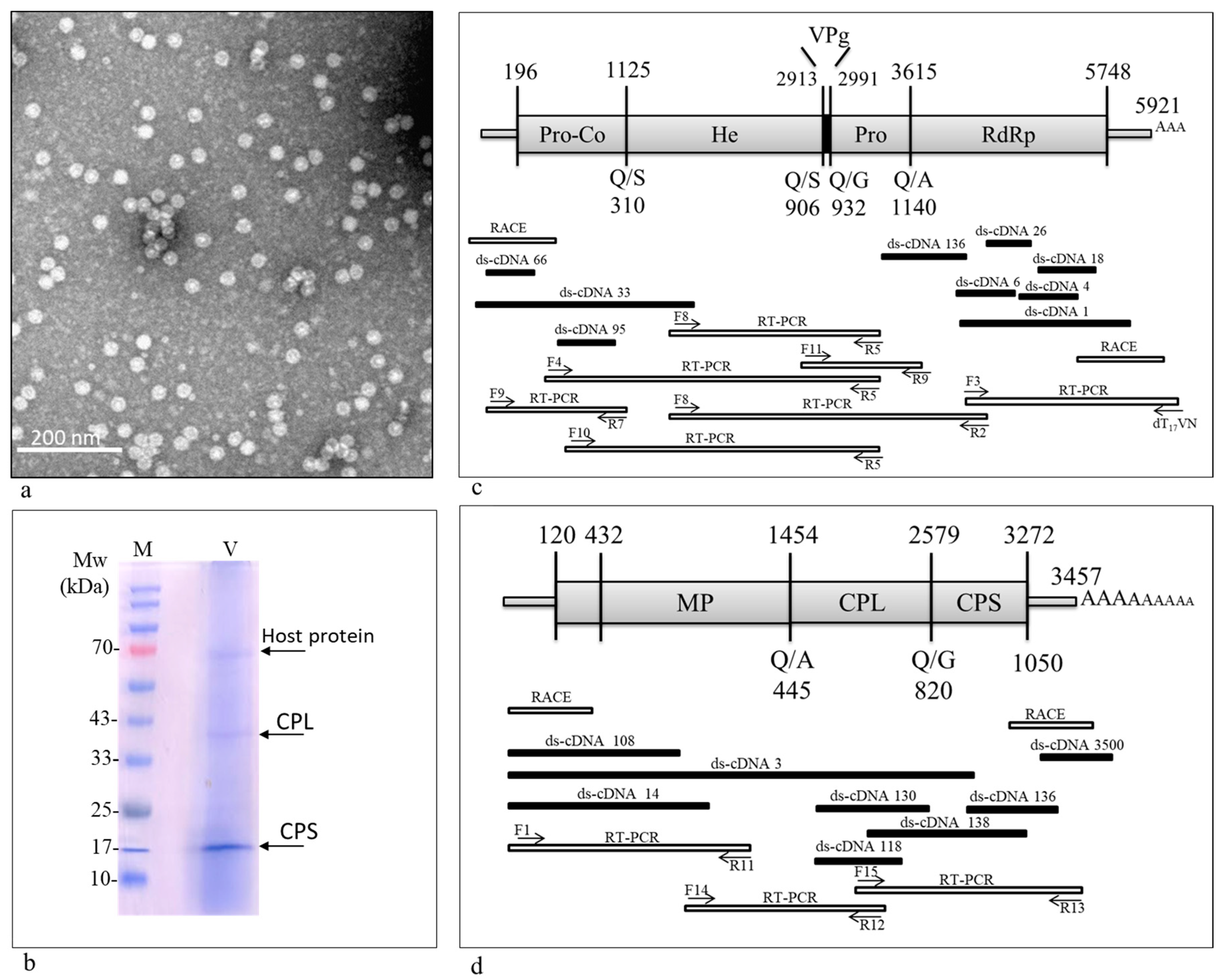
3.1. A Comovirus Was Identified in Asymptomatic Wild B. hirta Plants
3.2. Genome Organization
3.3. 3C-like Proteinase Cleavage Sites and Conserved Motifs in ArLV1-IL-Bh
3.4. A Host Range Analysis
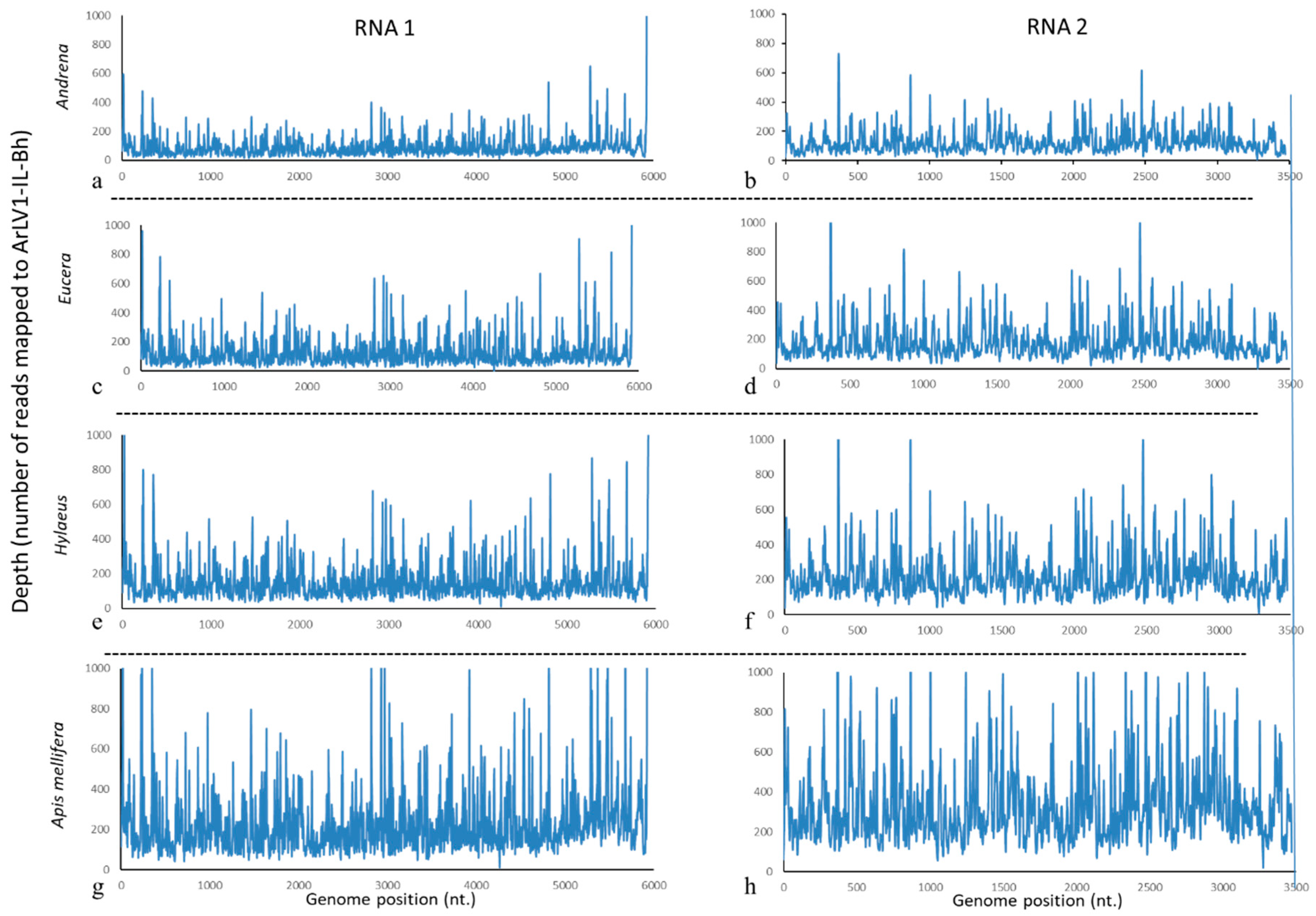
3.5. ArLV1-IL-Bh Identified in Managed and Wild Bee Populations
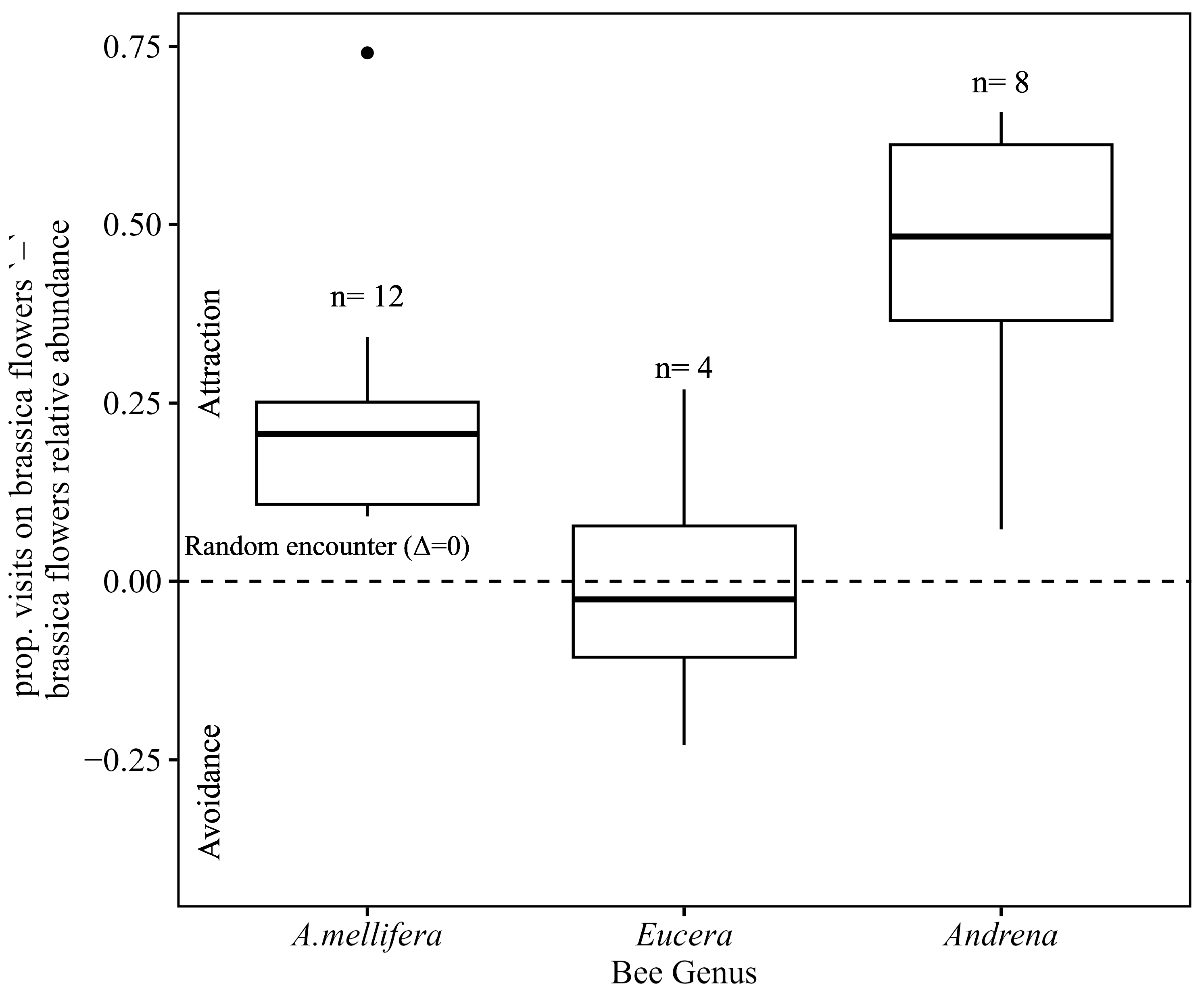
3.6. Bee Tendency to Visit Brassica Flowers
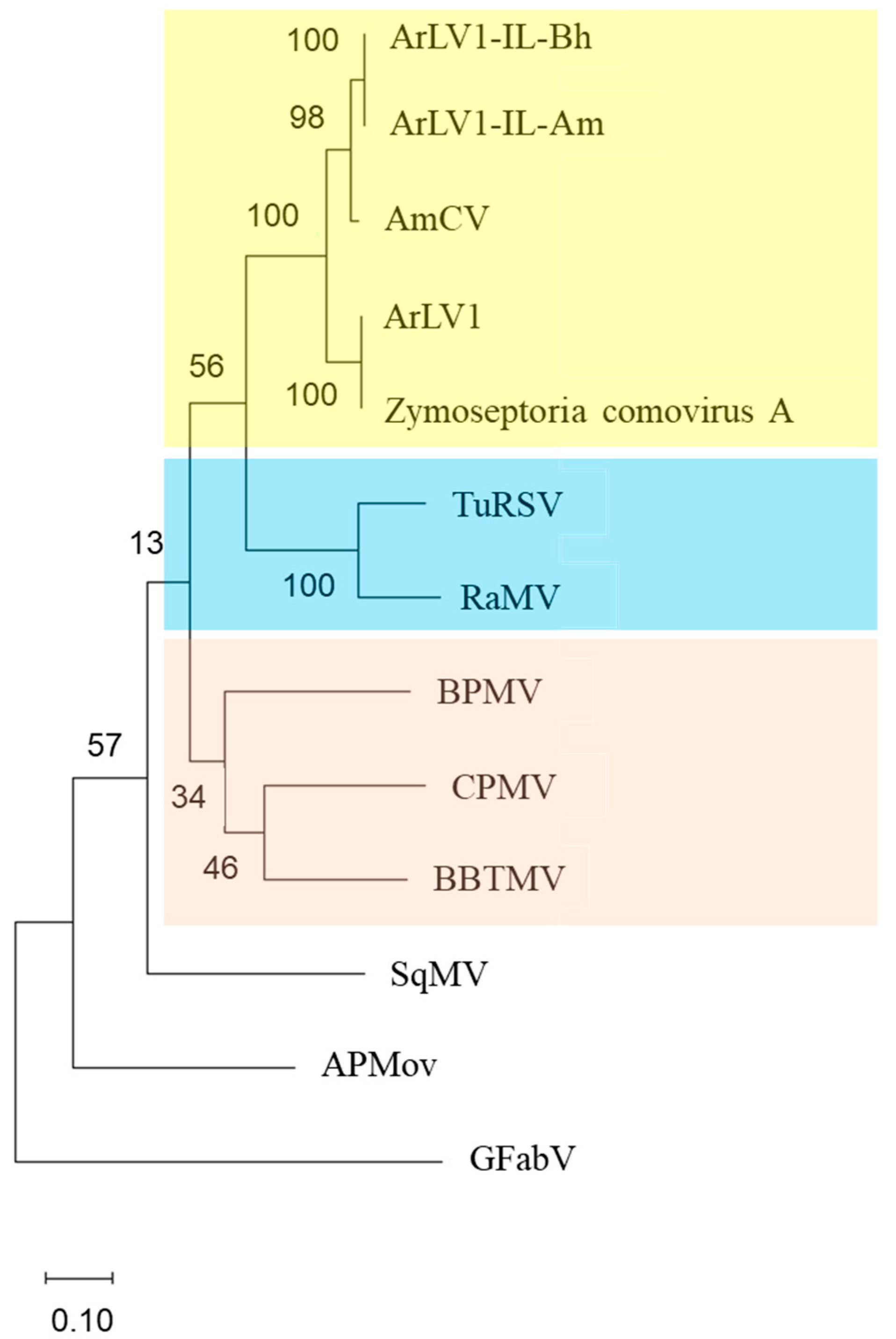
3.7. A Phylogenetic Tree Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Kammen, A. Plant viruses with a divided genome. Annu. Rev. Phytopathol. 1972, 10, 125–150. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.R.; Dasgupta, I.; Fuchs, M.; Iwanami, T.; Karasev, A.V.; Petrzik, K.; Sanfaçon, H.; Tzanetakis, I.; van der Vlugt, R.; Wetzel, T.; et al. ICTV virus taxonomy profile: Secoviridae. J. Gen. Virol. 2017, 98, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Lomonossoff, G.; Shanks, M. The nucleotide sequence of cowpea mosaic virus B RNA. EMBO J. 1983, 2, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- van Wezenbeek, P.; Verver, J.; Harmsen, J.; Vos, P.; van Kammen, A. Primary structure and gene organization of the middle-component RNA of cowpea mosaic virus. EMBO J. 1983, 2, 941–946. [Google Scholar] [CrossRef]
- Peters, S.; Voorhorst, W.; Wery, J.; Wellink, J.; van Kammen, A. A regulatory role for the 32K protein in proteolytic processing of cowpea mosaic virus polyproteins. Virology 1992, 191, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Wellink, J.; Van Kammen, A. Cell-to-cell transport of cowpea mosaic virus requires both the 58K/48K proteins and the capsid proteins. J. Gen. Virol. 1989, 70, 2279–2286. [Google Scholar] [CrossRef]
- Fulton, J.P.; Gergerich, R.C.; Scott, H.A. Beetle Transmission of Plant Viruses. Annu. Rev. Phytopathol. 1987, 25, 111–123. [Google Scholar] [CrossRef]
- Verhoeven, A.; Kloth, K.J.; Kupczok, A.; Oymans, G.H.; Damen, J.; Rijnsburger, K.; Jiang, Z.; Deelen, C.; Sasidharan, R.; van Zanten, M.; et al. Arabidopsis latent virus 1, a comovirus widely spread in Arabidopsis thaliana collections. New Phytol. 2023, 237, 1146–1153. [Google Scholar] [CrossRef]
