
Transcriptome Analysis Revealed the Key Genes and Pathways Involved in Seed Germination of Maize Tolerant to Deep-Sowing

 ,
, 
Abstract
:1. Introduction
2. Results
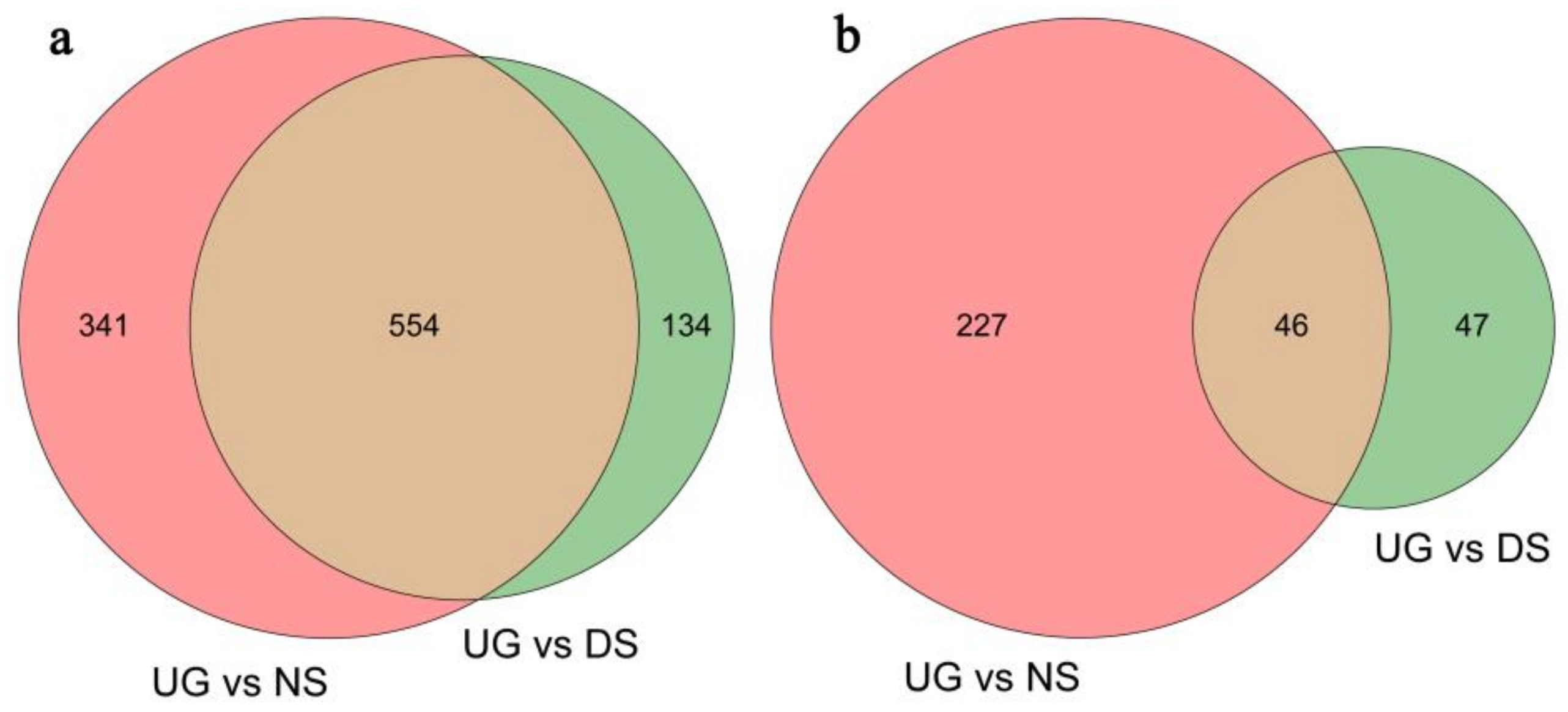
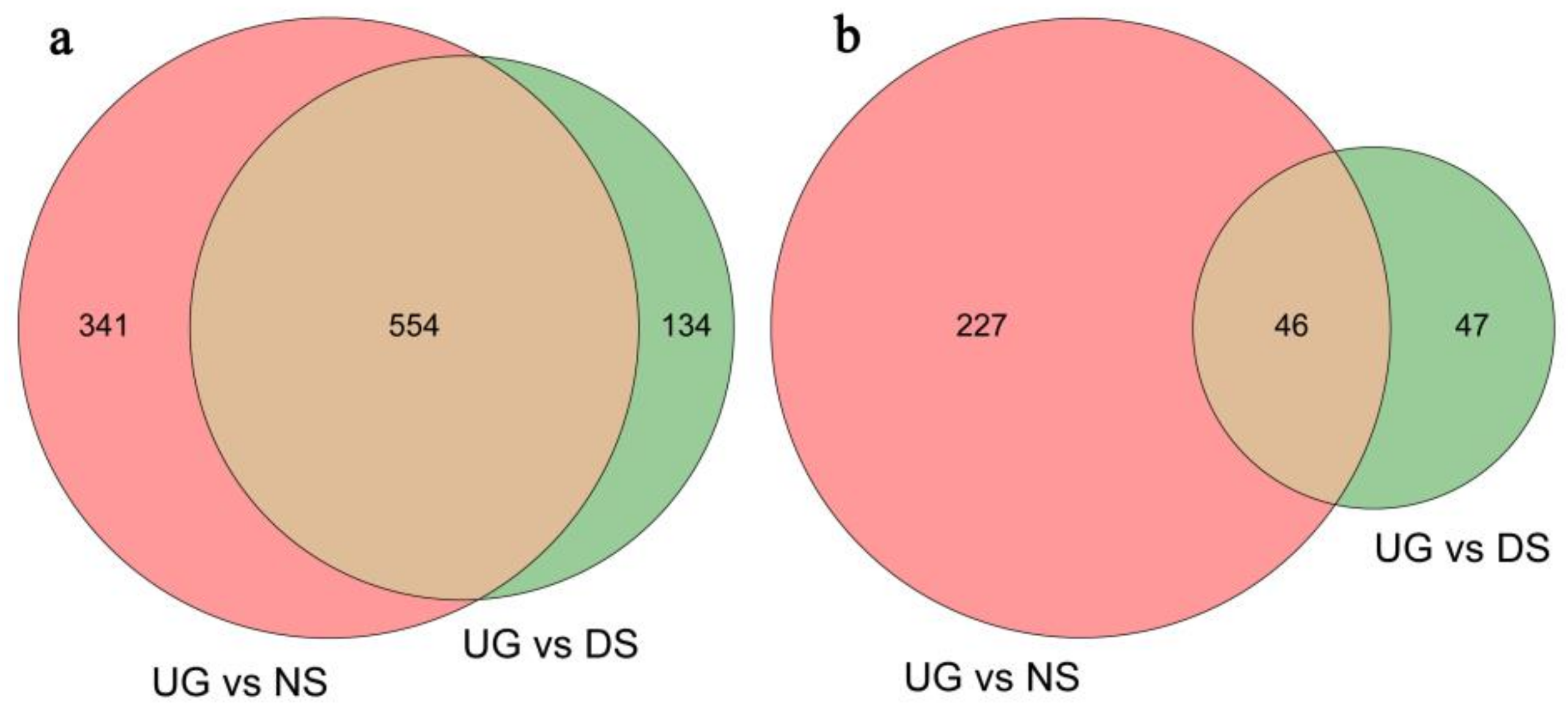
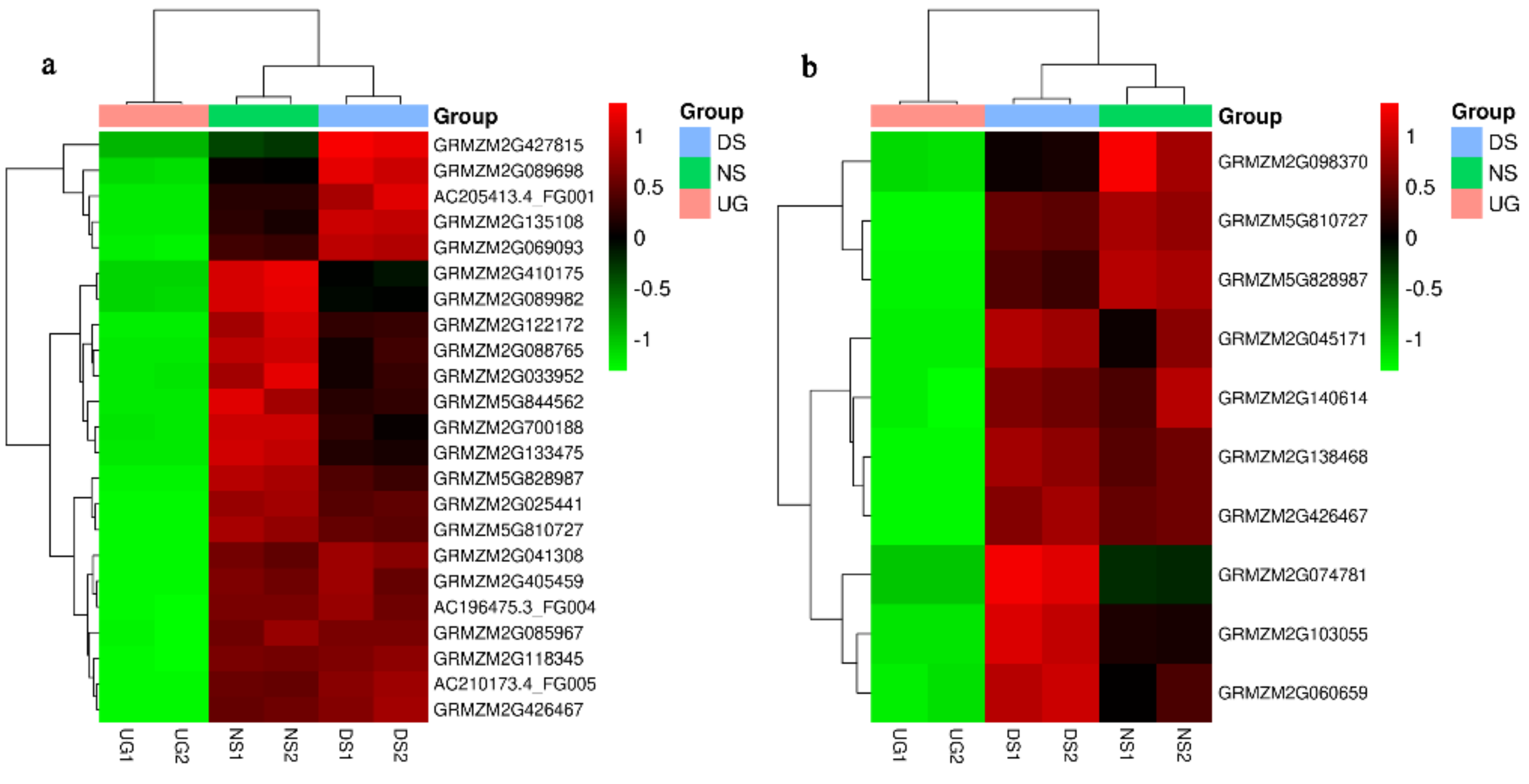
2.1. Deep-Sowing-Induced Transcriptome Changes in Maize
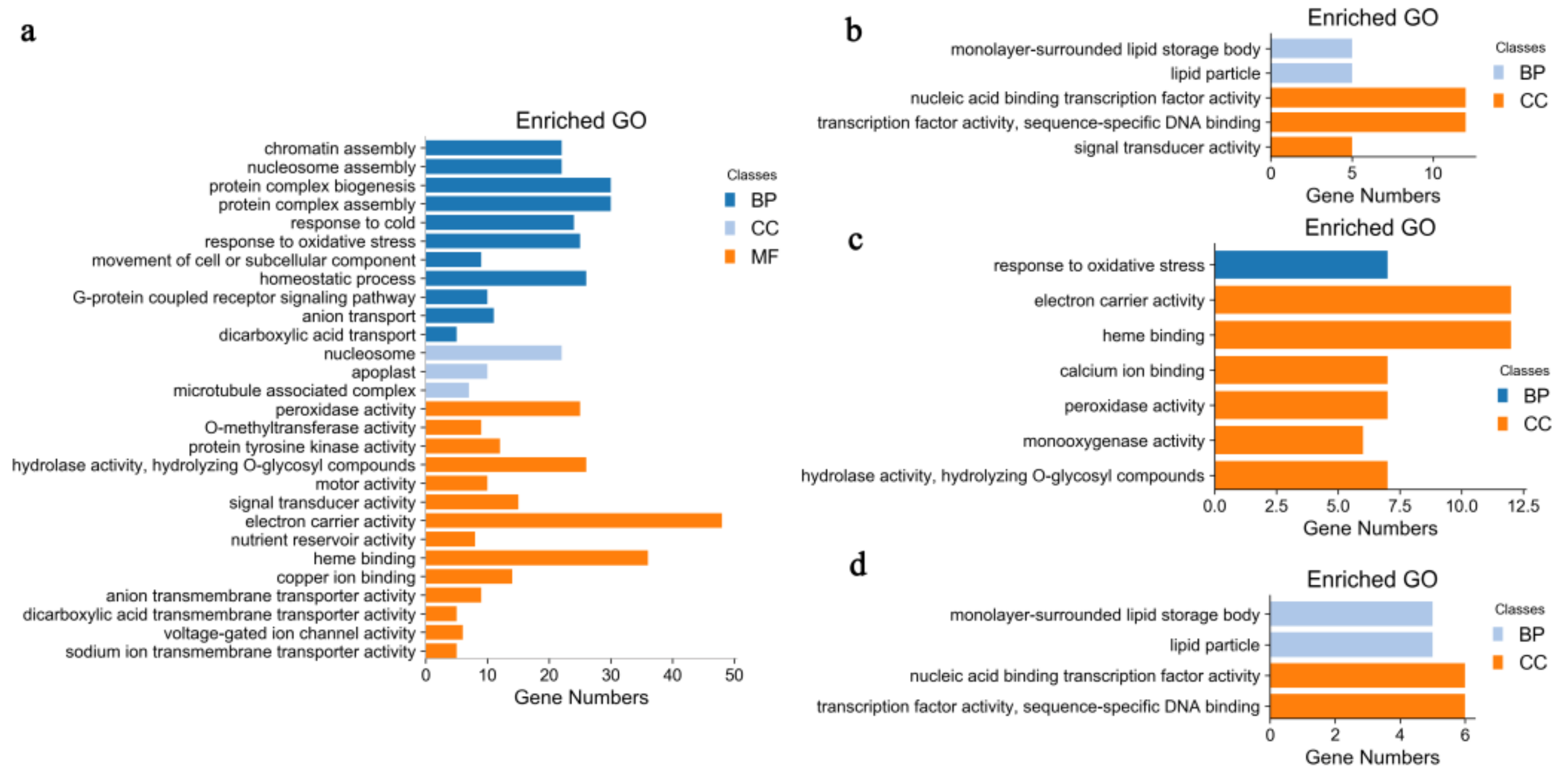
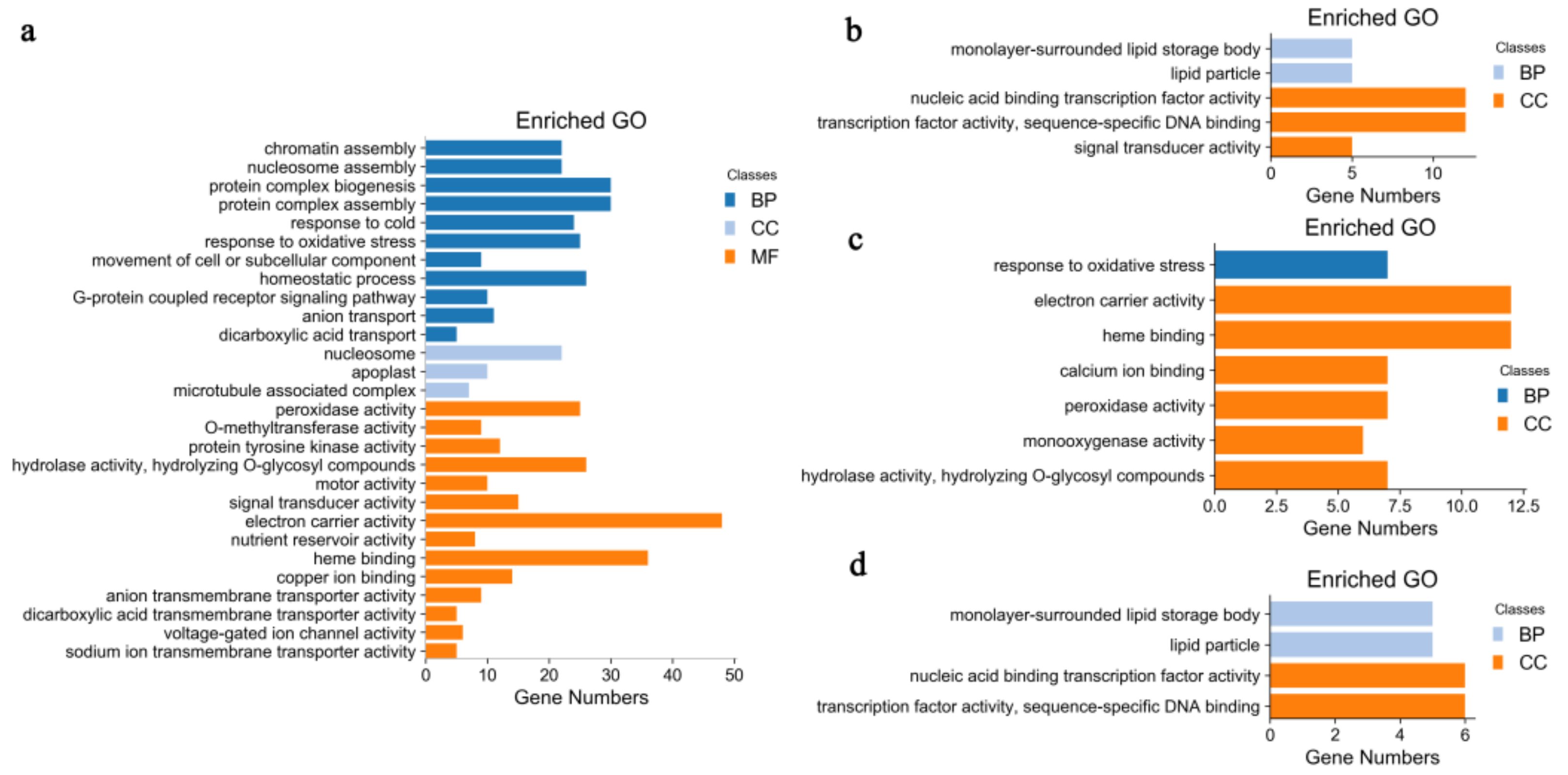
2.2. Identification and GO Functional Enrichment Analysis of DEGs
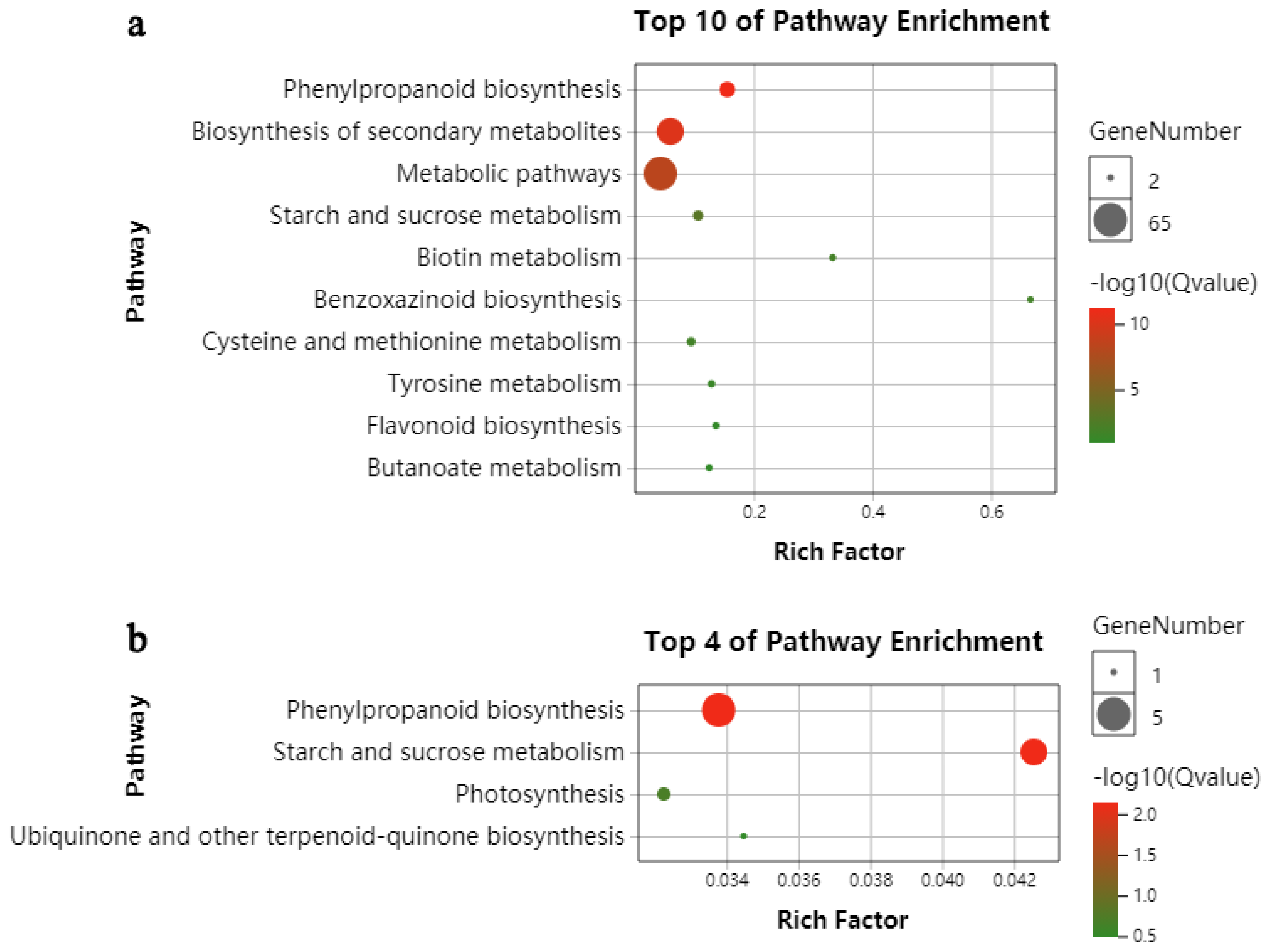
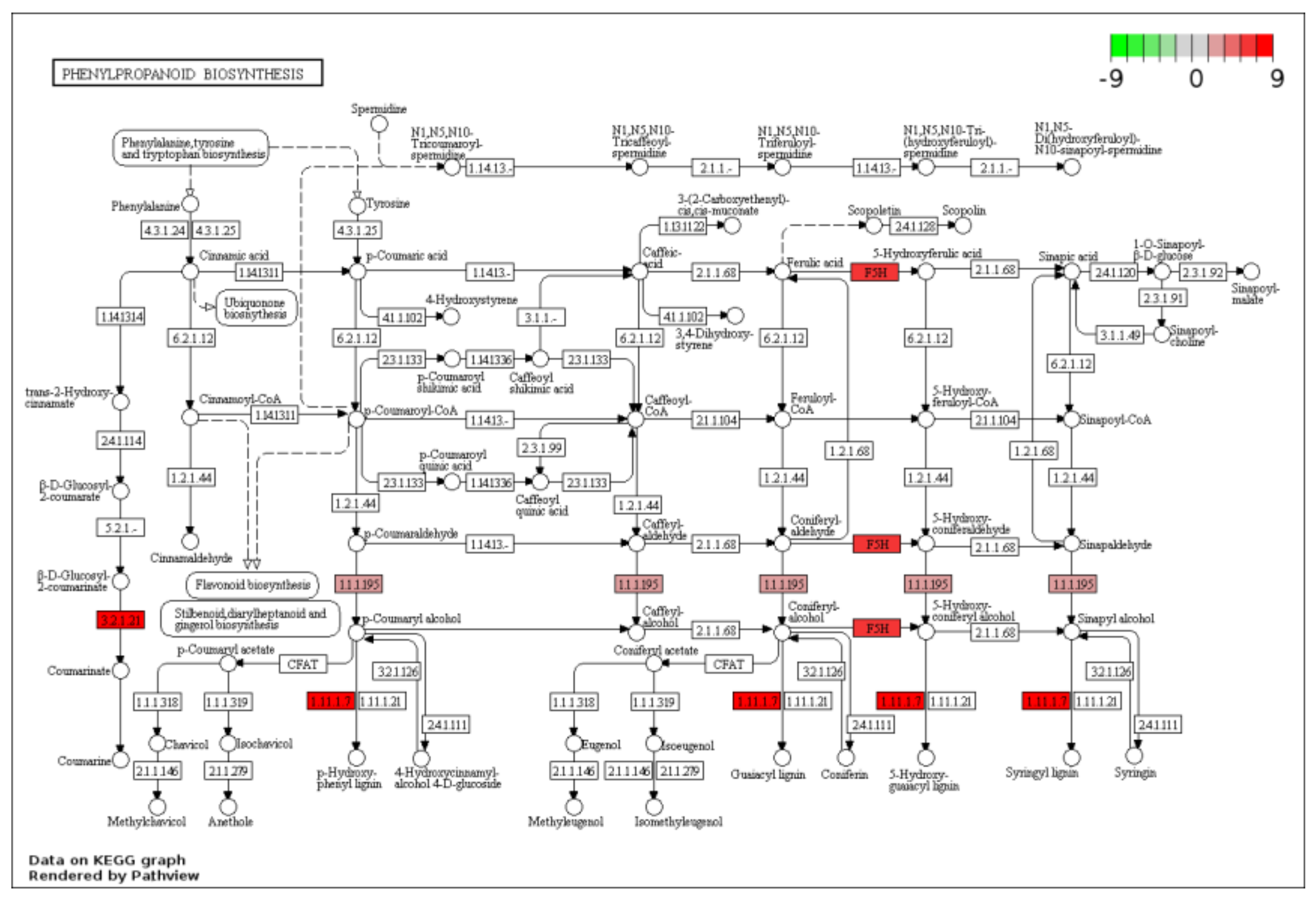
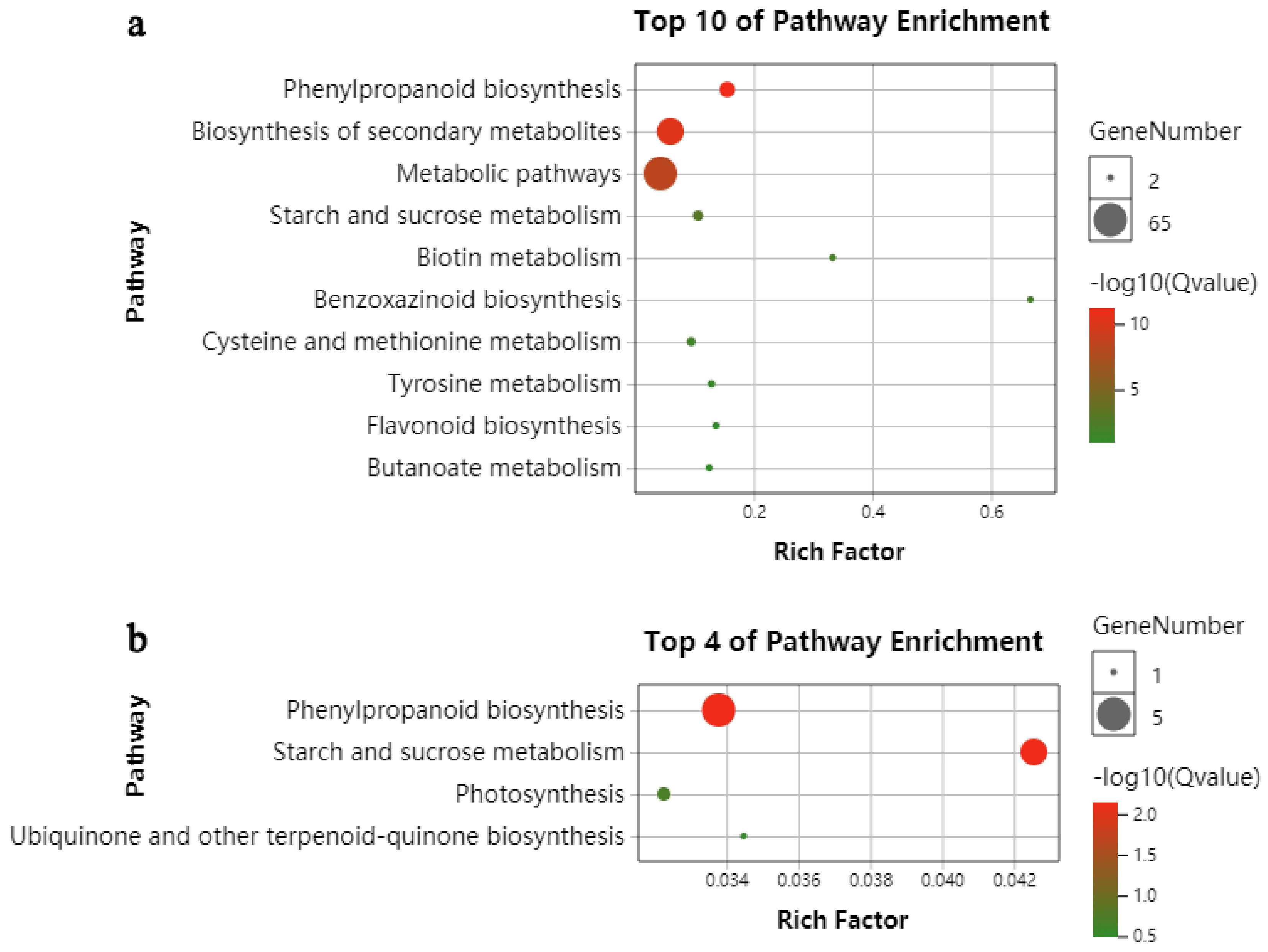
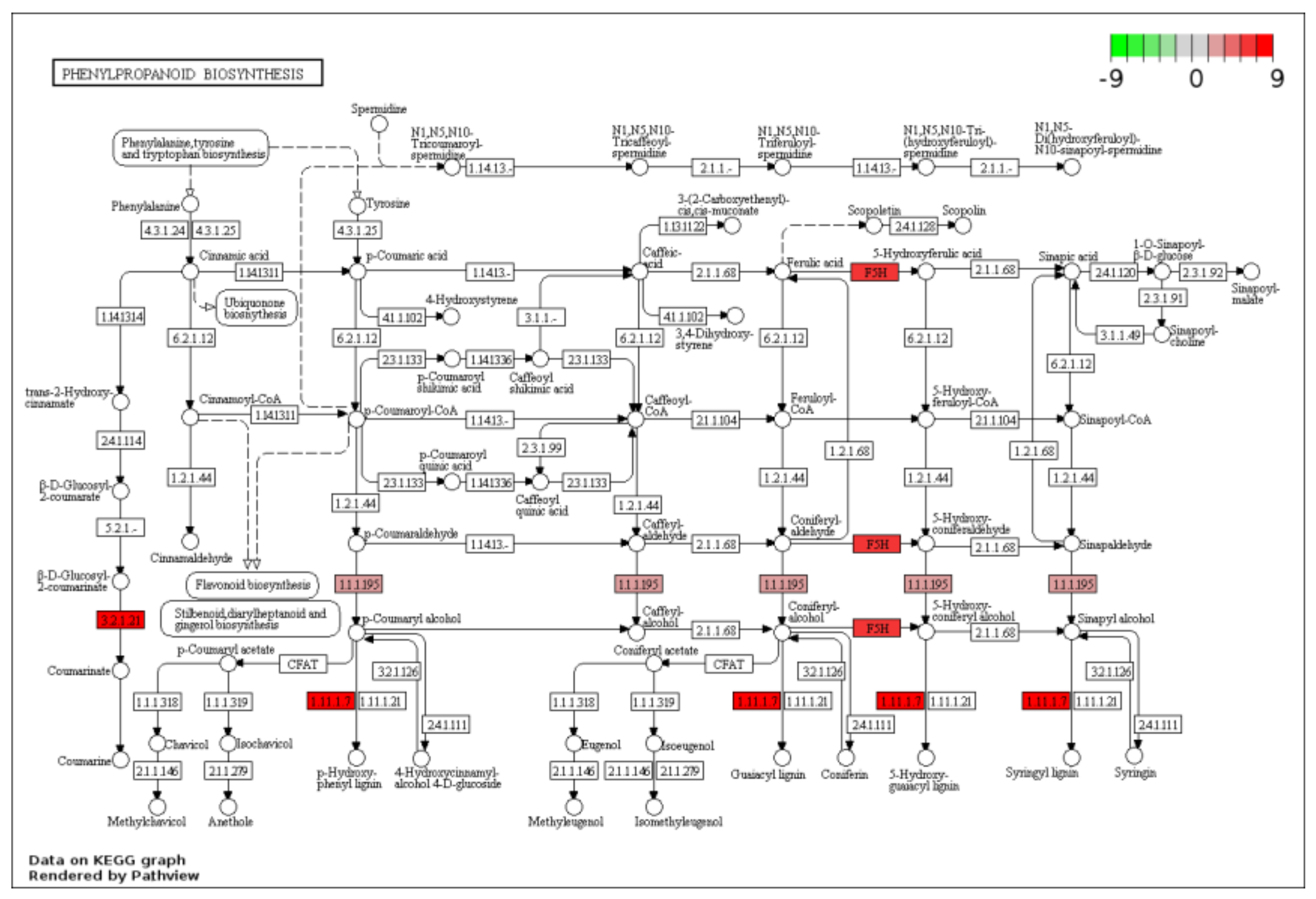
2.3. KEGG Functional-Enrichment Analysis of DEGs
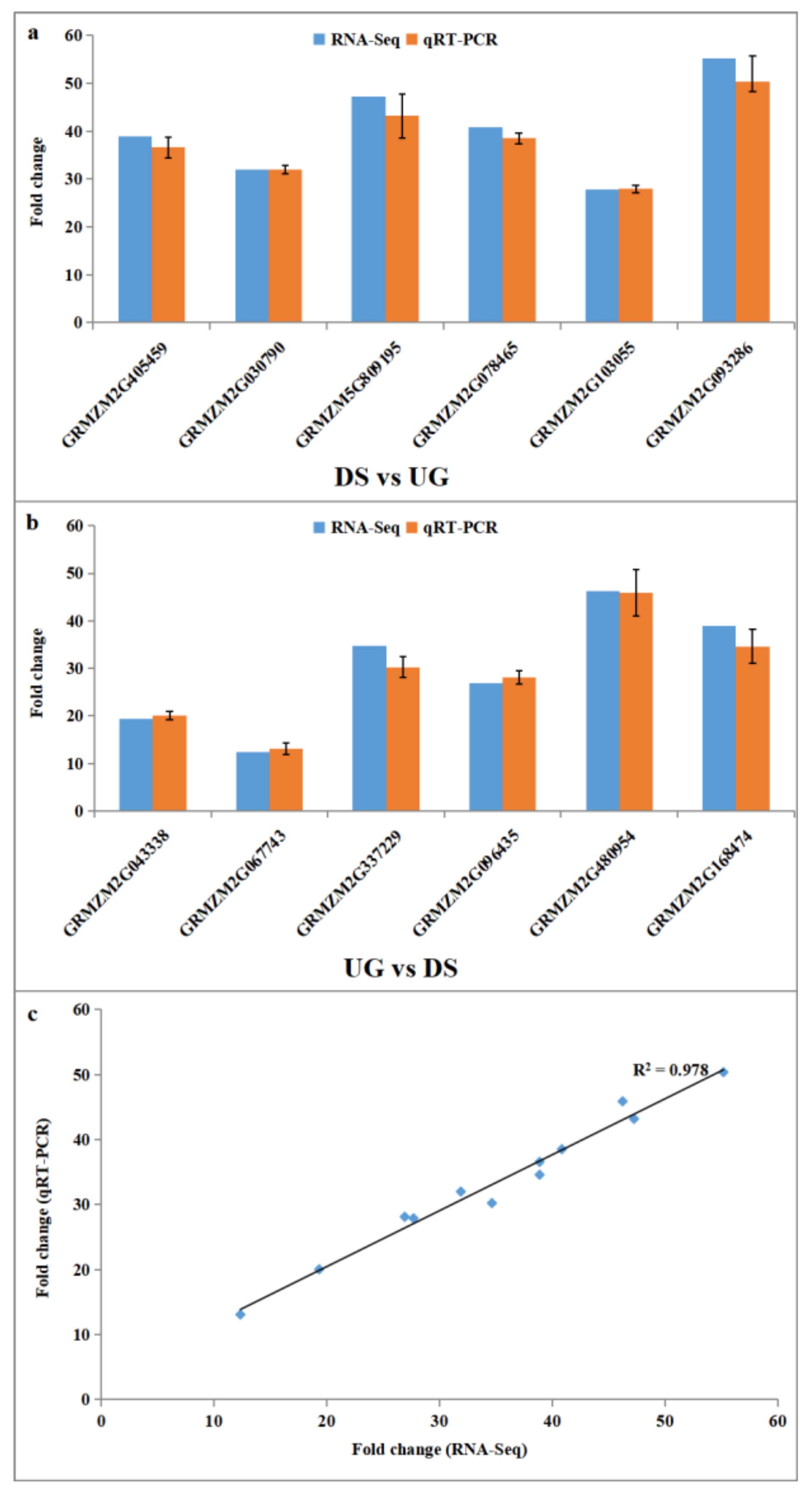
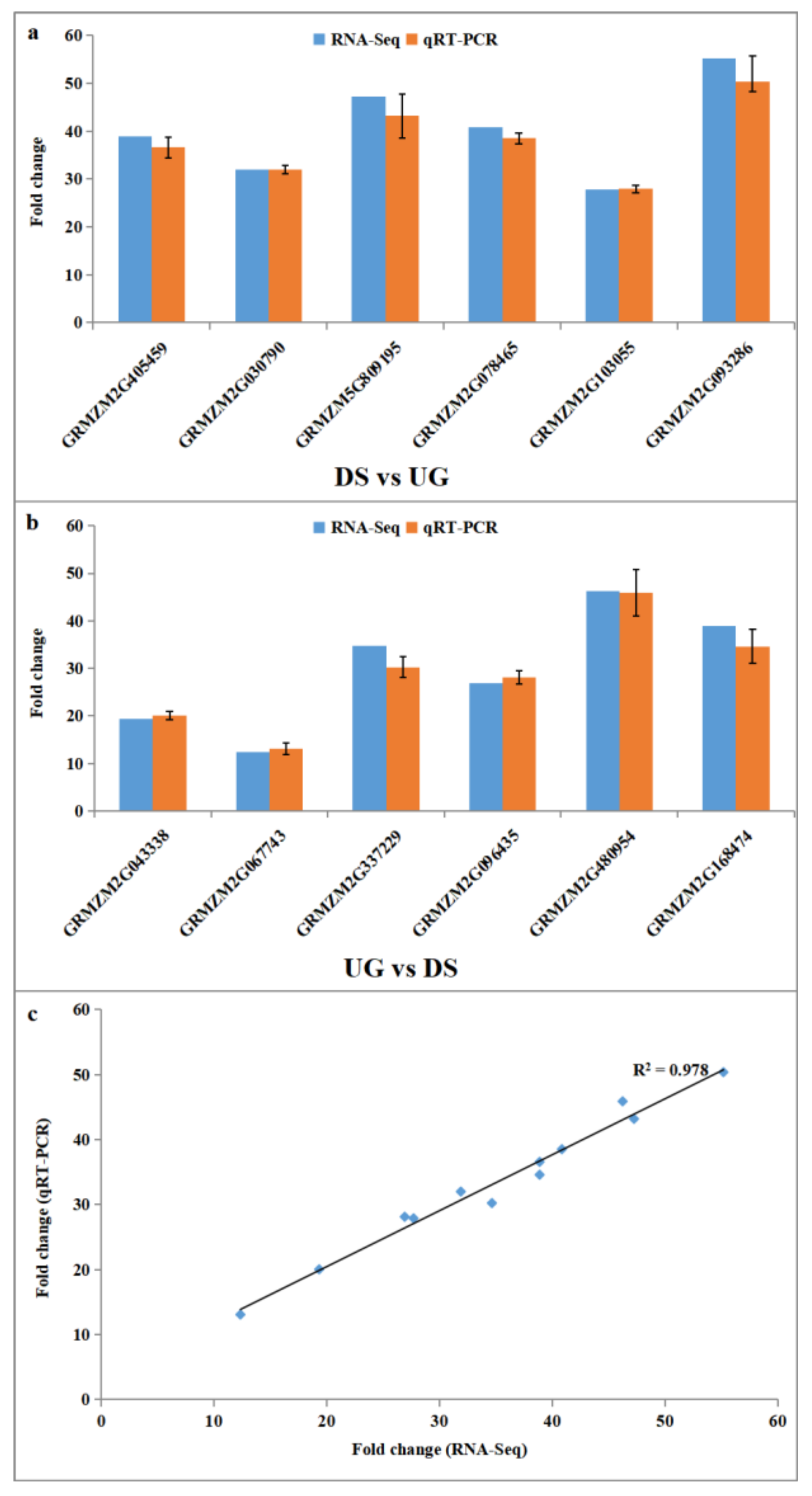
2.4. Experimental Validation of Differential Expressed Genes by qRT-PCR
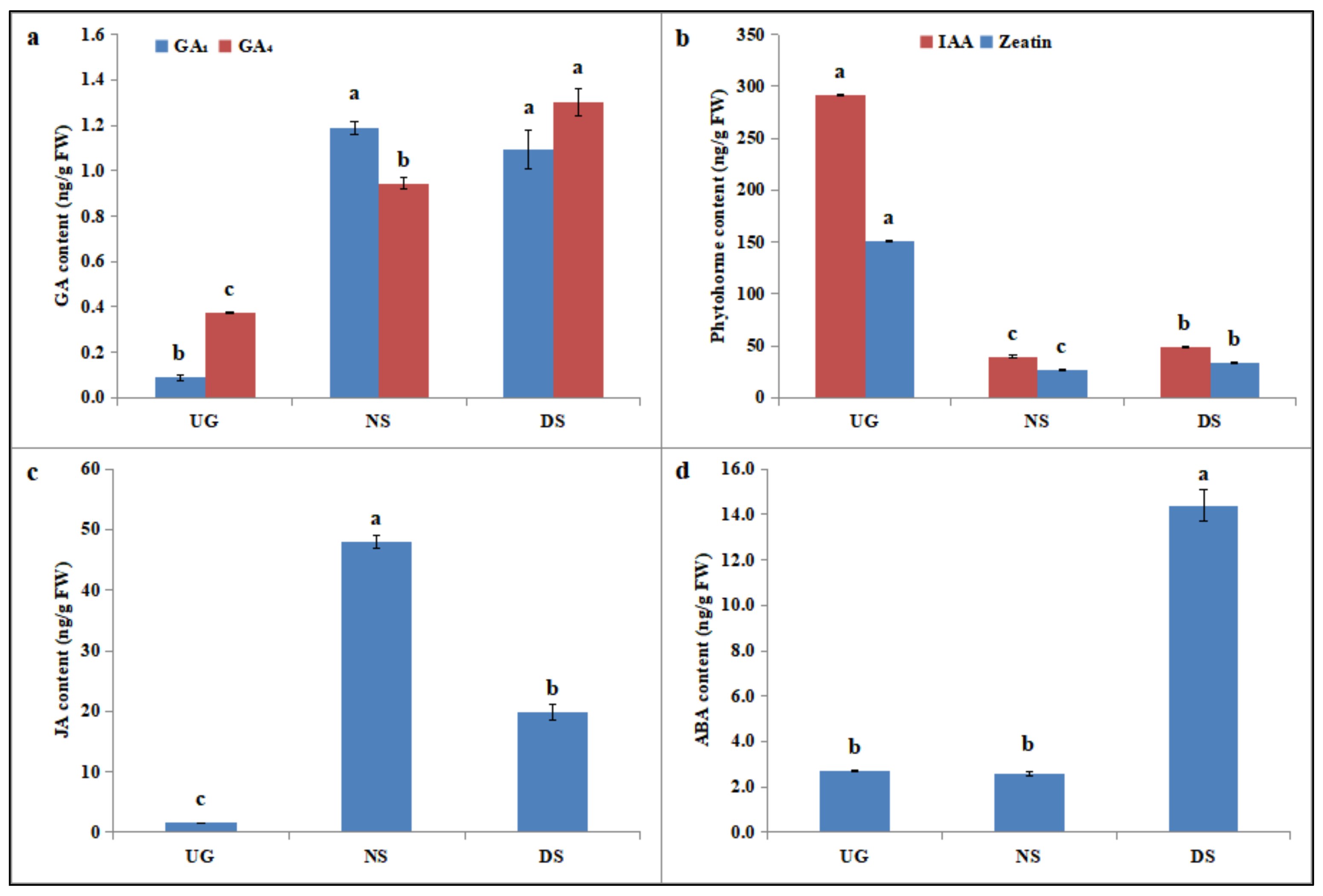
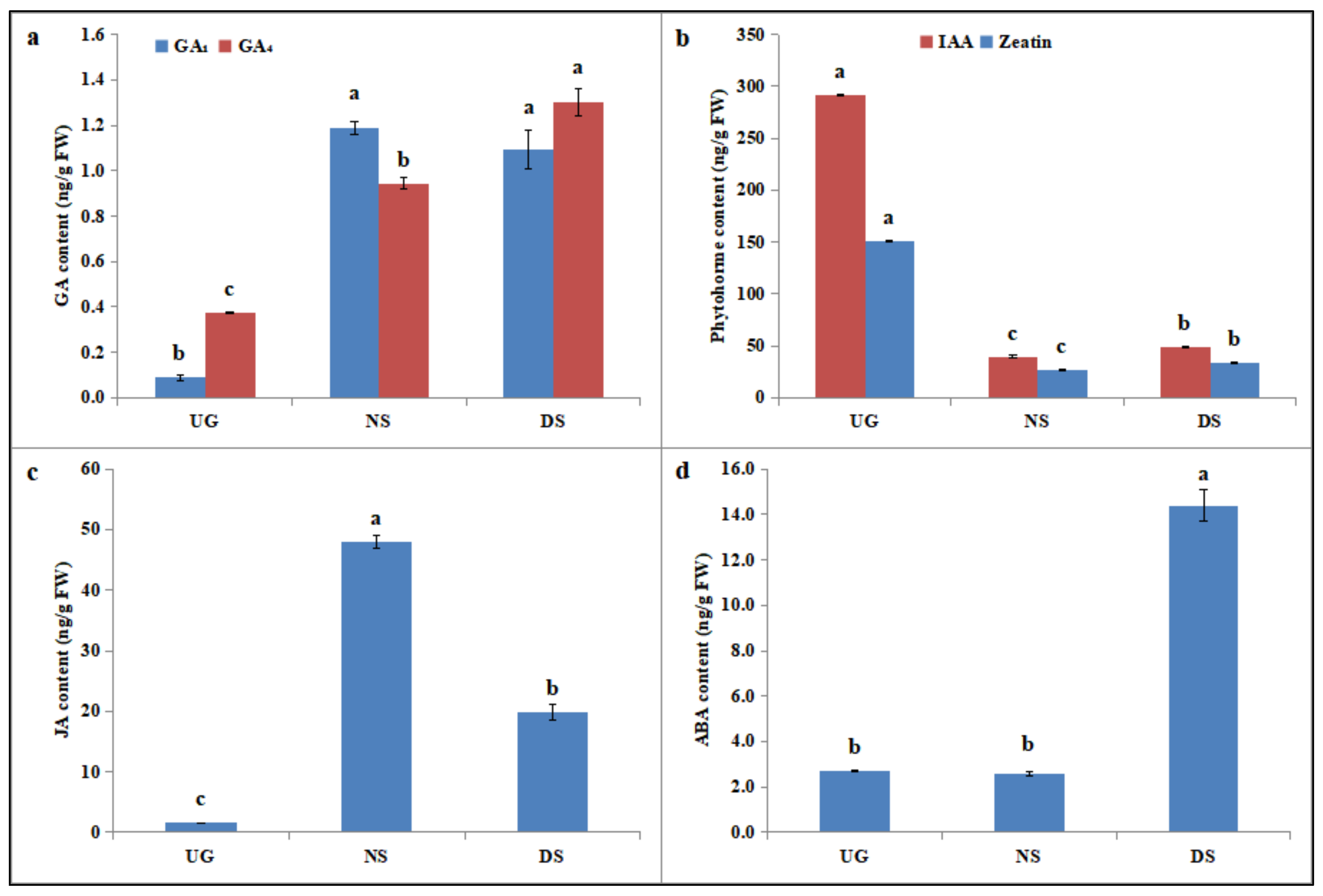
2.5. Deep-Sowing-Induced Changes in Phytohormone Levels in Maize
2.6. Change in Enzyme Activity, Malondialdehyde and Proline Content of Maize in Response to Deep Sowing
2.7. Expression Analysis of Candidate Genes of Different Deep-Sowing-Tolerant Maize Inbred Lines
3. Discussion
4. Materials and Methods
4.1. Preparation of Plant Material
4.2. Measurements of Enzyme Activity, Malondialdehyde and Proline Content
4.3. Determination of Phytohormone Content
4.4. RNA Isolation, cDNA Library Preparation and Transcriptome Sequencing
4.5. Gene Quantification and Differential-Expression Analysis
4.6. GO and KEGG Pathway-Enrichment Analyses for Differentially Expressed Genes
4.7. Validation of RNA-Seq Data by Real-Time Quantitative PCR
4.8. Quantitative Real-Time PCR Expression Analysis of Candidate Genes
