

Transcriptomic and iTRAQ-Based Quantitative Proteomic Analyses of inap CMS in Brassica napus L.

Abstract
:1. Introduction
2. Results
2.1. Phenotype of inap CMS and Its Maintainer
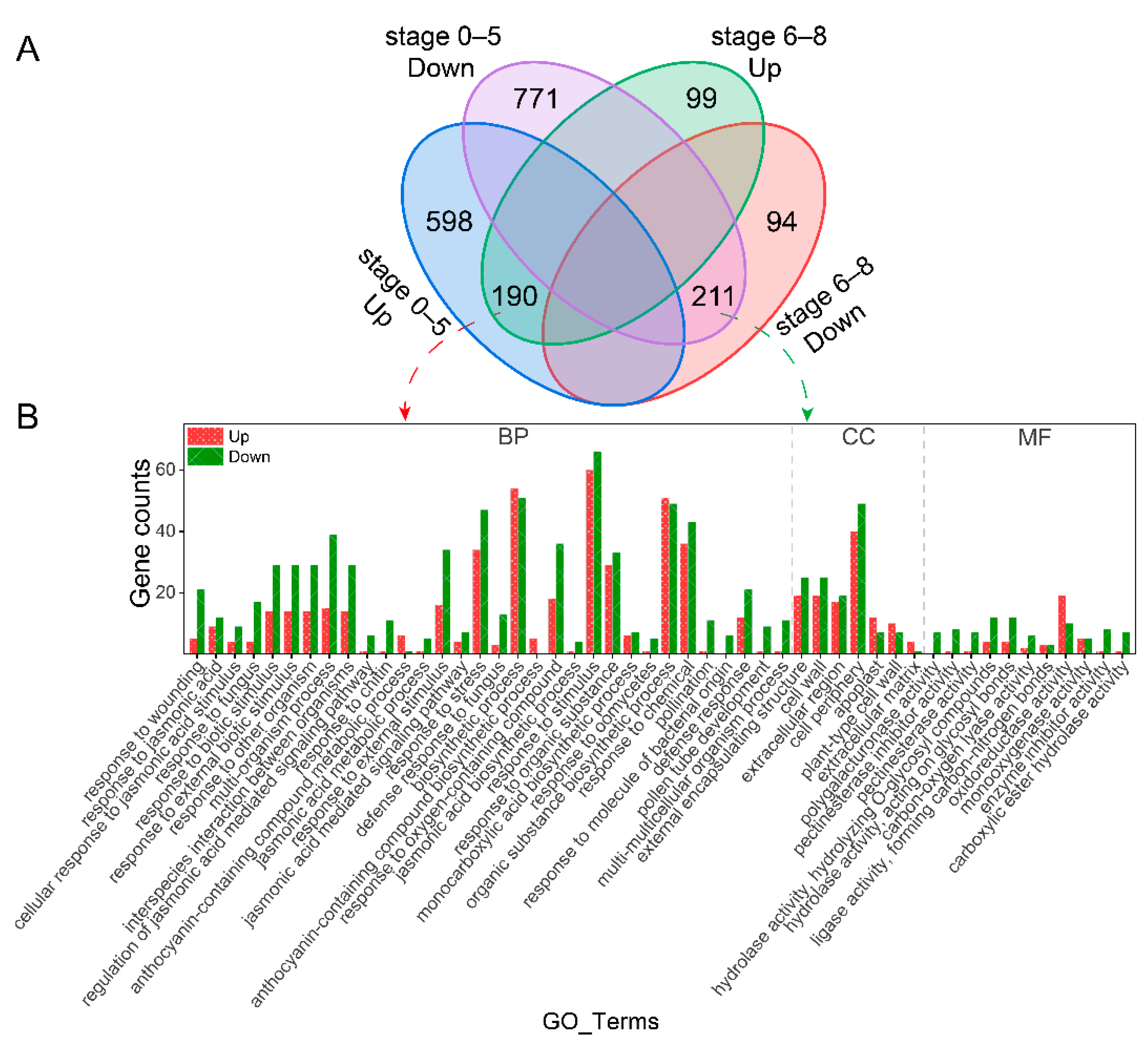
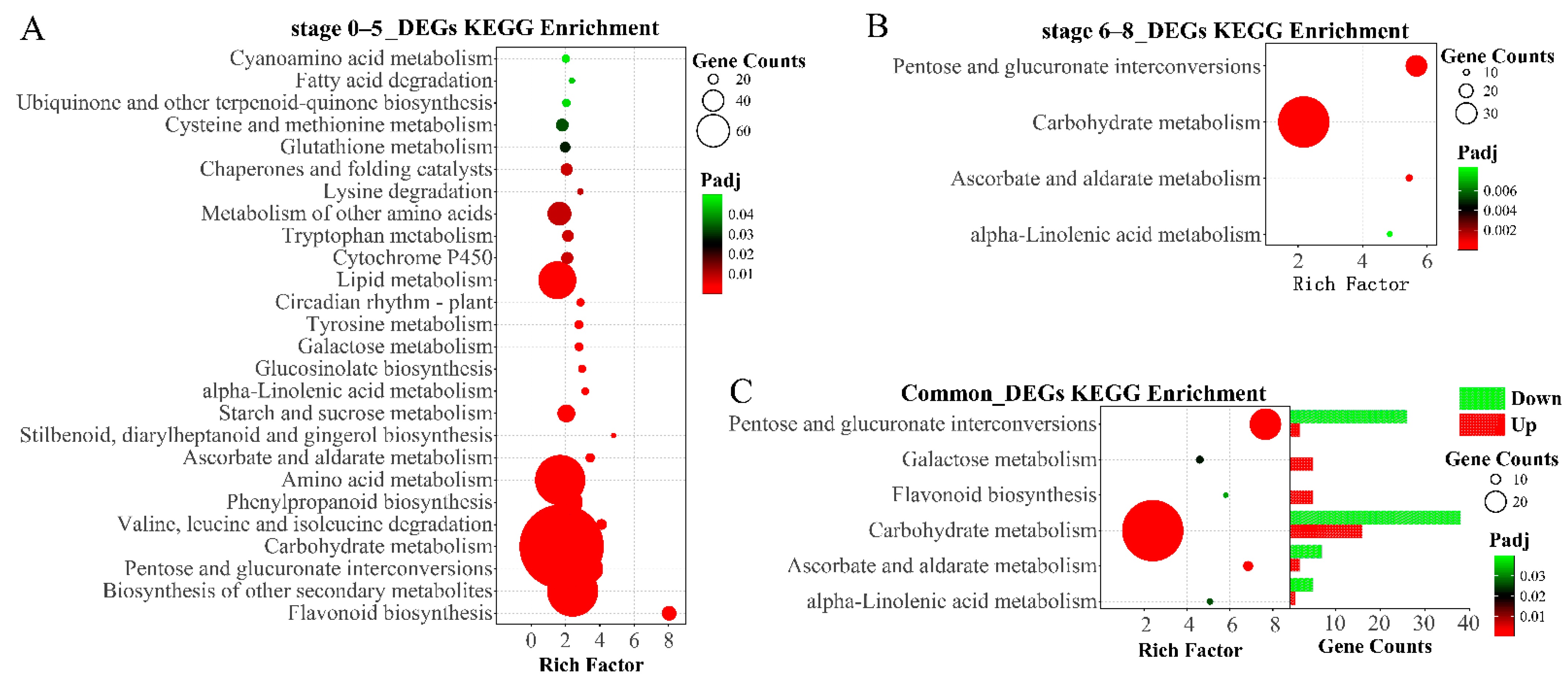
2.2. Differential Expressed Genes Analysis
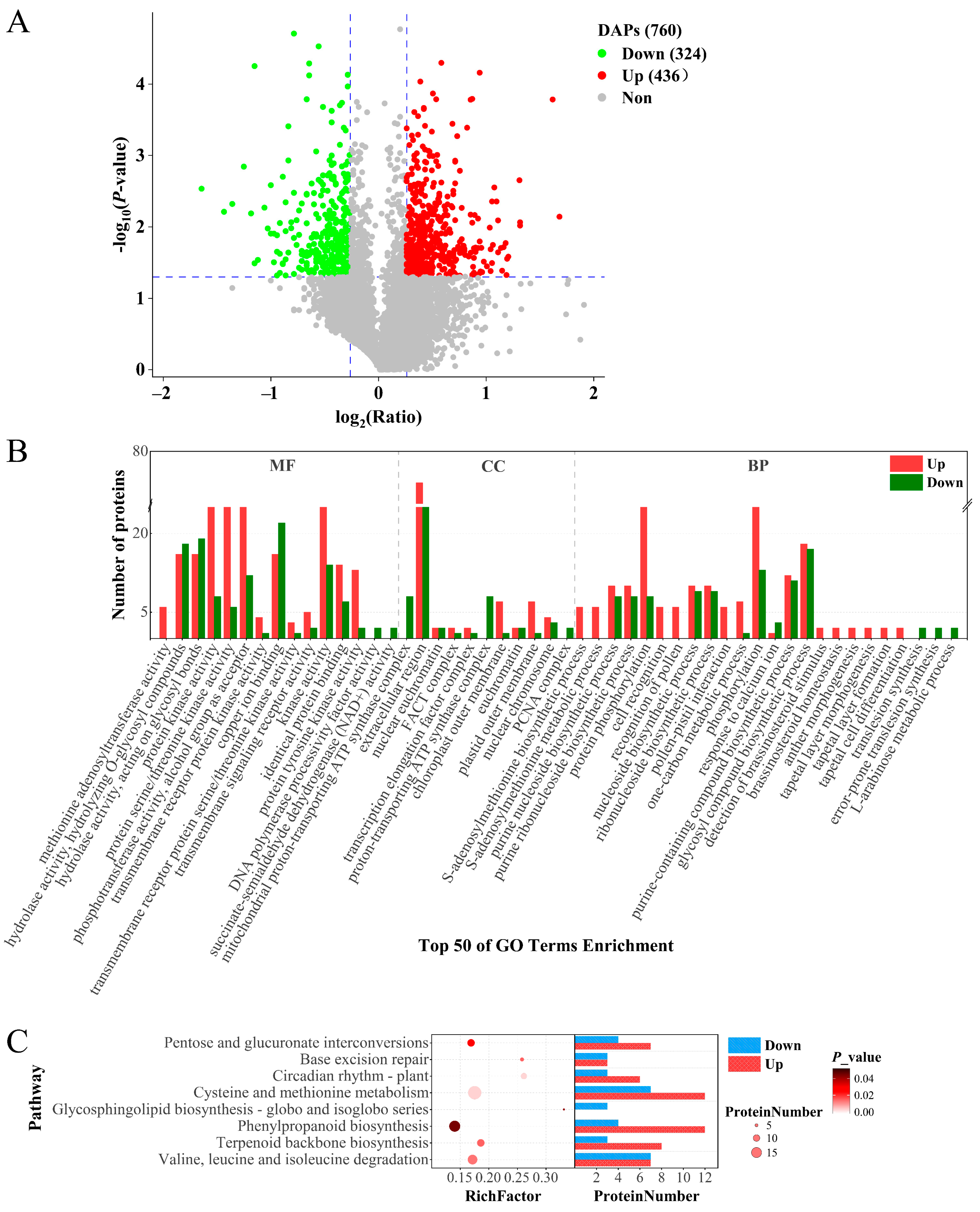
2.3. Differential Abundant Proteins Analysis
2.4. DAPs Related to Male Sterility in inap CMS
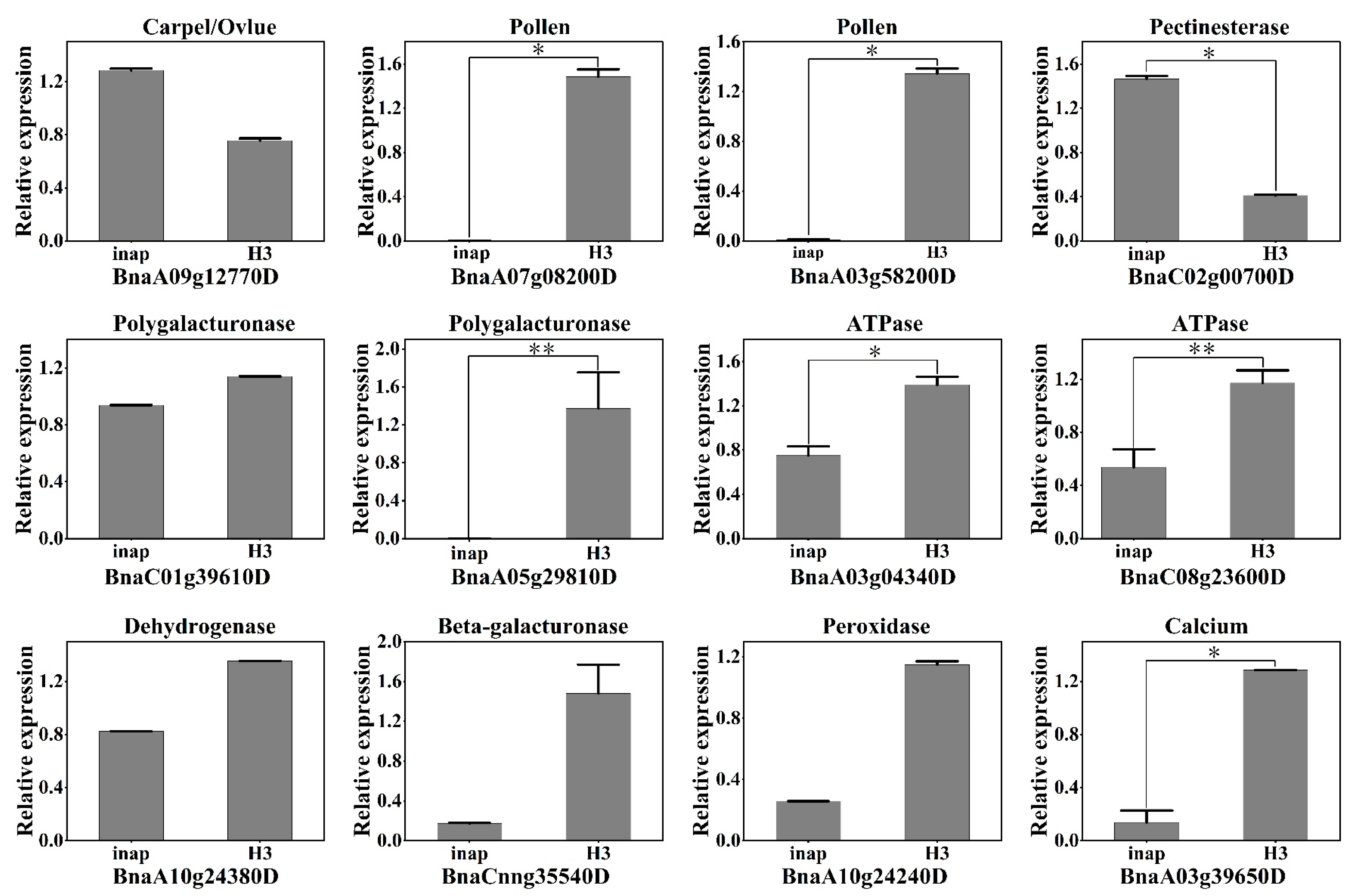
2.5. Validation of DAPs by qRT-PCR Analysis
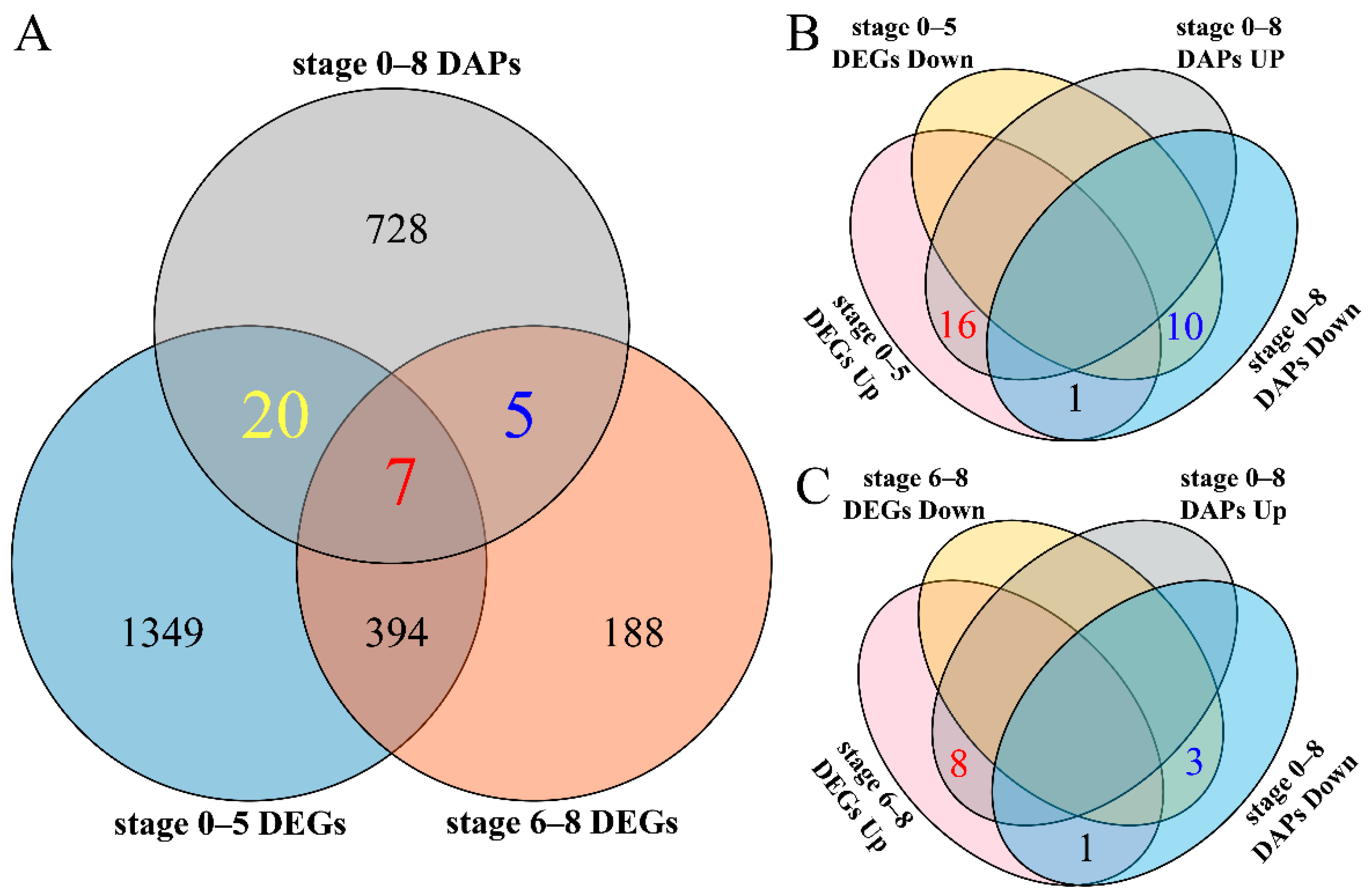
2.6. Conjoint Analysis of DEGs and DAPs
3. Discussion
3.1. PCD Related to Male Sterility in inap CMS
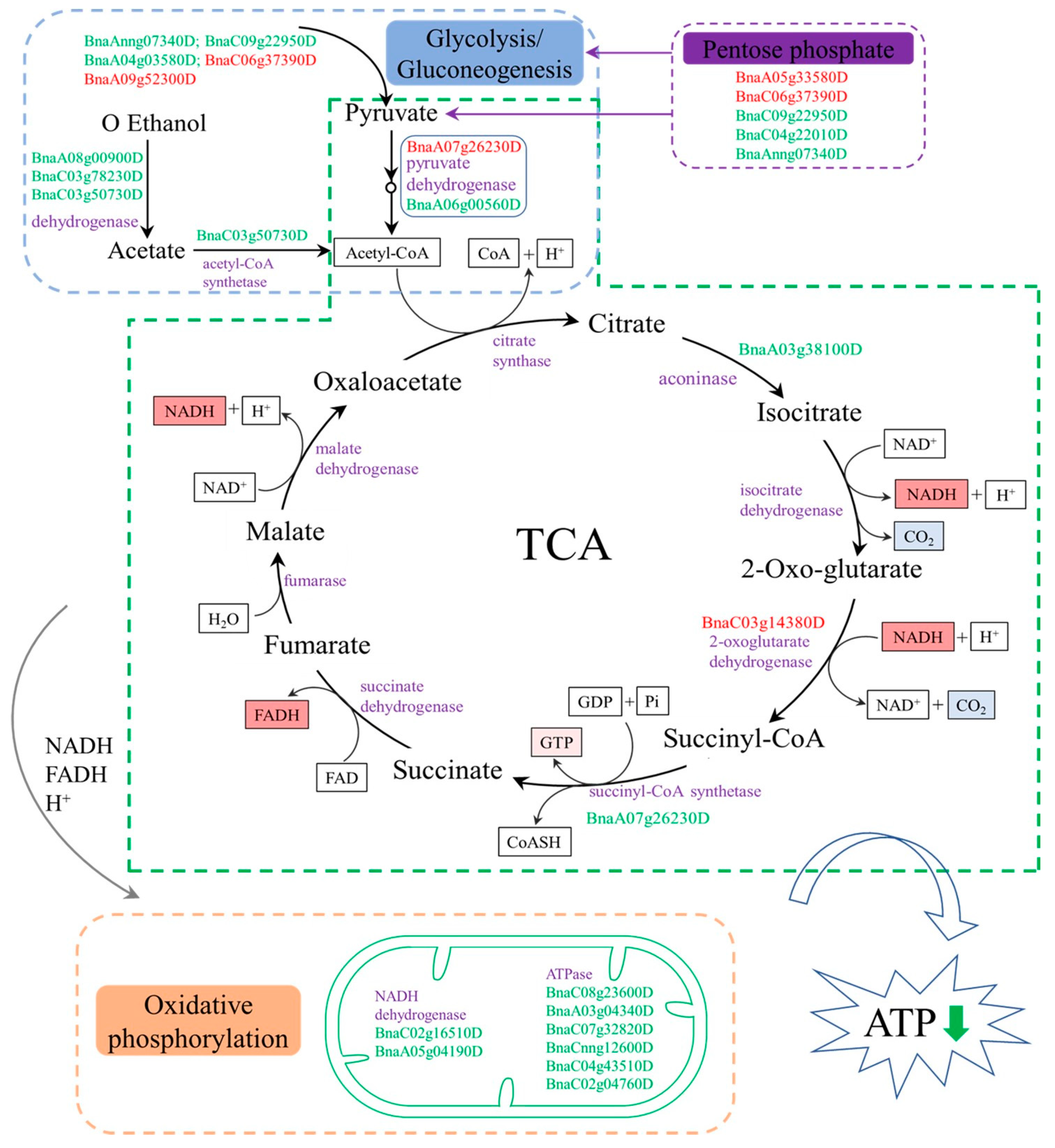
3.2. Energy Metabolism Related to Male Sterility in inap CMS
4. Materials and Methods
4.1. Plant Materials
4.2. Transmission Electron Microscopy
4.3. Transcriptome Analysis
4.4. Protein Extraction and iTRAQ Analysis
4.5. Quantitative RT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Laser, K.D.; Lersten, N.R. Anatomy and cytology of microsporogenesis in cytoplasmic male sterile angiosperms. Bot. Rev. 1972, 38, 425–454. [Google Scholar]
- Hanson, M.R.; Bentolila, S. Interaction of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16, S154–S169. [Google Scholar] [CrossRef] [PubMed]
- Bohra, A.; Jha, U.C.; Adhimoolam, P.; Bisht, D.; Singh, N.P. Cytoplasmic male sterility (CMS) in hybrid breeding in field crops. Plant Cell Rep. 2016, 35, 967–993. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, S.; Budar, F.; Férault, M.; Pelletier, G. A 2.5 kb NcoI fragment of Ogura radish mitochondrial DNA is correlated with cytoplasmic male-sterility in Brassica cybrids. Currt. Genet. 1991, 19, 121–127. [Google Scholar]
- Singh, M.; Brown, G.G. Characterization of expression of a mitochondrial gene region associated with the Brassica Polima CMS developmental influences. Curr. Genet. 1993, 24, 316–322. [Google Scholar]