- Bristow, P.R.; Martin, R.R. Transmission and the role of honeybees in field spread of blueberry shock ilarvirus, a pollen-borne virus of highbush blueberry. Phytopathology 1999, 89, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Kusakari, S.-I.; Kawaratani, M.; Negoro, J.-I.; Ohki, S.T.; Osaki, T. Tobacco mosaic virus is transmissible from tomato to tomato by pollinating bumblebees. J. Gen. Plant Pathol. 2000, 66, 71–74. [Google Scholar] [CrossRef]
- Shipp, J.; Buitenhuis, R.; Stobbs, L.; Wang, K.; Kim, W.; Ferguson, G. Vectoring of Pepino mosaic virus by bumble-bees in tomato greenhouses. Ann. Appl. Biol. 2008, 153, 149–155. [Google Scholar] [CrossRef]
- Darzi, E.; Smith, E.; Shargil, D.; Lachman, O.; Ganot, L.; Dombrovsky, A. The honeybee Apis mellifera contributes to Cucumber green mottle mosaic virus spread via pollination. Plant Pathol. 2018, 67, 244–251. [Google Scholar] [CrossRef]
- Levitzky, N.; Smith, E.; Lachman, O.; Luria, N.; Mizrahi, Y.; Bakelman, H.; Sela, N.; Laskar, O.; Milrot, E.; Dombrovsky, A. The bumblebee Bombus terrestris carries a primary inoculum of Tomato brown rugose fruit virus contributing to disease spread in tomatoes. PLoS ONE 2019, 14, e0210871. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, M.M.; Tran, L.; McKee, C.G.; Polo, R.O.; Newman, T.; Lansing, L.; Griffiths, J.S.; Bilodeau, G.J.; Rott, M.; Guarna, M.M. Honey bees as biomonitors of environmental contaminants, pathogens, and climate change. Ecol. Indic. 2022, 134, 108457. [Google Scholar] [CrossRef]
- Fetters, A.M.; Ashman, T. The pollen virome: A review of pollen-associated viruses and consequences for plants and their interactions with pollinators. Am. J. Bot. 2023, 110, e16144. [Google Scholar] [CrossRef]
- Granberg, F.; Vicente-Rubiano, M.; Rubio-Guerri, C.; Karlsson, O.E.; Kukielka, D.; Belák, S.; Sánchez-Vizcaíno, J.M. Metagenomic detection of viral pathogens in Spanish honeybees: Co-infection by aphid lethal paralysis, Israel acute paralysis and Lake Sinai viruses. PLoS ONE 2013, 8, e57459. [Google Scholar] [CrossRef]
- Schoonvaere, K.; De Smet, L.; Smagghe, G.; Vierstraete, A.; Braeckman, B.P.; de Graaf, D.C. Unbiased RNA shotgun metagenomics in social and solitary wild bees detects associations with eukaryote parasites and new viruses. PLoS ONE 2016, 11, e0168456. [Google Scholar] [CrossRef]