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, H.; Zhang, L.; Wang, J.; Li, C.; Zeng, X.; Xie, S.; Zhang, Y.; Liu, S.; Hu, S.; Wang, J.; et al. Quantitative trait locus analysis for deep-sowing germination ability in the Maize IBM Syn10 DH population. Front. Plant Sci. 2017, 8, 813. [Google Scholar] [CrossRef] [PubMed]
- Christov, N.K.; Christova, P.K.; Kato, H.; Liu, Y.; Sasaki, K.; Imai, R.Y. TaSK5, an abiotic stress-inducible GSK3/shaggy-like kinase from wheat, confers salt and drought tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2014, 84, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Du, L.G.; Jiang, H.Y.; Zhao, G.W.; Ren, J.Y. Gene cloning of ZmMYB59 transcription factor in maize and its expression during seed germination in response to deep sowing and exogenous hormones. Plant Breed. 2017, 6, 834–844. [Google Scholar] [CrossRef]
- Zhao, G.W.; Fu, J.J.; Wang, G.Y.; Ma, P.; Wu, L.W.; Wang, J.H. Gibberellin-induced mesocotyl elongation in deep-sowing tolerant maize inbred line 3681-4. Plant Breed. 2010, 129, 87–91. [Google Scholar] [CrossRef]
- Zhao, G.W.; Wang, J.H. Effect of gibberellin and uniconazole on mesocotyl elongation of darkness-grown maize under different seeding depths. Plant Prod. Sci. 2008, 11, 423–429. [Google Scholar] [CrossRef]
- Troyer, A.F. The location of genes governing long first internode of corn. Genetics 1997, 145, 1149–1154. [Google Scholar] [CrossRef]
- Zhao, G.W.; Ma, P.; Wang; J.H.; Wang, G.Y. Identification of deep-seeding tolerance in different maize inbred lines and their physiological response to deep-seeding condition. J. Maize Sci. 2009, 17, 9–13, (In Chinese with English summary). [Google Scholar]
- Zhang, H.W.; Ma, P.; Zhao, Z.N.; Zhao, G.W.; Tian, B.H.; Wang, J.H.; Wang, G.Y. Mapping QTL controlling maize deep-seeding tolerance-related traits and confirmation of a major QTL for mesocotyl length. Theor. Appl. Genet. 2012, 124, 223–232. [Google Scholar] [CrossRef]