- L’Homme, Y.; Stahl, R.J.; Li, X.Q.; Hameed, A.; Brown, G.G. Brassica nap cytoplasmic male sterility is associated with expression of a mtDNA region containing a chimeric gene similar to the pol CMS-associated orf224 gene. Curr. Genet. 1997, 31, 325–335. [Google Scholar] [CrossRef]
- Landgren, M.; Zetterstrand, M.; Sundberg, E.; Glimelius, K. Alloplasmic male-sterile Brassica lines containing B. tournefortii mitochondria express an ORF 3′ of the atp6 gene and a 32 kDa protein. Plant Mol. Biol. 1996, 32, 879–890. [Google Scholar]
- Sang, S.; Cheng, H.; Hao, M.; Ding, B.; Mei, D.; Wang, H.; Wang, W.; Liu, J.; Fu, L.; Liu, K.; et al. Mitochondrial localization of ORF346 causes pollen abortion in alloplasmic male sterility. Crop. J. 2021, 9, 879–890. [Google Scholar] [CrossRef]
- Sun, Q.; Hu, C.; Hu, J.; Li, S.; Zhu, Y. Quantitative proteomic analysis of CMS-related changes in Honglian CMS rice anther. Protein J. 2009, 28, 341–348. [Google Scholar]
- Nie, Z.; Chen, J.; Song, Y.; Fu, H.; Wang, H.; Niu, Q.; Zhu, W. Comparative transcriptome analysis of the anthers from the cytoplasmic male-sterile pepper Line HZ1A and its maintainer line HZ1B. Horticulturae 2021, 7, 580. [Google Scholar]
- Hao, M.; Yang, W.; Li, T.; Shoaib, M.; Sun, J.; Liu, D.; Li, X.; Nie, Y.; Tian, X.; Zhang, A. Combined transcriptome and proteome analysis of anthers of AL-type cytoplasmic male sterile line and its maintainer line reveals new insights into mechanism of male sterility in common wheat. Front. Genet. 2021, 12, 762332. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ding, X.; Han, S.; He, T.; Zhang, H.; Yang, L.; Yang, S.; Gai, J. Differential proteomics analysis to identify proteins and pathways associated with male sterility of soybean using iTRAQ-based strategy. J. Proteom. 