- Galbraith, D.A.; Fuller, Z.L.; Ray, A.M.; Brockmann, A.; Frazier, M.; Gikungu, M.W.; Martinez, J.F.I.; Kapheim, K.M.; Kerby, J.T.; Kocher, S.D.; et al. Investigating the viral ecology of global bee communities with high-throughput metagenomics. Sci. Rep. 2018, 8, 8879. [Google Scholar] [CrossRef]
- Roberts, J.M.K.; Ireland, K.B.; Tay, W.T.; Paini, D. Honey bee-assisted surveillance for early plant virus detection. Ann. Appl. Biol. 2018, 173, 285–293. [Google Scholar] [CrossRef]
- Kwon, M.; Jung, C.; Kil, E.-J. Metagenomic analysis of viromes in honey bee colonies (Apis mellifera; Hymenoptera: Apidae) after mass disappearance in Korea. Front. Cell. Infect. Microbiol. 2023, 13, 1124596. [Google Scholar] [CrossRef]
- Lee, E.; Vansia, R.; Phelan, J.; Lofano, A.; Smith, A.; Wang, A.; Bilodeau, G.J.; Pernal, S.F.; Guarna, M.M.; Rott, M.; et al. Area Wide Monitoring of Plant and Honey Bee (Apis mellifera) Viruses in Blueberry (Vaccinium corymbosum) Agroecosystems Facilitated by Honey Bee Pollination. Viruses 2023, 15, 1209. [Google Scholar] [CrossRef]
- Levitt, A.L.; Singh, R.; Cox-Foster, D.L.; Rajotte, E.; Hoover, K.; Ostiguy, N.; Holmes, E.C. Cross-species transmission of honey bee viruses in associated arthropods. Virus Res. 2013, 176, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Furst, M.A.; McMahon, D.P.; Osborne, J.L.; Paxton, R.J.; Brown, M.J.F. Disease associations between honeybees and bumblebees as a threat to wild pollinators. Nature 2014, 506, 364–366. [Google Scholar] [CrossRef]
- Michener, C.D. The Bees of the World; JHU Press: Baltimor, MD, USA; London, UK, 2000; Volume 1. [Google Scholar]
- Wood, T.J.; Roberts, S.P. An assessment of historical and contemporary diet breadth in polylectic Andrena bee species. Biol. Conserv. 2017, 215, 72–80. [Google Scholar] [CrossRef]
- Hennessy, G.; Goulson, D.; Ratnieks, F.L.W. Population assessment and foraging ecology of nest aggregations of the rare solitary bee, Eucera longicornis at Gatwick Airport, and implications for their management. J. Insect Conserv. 2020, 24, 947–960. [Google Scholar] [CrossRef]