- Han, Q.H.; Shen, Y.; Lv, L.; Lee, M.; Lübberstedt, T.; Zhao, G.W. QTL analysis of deep-sowing tolerance during seed germination in the maize IBM Syn4 RIL population. Plant Breed. 2020, 139, 1125–1134. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Zhao, X.Q.; Zhong, Y.; Zhou, W.Q. Molecular mechanisms of mesocotyl elongation induced by brassinosteroid in maize under deep-seeding stress by RNA-sequencing, microstructure observation, and physiological metabolism. Genomics 2021, 113, 3565–3581. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.J.; Wu, Z.K.; Liu, H.; Wu, X.L.; Wang, W. 2-DE-based proteomic analysis of protein changes associated with etiolated mesocotyl growth in Zea mays. BMC Genom. 2019, 20, 758. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.J.; Hao, R.Q.; Wu, X.L.; Wang, W. Maize mesocotyl: Role in response to stress and deep-sowing tolerance. Plant Breed. 2020, 139, 466–473. [Google Scholar] [CrossRef]
- Ohno, H.; Banayo, N.; Bueno, C.; Kashiwagi, J.; Nakashima, T.; Corales, A.; Garcia, R.; Sandhu, N.; Kumar, A.; Kato, Y. Longer mesocotyl contributes to quick seedling establishment, improved root anchorage, and early vigor of deep-sown rice. Field Crop Res. 2018, 228, 84–92. [Google Scholar] [CrossRef]
- Polthanee, A. Effect of seeding depth and soil mulching on growth and yield of peanut grown after rice in the post-monsoon season of northeastern Thailand. Plant Prod. Sci. 2001, 4, 235–240. [Google Scholar] [CrossRef]
- Brown, P.R.; Singleton, G.R.; Tann, C.R.; Mock, I. Increasing sowing depth to reduce mouse damage to winter crops. Crop Prot. 2003, 22, 653–660. [Google Scholar] [CrossRef]
- VanDelft, G.J.; Graves, J.D.; Fitter, A.H.; Van, A.A. Striga seed avoidance by deep planting and no-tillage in sorghum and maize. Int. J. Pest Manag. 2000, 46, 251–256. [Google Scholar] [CrossRef]