2016, 138, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Liu, Q.; Wu, X.; Jiang, J.; Wu, J.; Fang, Y.; Li, A.; Wang, Y. Morphological Structure and Transcriptome Comparison of the Cytoplasmic Male Sterility Line in Brassica napus (SaNa-1A) Derived from Somatic Hybridization and Its Maintainer Line SaNa-1B. Front. Plant Sci. 2016, 7, 1313. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Song, L.; Liu, Y.; Han, F.; Liu, W. Electrophysiological, Morphologic, and Transcriptomic Profiling of the Ogura-CMS, DGMS and Maintainer Broccoli Lines. Plants 2022, 11, 561. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, Q.; Zhang, L.; Li, Z.; Wang, C.; Zhang, G. Comparative proteomic analysis of developmental changes in P-Type cytoplasmic male sterile and maintainer anthers in wheat. Int. J. Mol. Sci. 2021, 22, 2012. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Shi, F.; Zhou, L.; Zhou, Y.; Liu, Z.; Ji, R.; Feng, H. iTRAQ-based proteomic analysis of fertile and sterile flower buds from a genetic male sterile line ‘AB01’ in Chinese cabbage (Brassica campestris L. ssp. pekinensis). J. Proteom. 2019, 204, 103395. [Google Scholar] [CrossRef]
- Han, F.; Zhang, X.; Yang, L.; Zhuang, M.; Zhang, Y.; Li, Z.; Fang, Z.; Lv, H. iTRAQ-based proteomic analysis of Ogura-CMS cabbage and its maintainer line. Int. J. Mol. Sci. 2018, 19, 3180. [Google Scholar]
- Wan, L.; Zha, W.; Cheng, X.; Liu, C.; Lv, L.; Liu, C.; Wang, Z.; Du, B.; Chen, R.; Zhu, L.; et al. A rice beta-1,3-glucanase gene Osg1 is required for callose degradation in pollen development. Planta 2011, 233, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liao, J.; Luo, M.; Kirchoff, B.K. Calcium distribution and Function during Anther Development of Torenia fournieri L. Ann. Bot. Fenn. 2008, 45, 195–203. [Google Scholar]