- Müller, A. The hidden diet—Examination of crop content reveals distinct patterns of pollen host use by Central European bees of the genus Hylaeus (Hymenoptera, Colletidae). Alp. Èntomol. 2023, 7, 21–35. [Google Scholar] [CrossRef]
- Gathmann, A.; Tscharntke, T. Foraging ranges of solitary bees. J. Anim. Ecol. 2002, 71, 757–764. [Google Scholar] [CrossRef]
- Zurbuchen, A.; Landert, L.; Klaiber, J.; Müller, A.; Hein, S.; Dorn, S. Maximum foraging ranges in solitary bees: Only few individuals have the capability to cover long foraging distances. Biol. Conserv. 2010, 143, 669–676. [Google Scholar] [CrossRef]
- Hibi, T.; Furuki, I. Melon Necrotic Spot Virus; Descriptions of Plants Viruses No. 302; Association of Applied Biologists: Wellesbourne, UK, 1985. [Google Scholar]
- Dombrovsky, A.; Pearlsman, M.; Lachman, O.; Antignus, Y. Characterization of a new strain of Eggplant mottled crinkle virus (EMCV) infecting eggplants in Israel. Phytoparasitica 2009, 37, 477–483. [Google Scholar] [CrossRef]
- Rosner, A.; Ginzburg, I.; Bar-Joseph, M. Molecular cloning of complementary DNA sequences of citrus tristeza virus RNA. J. Gen. Virol. 1983, 64, 1757–1763. [Google Scholar] [CrossRef]
- Khandekar, S.; He, J.; Leisner, S. Complete nucleotide sequence of the Toledo isolate of turnip ringspot virus. Arch. Virol. 2009, 154, 1917–1922. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Gao, S.; Padmanabhan, C.; Li, R.; Galvez, M.; Gutierrez, D.; Fuentes, S.; Ling, K.-S.; Kreuze, J.; Fei, Z. VirusDetect: An automated pipeline for efficient virus discovery using deep sequencing of small RNAs. Virology 2017, 500, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R. Using the velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinform. 2010, 31, 11.5.1–11.5.12. [Google Scholar] [CrossRef] [PubMed]
- Houtgast, E.J.; Sima, V.-M.; Bertels, K.; Al-Ars, Z. Hardware acceleration of BWA-MEM genomic short read mapping for longer read lengths. Comput. Biol. Chem. 2018, 75, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Sanfaçon, H.; Wellink, J.; Le Gall, O.; Karasev, A.; van der Vlugt, R.; Wetzel, T. Secoviridae: A proposed family of plant viruses within the order Picornavirales that combines the families Sequiviridae and Comoviridae, the unassigned genera Cheravirus and Sadwavirus, and the proposed genus Torradovirus. Arch. Virol. 2009, 154, 899–907. [Google Scholar] [CrossRef]