- Anderson, M.D.; Prasad, T.K.; Stewart, C.R. Changes in isozyme profiles of catalase, peroxidase, and glutathione reductase during acclimation to chilling in mesocotyls of maize seedlings. Plant Physiol. 1995, 109, 1247–1257. [Google Scholar] [CrossRef]
- Lamichaney, A.; Kudekallu, S.; Kamble, U.; Sarangapany, N.; Katiyar, P.K.; Bohra, A. Differences in seed vigour traits between desi (pigmented) and kabuli (non-pigmented) ecotypes of chickpea (Cicer arietinum) and its association with field emergence. J. Environ. Biol. 2017, 38, 735–742. [Google Scholar] [CrossRef]
- Ahamd, I.; Kamran, M.; Ali, S.; Bilegjargal, B.; Cai, T.; Ahmad, S.; Meng, X.P.; Su, W.N.; Liu, T.N.; Han, Q.F. Uniconazole application strategies to improve lignin biosynthesis, lodging resistance and production of maize in semiarid regions. Field Crop. Res. 2018, 222, 66–77. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, X.G.; Yang, J.; Liu, W.W.; Du, Q.G.; Wang, H.Q.; Fu, C.X.; Li, W.X. MicroRNA528 affects lodging resistance of maize by regulating lignin biosynthesis under nitrogen-luxury conditions. Mol. Plant 2018, 11, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Del Buono, D.; Luzi, F.; Puglia, D. Lignin nanoparticles: A promising tool to improve maize physiological, biochemical, and chemical traits. Nanomaterials 2021, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- Njeri, R.; Lin, Z.; Yang, P.; He, D. The rice alpha-amylase, conserved regulator of seed maturation and germination. Int. J. Mol. Sci. 2019, 20, 450. [Google Scholar]
- Yu, Y.; Guo, G.; Lv, D.; Hu, Y.; Li, J.; Li, X.; Yan, Y. Transcriptome analysis during seed germination of elite Chinese bread wheat cultivar Jimai 20. BMC Plant Biol. 2014, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Zarattini, M.; Forlani, G. Toward unveiling the mechanisms for transcriptional regulation of proline biosynthesis in the plant cell response to biotic and abiotic stress conditions. Front. Plant Sci. 2017, 8, 927. [Google Scholar] [CrossRef] [PubMed]