- Tian, H.Q.; Kuang, A.; Musgrave, M.E.; Russell, S.D. Calcium distribution in fertile and sterile anthers of a photoperiod-sensitive genic male-sterile rice. Planta 1998, 204, 183–192. [Google Scholar] [CrossRef]
- Kang, L.; Li, P.; Wang, A.; Ge, X.; Li, Z. A Novel Cytoplasmic Male Sterility in Brassica napus (inap CMS) with Carpelloid Stamens via Protoplast Fusion with Chinese Woad. Front. Plant Sci. 2017, 8, 529. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, Y.; Futami, K.; Tsuji, K.; Murai, K. Alloplasmic wheats with Aegilops crassa cytoplasm which express photoperiod-sensitive homeotic transformations of anthers, show alterations in mitochondrial DNA structure and transcription. Mol. Gen Genet. 1997, 255, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Farbos, I.; Mouras, A.; Bereterbide, A.; Glimelius, K. Defective cell proliferation in the floral meristem of alloplasmic plants of Nicotiana tabacum leads to abnormal floral organ development and male sterility. Plant J. 2001, 26, 131–142. [Google Scholar]
- Linke, B.; Nothnagel, T.; Börner, T. Flower development in carrot CMS plants mitochondria affect the expression of MADS box genes homologous to GLOBOSA and deficiens. Plant J. 2003, 34, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Leino, M.; Teixeira, R.; Landgren, M.; Glimelius, K. Brassica napus lines with rearranged Arabidopsis mitochondria display CMS and a range of developmental aberrations. Theor. Appl. Genet. 2003, 106, 1156–1163. [Google Scholar] [CrossRef]
- Kaminski, P.; Dyki, B.; Stepowska, A.A. Improvement of Cauliflower Male Sterile Lines with Brassica Nigra Cytoplasm, Phenotypic Expression and Possibility of Practical Application. J. Agric. Sci. 2012, 4, 190–200. [Google Scholar] [CrossRef]
- Shu, J.; Liu, Y.; Li, Z.; Zhang, L.; Fang, Z.; Yang, L.; Zhuang, M.; Zhang, Y.; Lv, H. Organelle Simple Sequence Repeat Markers Help to Distinguish Carpelloid Stamen and Normal Cytoplasmic Male Sterile Sources in Broccoli. PLoS ONE 2015, 10, e0138750. [Google Scholar]