- Ritzenthaler, C.; Viry, M.; Pinck, M.; Margis, R.; Fuchs, M.; Pinck, L. Complete nucleotide sequence and genetic organization of grapevine fanleaf nepovirus RNA1. J. Gen. Virol. 1991, 72, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Blinov, V.M.; Donchenko, A.P.; Koonin, E.V. An NTP-binding motif is the most conserved sequence in a highly diverged monophyletic group of proteins involved in positive strand RNA viral replication. J. Mol. Evol. 1989, 28, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Mayoa, M.; Fritsch, C. A possible consensus sequence for VPg of viruses in the family Comoviridae. FEBS Lett. 1994, 354, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.F.; Fletterick, R.J. Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: Structural and functional implications. Proc. Natl. Acad. Sci. USA 1988, 85, 7872–7876. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Donchenko, A.P.; Blinov, V.M.; Koonin, E.V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases: A distinct protein superfamily with a common structural fold. FEBS Lett. 1989, 243, 103–114. [Google Scholar] [CrossRef]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar] [CrossRef]
- Koonin, E.V.; Mushegian, A.R.; Ryabov, E.V.; Dolja, V.V. Diverse groups of plant RNA and DNA viruses share related movement proteins that may possess chaperone-like activity. J. Gen. Virol. 1991, 72, 2895–2903. [Google Scholar] [CrossRef]
- Kobayashi, Y.O.; Kobayashi, A.K.Y.O.; Hagiwara, K.; Uga, H.; Mikoshiba, Y.; Naito, T.; Honda, Y.; Omura, T. Gentian mosaic virus: A new species in the genus Fabavirus. Phytopathology 2005, 95, 192–197. [Google Scholar] [CrossRef][Green Version]
- Sainsbury, F.; Cañizares, M.C.; Lomonossoff, G.P. Cowpea mosaic virus: The plant virus–based biotechnology workhorse. Annu. Rev. Phytopathol. 2010, 48, 437–455. [Google Scholar] [CrossRef]
- Sicard, A.; Yvon, M.; Timchenko, T.; Gronenborn, B.; Michalakis, Y.; Gutierrez, S.; Blanc, S. Gene copy number is differentially regulated in a multipartite virus. Nat. Commun. 2013, 4, 2248. [Google Scholar] [CrossRef]