- Biju, S.; Fuentes, S.; Gupta, D. Silicon improves seed germination and alleviates drought stress in lentil crops by regulating osmolytes, hydrolytic enzymes and antioxidant defense system. Plant Physiol. Biochem. 2017, 119, 250–264. [Google Scholar] [CrossRef]
- Fang, Y.J.; Jian, L.; Jiang, J.; Geng, Y.; Wang, Y. Physiological and epigenetic analyses of Brassica napus seed germination in response to salt stress. Acta Physiol. Plant. 2017, 39, 128. [Google Scholar] [CrossRef]
- Yin, M.Q.; Song, W.J.; Guo, G.Y.; Li, F.; Sheteiwy, M.S.; Pan, R.H.; Hu, J.; Guan, Y.J. Starchy degradation is related with radicle emergence during wheat seed germination. Seed Sci. Technol. 2018, 46, 59–64. [Google Scholar] [CrossRef]
- Zhao, G.W.; Wang, J.H. Effect of auxin on mesocotyl elongation of dark-grown maize under different seeding depths. Russ. J. Plant Physiol. 2010, 57, 79–86. [Google Scholar] [CrossRef]
- Pan, B.R.; Zhong, T.L.; Zhao, G.W. Promoting deep-sowing germinability of corn (Zea mays) by seed soaking with gibberellic acid. Arch. Agron. Soil. Sci. 2017, 63, 1314–1323. [Google Scholar] [CrossRef]
- Hu, Z.Y.; Yamauchi, T.; Yang, J.; Jikumaru, Y.; Tsuchida-Mayama, T.; Ichikawa, I.; Takamure, I.; Nagamura, Y.; Tsutsumi, N.; Yamaguchi, S.; et al. Strigolactone and cytokinin act antagonistically in regulating rice mesocotyl elongation in darkness. Plant Cell Physiol. 2014, 55, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.J.; Mei, H.W.; Fan, P.Q.; Li, Y.N.; Xu, X.Y.; Wei, H.B.; Yan, M.; Luo, L.J. Dynamic transcriptome and phytohormone profiling along the time of light exposure in the mesocotyl of rice seedling. Sci. Rep. 2017, 7, 11961. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Ma, B.; Lu, X.; Huang, Y.H.; He, S.J.; Yang, C.; Yin, C.C.; Zhao, H.; Zhou, Y.; Zhang, W.K.; et al. Ethylene-inhibited jasmonic acid biosynthesis promotes mesocotyl/coleoptile elongation of etiolated rice seedlings. Plant Cell 2017, 29, 1053–1072. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Takahashi, K.; Saigusa, M. Morphological and anatomical effects of abscisic acid (ABA) and fluridone (FLU) on the growth of rice mesocotyls. Plant Growth Regul. 2001, 34, 273–275. [Google Scholar] [CrossRef]
- Cui, W.T.; Zhu, D.; Shen, W.B.; Mei, Y.D.; Hu, D.K.; Shi, Y.J.; Ren, Y.; Shen, W.; Gu, Q.; Xu, D.K.; et al. Hydrogen peroxide is involved in beta-Cyclodextrin-hemin complex-induced lateral root formation in tomato seedlings. Front. Plant Sci. 2017, 8, 1445. [Google Scholar] [CrossRef]
- Hong, L.W.; Ye, C.T.; Lin, J.C.; Fu, H.H.; Wu, X.H.; Li, Q.S. Alternative polyadenylation is involved in auxin-based plant growth and development. Plant J. 2018, 93, 246–258. [Google Scholar] [CrossRef]
- Ostrowski, M.; Hetmann, A.; Jakubowska, A. Indole-3-acetic acid UDP-glucosyltransferase from immature seeds of pea is involved in modification of glycoproteins. Phytochemistry 2015, 117, 25–33. [Google Scholar] [CrossRef]
- Hu, J.; Xie, X.J.; Wang, Z.F.; Song, W.J. Sand priming improves alfalfa germination under high-salt concentration stress. Seed Sci. Technol. 2006, 34, 199–204. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, J.; Qin, G.C.; Cui, H.W.; Wang, Q.T. Salicylic acid analogues with biological activity may induce chilling tolerance of maize (Zea mays) seeds. Botany 2012, 90, 845–855. [Google Scholar] [CrossRef]
- Pinhero, R.G.; Rao, M.V.; Paliyath, G.; Murr, D.P.; Fletcher, R.A. Changes in activities of antioxidant enzymes and their relationship to genetic and paclobutrazol-induced chilling tolerance of maize seedlings. Plant Physiol. 1997, 114, 695–704. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, T.T.; Huang, Y.T.; Guan, Y.J.; Hu, J. Salicylic acid biosynthesis inhibitors increase chilling injury to maize (Zea mays L.) seedlings. Plant Growth Regul. 2018, 86, 11–21. [Google Scholar] [CrossRef]