- Hama, E.; Takumi, S.; Ogihara, Y.; Murai, K. Pistillody is caused by alterations to the class-B MADS-box gene expression pattern in alloplasmic wheats. Planta 2004, 218, 712–720. [Google Scholar]
- Carlsson, J.; Lagercrantz, U.; Sundstrom, J.; Teixeira, R.; Wellmer, F.; Meyerowitz, E.M.; Glimelius, K. Microarray analysis reveals altered expression of a large number of nuclear genes in developing cytoplasmic male sterile Brassica napus flowers. Plant J. 2007, 49, 452–462. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, M.; Yu, J. Mitochondrial retrograde regulation tuning fork in nuclear genes expressions of higher plants. J. Genet. Genom. 2008, 35, 65–71. [Google Scholar] [CrossRef]
- Romanel, E.; Das, P.; Amasino, R.M.; Traas, J.; Meyerowitz, E.; Alves-Ferreira, M. Reproductive Meristem22 is a unique marker for the early stages of stamen development. Int. J. Dev. Biol. 2011, 55, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef]
- Luo, D.; Xu, H.; Liu, Z.; Guo, J.; Li, H.; Chen, L.; Fang, C.; Zhang, Q.; Bai, M.; Yao, N.; et al. A detrimental mitochondrial-nuclear interaction causes cytoplasmic male sterility in rice. Nat. Genet. 2013, 45, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Moons, A. Osgstu3 and osgtu4, encoding tau class glutathione S-transferases, are heavy metal- and hypoxic stress-induced and differentially salt stress-responsive in rice. FEBS Lett. 2003, 553, 427–432. [Google Scholar] [CrossRef]