- Moreau, Y.; Gil, P.; Exbrayat, A.; Rakotoarivony, I.; Bréard, E.; Sailleau, C.; Viarouge, C.; Zientara, S.; Savini, G.; Goffredo, M. The genome segments of bluetongue virus differ in copy number in a host-specific manner. J. Virol. 2020, 95, 1110–1128. [Google Scholar] [CrossRef]
- Roossinck, M.J. A new look at plant viruses and their potential beneficial roles in crops. Mol. Plant Pathol. 2015, 16, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, E.; Lozano-Duran, R. Plant viruses as probes to engineer tolerance to abiotic stress in crops. Stress Biol. 2022, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Sáez, C.; Pagán, I. When plants are Trojan horses for viruses. New Phytol. 2023, 237, 1071–1073. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. and Orientation | RNA Partite | Position (bp) | Sequence (5′–3′) |
|---|---|---|---|
| F1 | II | 1 | TCCGCCAGTACTGGGGAG |
| F3 | I | 4634 | GTGGAATACCTTCTGGATTTCC |
| F4 | I | 668 | AAGCTATCGATTGGACAGTTG |
| F5 | I | 4223 | GTCCAAAGGATGAAAAACTGC |
| F6 | II | 1948 | TATCTATGACTATAGATTGGTTT |
| F7 | II | 461 | AGCTCAAGCACTGCATTTGAA |
| F8 | I | 2104 | GAATTTCATTCGTATGGTGAT |
| F9 | I | 4 | GAACAGGACCAGGGTCCGC |
| F10 | I | 1092 | AGCATTTGGTTGTCCCACTATCATTG |
| F11 | I | 2222 | TCGATAAATTTGAGCATCTACTG |
| F14 | II | 689 | TGGTGAAAATGAAGTGGTTCAC |
| F15 | II | 2120 | AGGAGGCACTGGAGTAG |
| R1 | I | 748 | TAGGGCAATATTTTTCAACCAC |
| R2 | I | 4062 | GGGAAATCCTTCGGATGTG |
| R3 | I | 4598 | AAGTCGGGAACAGCAAGCTA |
| R5 | I | 3231 | ACAGAGCTCACTATTTTCAAAA |
| R7 | I | 1302 | CTGCCAAACAAAATTTTGCAAGC |
| R8 | I | 2329 | GAACAAATTGCGCCTCCTGT |
| R9 | I | 3520 | CAACTTTAGCTACAACCAGAGA |
| R10 | I | 5921 | GAAAATATCATAACGCGACATATAAC |
| R11 | II | 1287 | TAGAACCAATGGCAGGAAGGT |
| R12 | II | 2168 | AGAACTCAAAGCGTTAGGCA |
| R13 | II | 3445 | ATGCGATATGATAAATCAAAATAC |
| Plant | Family | Symptoms | RT-PCR |
|---|---|---|---|
| Brassica perviridis | Brassicaceae | Ringspot | Positive |
| B. rapa | Brassicaceae | No symptoms | Negative |
| Raphanus sativus | Brassicaceae | No symptoms | Positive |
| B. oleracea | Brassicaceae | No symptoms | Positive |
| Nicotiana benthamiana | Solanaceae | Mosaic | Positive |
| N. glutinosa | Solanaceae | No symptoms | Negative |
| Datura stramonium | Solanaceae | No symptoms | Positive |