- Zhang, X.; Shen, L.; Li, F.; Zhang, Y.; Meng, D.; Sheng, J. Up regulating arginase contributes to amelioration of chilling stress and the antioxidant system in cherry tomato fruits. J. Sci. Food Agric. 2010, 90, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, J.; Gao, Y.; Wang, C.; Guo, G.; Luo, Y.; Huang, Y.; Hu, W.; Sheteiwy, M.S.; Guan, Y.; et al. The synergistic priming effect of exogenous salicylic acid and H2O2 on chilling tolerance enhancement during maize (Zea mays L.) seed germination. Front. Plant Sci. 2017, 8, 1153. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, Y.L.; Lin, W.H.; Tong, J.H.; Huang, Z.G.; Xiao, L.T. Determination of both jasmonic acid and methyl jasmonate in plant samples by liquid chromatography tandem mass spectrometry. Chin. Sci. Bull. 2010, 55, 2231–2235. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of Next Generation Sequencing Data. PLoS ONE 2012, 7, e3061. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Yue, L.; Heng, Y.Y.; Qi, Y.; Xin, Y.; Zhou, D.; Wen, Y.X.; Zhen, S. AgriGO v2.0: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2010, 39, W316–W322. [Google Scholar] [CrossRef]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W1. [Google Scholar] [CrossRef]
- Lin, Y.A.; Zhang, C.L.; Lan, H.; Gao, S.B.; Liu, H.L.; Liu, J.; Cao, M.J.; Pan, G.T.; Rong, T.Z.; Zhang, S.Z. Validation of potential reference genes for qPCR in maize across abiotic stresses, hormone treatments, and tissue types. PLoS ONE 2014, 9, e95445. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | SOD (U/g FW) | CAT (U/g FW) | POD (U/g FW) | MDA (μmol/L) | Pro (μg/g FW) | α-Amylase (mg/(g·min)) |