- Bartling, D.; Radzio, R.; Steiner, U.; Weiler, E.W. A glutathione S-transferase with glutathione-peroxidase activity from Arabidopsis thaliana. Eur. J. Biochem. 1993, 216, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Hosoi, F.; Yamaguchi-Iwai, Y.; Nakamura, H.; Masutani, H.; Ueda, S.; Nishiyama, A.; Takeda, S.; Wada, H.; Spyrou, G.; et al. Thioredoxin-2 (TRX-2) is an essential gene regulating mitochondria-dependent apoptosis. EMBO J. 2002, 21, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2593–2606. [Google Scholar] [CrossRef]
- Meo, I.D.; Fagiolari, G.; Prelle, A.; Viscomi, C.; Zeviani, M.; Tiranti, V. Chronic exposure to sulfide causes accelerated degradation of cytochrome c oxidase in ethylmalonic encephalopathy. Antioxid Redox Signal. 2011, 15, 353–362. [Google Scholar] [CrossRef]
- Zhang, H.; Li, S.; Yi, P.; Wan, C.; Chen, Z.; Zhu, Y. A Honglian CMS line of rice displays aberrant F0 of F0F1-ATPase. Plant Cell Rep. 2007, 26, 1065–1071. [Google Scholar] [CrossRef]
- Smyth, D.; Bowman, J.; Meyerowitz, E. Early Flower Development in Arabidopsis. Plant Cell 1990, 8, 755–767. [Google Scholar]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.B.; Fang, Y.N.; Pan, Z.Y.; Sun, L.; Deng, X.X.; Grosser, J.W.; Guo, W.W. iTRAQ-based quantitative proteomics analysis revealed alterations of carbohydrate metabolism pathways and mitochondrial proteins in a male sterile cybrid pummelo. J. Proteome. Res. 2014, 13, 2998–3015. [Google Scholar] [CrossRef]
- Wen, B.; Zhou, R.; Feng, Q.; Wang, Q.; Wang, J.; Liu, S. IQuant: An automated pipeline for quantitative proteomics based upon isobaric tags. Proteomics 2014, 14, 2280–2285. [Google Scholar] [CrossRef]
- Brosch, M.; Yu, L.; Hubbard, T.; Choudhary, J. Accurate and sensitive peptide identification with MASCOT percolator. J. Proteome Res. 2009, 8, 3176–3181. [Google Scholar] [CrossRef]
- Xiong, Q.; Song, N.; Li, P.; Fischer, S.; Konertz, R.; Wagle, P.; Glöckner, G.; Wu, C.; Eichinger, L. RNAseq and quantitative proteomic analysis of Dictyostelium knock-out cells lacking the core autophagy proteins ATG9 and/or ATG16. BMC Genom. 2021, 22, 444. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DAPs_ID | Description | Mean_Ratio | p-Value | TAIR_ID |