| Vicia faba | Fabaceae | No symptoms | Positive |
| Vigna unguiculata | Fabaceae | No symptoms | Positive |
| Chenopodium amaranticolor | Amaranthacea | No symptoms | Positive |
| C. murale | Amaranthacea | No symptoms | Positive |
| C. quinoa | Amaranthacea | No symptoms | Positive |
| Gomphrena globosa | Amaranthacea | No symptoms | Positive |
| Library | Total Number of Reads | Total Number of Reads after Cleaning | * Number of Reads Mapped to RNA1 | * Number of Reads Mapped to RNA2 |
|---|---|---|---|---|
| Andrena | 18,749,336 | 18,740,291 (99.95%) | 75,028 (0.40%) | 63,370 (0.34%) |
| Eucera | 27,055,141 | 27,039,024 (99.94%) | 116,760 (0.43%) | 98,010 (0.36%) |
| Apis mellifera | 45,167,229 | 45,140,094 (99.94%) | 199,587 (0.44%) | 168,906 (0.37%) |
| Hylaeus | 33,800,839 | 33,784,842 (99.95%) | 139,155 (0.41%) | 115,759 (0.34%) |
| Honey bee (Apis mellifera) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Contig | RNA | Position (nt) | Contig Size (bp) | * Mismatch | * INDELs | % ArLV1-IL-Bh aa | % ArLV1 aa | % Zymoseptoria Comovirus A aa | % AmCV aa |
| 2688 | 1 | 124–492 | 368 | 7 | 0 | 99 | 82 | 82 | 98 |
| 3388 | 1 | 1327–1647 | 320 | 3 | 0 | 100 | 89 | 89 | 98 |
| 2241 | 1 | 1645–4430 | 2785 | 19 | 0 | 99 | 90 | 90 | 97 |
| 39_1 | 1 | 4362–5907 | 1545 | 26 | 0 | 99 | 83 | 83 | 94 |
| 39_3 | 2 | 1–3378 | 3378 | 56 | 4 | 99.8 | 79.4 | 79.9 | 92.19 |
| Mining bee (Andrena) | |||||||||
| 3396 | 1 | 2715–3376 | 661 | 6 | 0 | 99.5 | 90.4 | 90.4 | 97.2 |
| 2094 | 1 | 3361–3624 | 263 | 2 | 0 | 100 | 89.7 | 89.7 | 98.8 |
| 3946 | 1 | 3642–4061 | 419 | 4 | 0 | 100 | 87.1 | 87.1 | 96.4 |
| 67_2 | 1 | 4164–5892 | 1728 | 30 | 1 | 99.6 | 85.2 | 85.2 | 94.6 |
| 2253 | 2 | 182–744 | 562 | 5 | 0 | 99 | 67 | 67 | 86 |
| 1218 | 2 | 959–1199 | 240 | 4 | 0 | 79 | 76 | 76 | 79 |
| 4073 | 2 | 1184–1501 | 317 | 6 | 0 | 99 | 66 | 68 | 90 |
| 67_1 | 2 | 1563–3443 | 1880 | 25 | 3 | 99 | 82 | 82 | 93 |
| Long-horned bee (Eucera) | |||||||||
| 4418 | 1 | 4559–4820 | 261 | 7 | 0 | 99 | 92 | 92 | 98 |
| 2837 | 1 | 5051–5784 | 733 | 9 | 0 | 99 | 78 | 78 | 92 |
| 3346 | 2 | 498–744 | 246 | 2 | 0 | 99 | 92 | 93 | 95 |
| 2617 | 2 | 1057–1328 | 271 | 4 | 0 | 100 | 90 | 91 | 98 |