|---|---|---|---|---|---|---|
| UG | 128.83 ± 0.61 c a | 6.00 ± 0.06 c | 62.5 ± 12.5 c | 0.16 ± 0.01 c | 5.63 ± 0.03 b | 10.54 ± 0.75 c |
| NS | 155.69 ± 3.34 b | 8.94 ± 0.25 b | 125.1 ± 10.1 b | 0.19 ± 0.01 b | 5.89 ± 0.01 b | 54.84 ± 2.43 b |
| DS | 201.50 ± 0.60 a | 15.56 ± 0.06 a | 225.2 ± 25 a | 0.36 ± 0.02 a | 13.59 ± 0.11 a | 103.71 ± 5.49 a |
| Inbred Line | GRMZM2G103055 | GRMZM5G809195 | GRMZM2G030790 | GRMZM2G405459 | GRMZM2G043338 | GRMZM2G337229 |
|---|---|---|---|---|---|---|
| B73 | 5.18 ± 0.14 a a | 5.5 ± 0.34 a | 11.71 ± 1.06 a | 11.09 ± 0.31 a | 3.44 ± 0.28 a | 7.55 ± 0.97 a |
| Mo17 | 2.69 ± 0.08 b | 1.12 ± 0.02 b | 10.69 ± 0.26 a | 1.65 ± 0.01 b | 0.52 ± 0.03 b | 0.39 ± 0.04 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; He, J.; Ye, H.; Ding, M.; Xu, F.; Wu, R.; Zhao, F.; Zhao, G. Transcriptome Analysis Revealed the Key Genes and Pathways Involved in Seed Germination of Maize Tolerant to Deep-Sowing. Plants 2022, 11, 359. https://doi.org/10.3390/plants11030359
Wang Y, He J, Ye H, Ding M, Xu F, Wu R, Zhao F, Zhao G. Transcriptome Analysis Revealed the Key Genes and Pathways Involved in Seed Germination of Maize Tolerant to Deep-Sowing. Plants. 2022; 11(3):359. https://doi.org/10.3390/plants11030359
Chicago/Turabian StyleWang, Yang, Jinna He, Haotian Ye, Mingquan Ding, Feiwang Xu, Rong Wu, Fucheng Zhao, and Guangwu Zhao. 2022. "Transcriptome Analysis Revealed the Key Genes and Pathways Involved in Seed Germination of Maize Tolerant to Deep-Sowing" Plants 11, no. 3: 359. https://doi.org/10.3390/plants11030359
APA StyleWang, Y., He, J., Ye, H., Ding, M., Xu, F., Wu, R., Zhao, F., & Zhao, G. (2022). Transcriptome Analysis Revealed the Key Genes and Pathways Involved in Seed Germination of Maize Tolerant to Deep-Sowing. Plants, 11(3), 359. https://doi.org/10.3390/plants11030359