|---|---|---|---|---|
| Carpel and ovule development | ||||
| BnaA09g12770D | Elongation factor G, chloroplastic | 1.53 | 0.0206 | AT1G62750 |
| BnaA05g11640D | Transcription factor TCP10 | 1.43 | 0.0067 | AT2G31070 |
| BnaC06g12160D | O-acyltransferase WSD1 | 1.37 | 0.0196 | AT5G37300 |
| BnaC05g44320D | Arogenate dehydratase/prephenate dehydratase 2, chloroplastic | 1.31 | 0.0001 | AT3G07630 |
| BnaC03g36730D | ABC transporter I family member 6, chloroplastic | 1.29 | 0.0458 | AT3G10670 |
| Anther and pollen development | ||||
| BnaAnng09150D | Protein SHI RELATED SEQUENCE 5 | 0.74 | 0.0462 | AT1G75520 |
| BnaC05g00110D | Squamosa promoter-binding-like protein 8 | 0.72 | 0.0031 | AT4G39400 |
| BnaC01g09870D | Piriformospora indica-insensitive protein 2 | 0.56 | 0.0012 | AT1G13230 |
| BnaA07g08200D | Pollen-specific protein-like | 0.48 | 0.0054 | AT4G18596 |
| BnaA03g58200D | Pollen-specific protein-like | 0.37 | 0.0061 | AT4G18596 |
| Pectinesterase | ||||
| BnaC02g00700D | Probable pectinesterase/pectinesterase inhibitor 51 | 0.8 | 0.0004 | AT5G09760 |
| BnaA02g00180D | Probable pectinesterase/pectinesterase inhibitor 51 | 0.72 | 0.0419 | AT5G09760 |
| Polygalacturonase | ||||
| BnaA05g29810D | Exopolygalacturonase clone GBGA483 | 0.63 | 0.0002 | AT3G07850 |
| BnaA05g26870D | Polygalacturonase inhibitor 1 | 0.65 | 0.0142 | AT5G06860 |
| Oxidative phosphorylation | ||||
| BnaA05g04190D | Acyl carrier protein 1, mitochondrial | 0.82 | 0.0247 | AT2G44620 |
| BnaC02g16510D | Acyl carrier protein 2, mitochondrial | 0.81 | 0.0217 | AT1G65290 |
| BnaC02g04760D | ATP synthase subunit O, mitochondrial | 0.7 | 0.0279 | AT5G13450 |
| BnaC04g43510D | ATP synthase subunit gamma, mitochondrial | 0.68 | 0.0232 | AT2G33040 |