| 2924_1 | 2 | 1429–1634 | 205 | 5 | 0 | 97 | 53 | 77 | 90 |
| 2924_0 | 2 | 1612–2155 | 543 | 14 | 0 | 74 | 68 | 69 | 71 |
| 1924 | 2 | 2313–2926 | 613 | 9 | 0 | 97 | 79 | 79 | 95 |
| 4230 | 2 | 2912–3317 | 405 | 6 | 2 | 100 | 66 | 66 | 84 |
| Masked bee (Hylaeus) | |||||||||
| 7428 | 1 | 3531–3924 | 393 | 6 | 0 | 85 | 73 | 73 | 80 |
| 9174 | 1 | 4032–4347 | 315 | 4 | 0 | 99 | 83 | 83 | 94 |
| 12,606 | 1 | 4704–4973 | 269 | 10 | 0 | 99 | 96 | 96 | 96 |
| 5150 | 1 | 5054–5597 | 543 | 7 | 0 | 100 | 90 | 90 | 99 |
| 12,033 | 2 | 822–1117 | 295 | 13 | 0 | 99 | 98 | 98 | 99 |
| 12,764 | 2 | 1548–1818 | 270 | 8 | 0 | 98 | 87 | 88 | 91 |
| 1842 | 2 | 1970–2208 | 238 | 2 | 0 | 100 | 86 | 86 | 94 |
| 4413 | 2 | 2193–3254 | 1061 | 23 | 1 | 98 | 78 | 89 | 93 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reingold, V.; Eliyahu, A.; Luria, N.; Leibman, D.; Sela, N.; Lachman, O.; Smith, E.; Mandelik, Y.; Sadeh, A.; Dombrovsky, A. A Distinct Arabidopsis Latent Virus 1 Isolate Was Found in Wild Brassica hirta Plants and Bees, Suggesting the Potential Involvement of Pollinators in Virus Spread. Plants 2024, 13, 671. https://doi.org/10.3390/plants13050671
Reingold V, Eliyahu A, Luria N, Leibman D, Sela N, Lachman O, Smith E, Mandelik Y, Sadeh A, Dombrovsky A. A Distinct Arabidopsis Latent Virus 1 Isolate Was Found in Wild Brassica hirta Plants and Bees, Suggesting the Potential Involvement of Pollinators in Virus Spread. Plants. 2024; 13(5):671. https://doi.org/10.3390/plants13050671
Chicago/Turabian StyleReingold, Victoria, Avi Eliyahu, Neta Luria, Diana Leibman, Noa Sela, Oded Lachman, Elisheva Smith, Yael Mandelik, Asaf Sadeh, and Aviv Dombrovsky. 2024. "A Distinct Arabidopsis Latent Virus 1 Isolate Was Found in Wild Brassica hirta Plants and Bees, Suggesting the Potential Involvement of Pollinators in Virus Spread" Plants 13, no. 5: 671. https://doi.org/10.3390/plants13050671
APA StyleReingold, V., Eliyahu, A., Luria, N., Leibman, D., Sela, N., Lachman, O., Smith, E., Mandelik, Y., Sadeh, A., & Dombrovsky, A. (2024). A Distinct Arabidopsis Latent Virus 1 Isolate Was Found in Wild Brassica hirta Plants and Bees, Suggesting the Potential Involvement of Pollinators in Virus Spread. Plants, 13(5), 671. https://doi.org/10.3390/plants13050671