| BnaC07g32820D | ATP synthase subunit d, mitochondrial | 0.67 | 0.0311 | AT3G52300 |
| BnaA03g04340D | ATP synthase subunit O, mitochondrial | 0.56 | 0.0229 | AT5G13450 |
| BnaC08g23600D | ATP synthase subunit d, mitochondrial | 0.53 | 0.0239 | AT3G52300 |
| PCD | ||||
| BnaA07g09120D | Glutathione S-transferase U13 | 0.75 | 0.0199 | AT1G27130 |
| BnaC03g69760D | Persulfide dioxygenase ETHE1 homolog, mitochondrial | 0.75 | 0.0050 | AT1G53580 |
| BnaC09g09340D | Thioredoxin reductase 2 | 0.72 | 0.0176 | AT2G17420 |
| BnaC01g25910D | Thioredoxin M3, chloroplastic | 0.68 | 0.0022 | AT2G15570 |
| BnaC08g42050D | Thioredoxin-like protein CXXS1 | 0.52 | 0.0131 | AT1G11530 |
| BnaA09g54380D | Peroxidase 22 | 0.75 | 0.0320 | AT2G38380 |
| BnaA10g24240D | Peroxidase 53 | 0.61 | 0.0217 | AT5G06720 |
| BnaC08g24170D | Peroxiredoxin-2E, chloroplastic | 0.74 | 0.0111 | AT3G52960 |
| In order of Mean_Ratio (inap-vs-H3), and the DAPs were identified at level of p-value < 0.05. | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, A.; Kang, L.; Yang, G.; Li, Z. Transcriptomic and iTRAQ-Based Quantitative Proteomic Analyses of inap CMS in Brassica napus L. Plants 2022, 11, 2460. https://doi.org/10.3390/plants11192460
Wang A, Kang L, Yang G, Li Z. Transcriptomic and iTRAQ-Based Quantitative Proteomic Analyses of inap CMS in Brassica napus L. Plants. 2022; 11(19):2460. https://doi.org/10.3390/plants11192460
Chicago/Turabian StyleWang, Aifan, Lei Kang, Guangsheng Yang, and Zaiyun Li. 2022. "Transcriptomic and iTRAQ-Based Quantitative Proteomic Analyses of inap CMS in Brassica napus L." Plants 11, no. 19: 2460. https://doi.org/10.3390/plants11192460
APA StyleWang, A., Kang, L., Yang, G., & Li, Z. (2022). Transcriptomic and iTRAQ-Based Quantitative Proteomic Analyses of inap CMS in Brassica napus L. Plants, 11(19), 2460. https://doi.org/10.3390/plants11192460