
Pleiotropic Effect of the compactum Gene and Its Combined Effects with Other Loci for Spike and Grain-Related Traits in Wheat


 ,
,  and
and
Abstract
:1. Introduction
2. Results
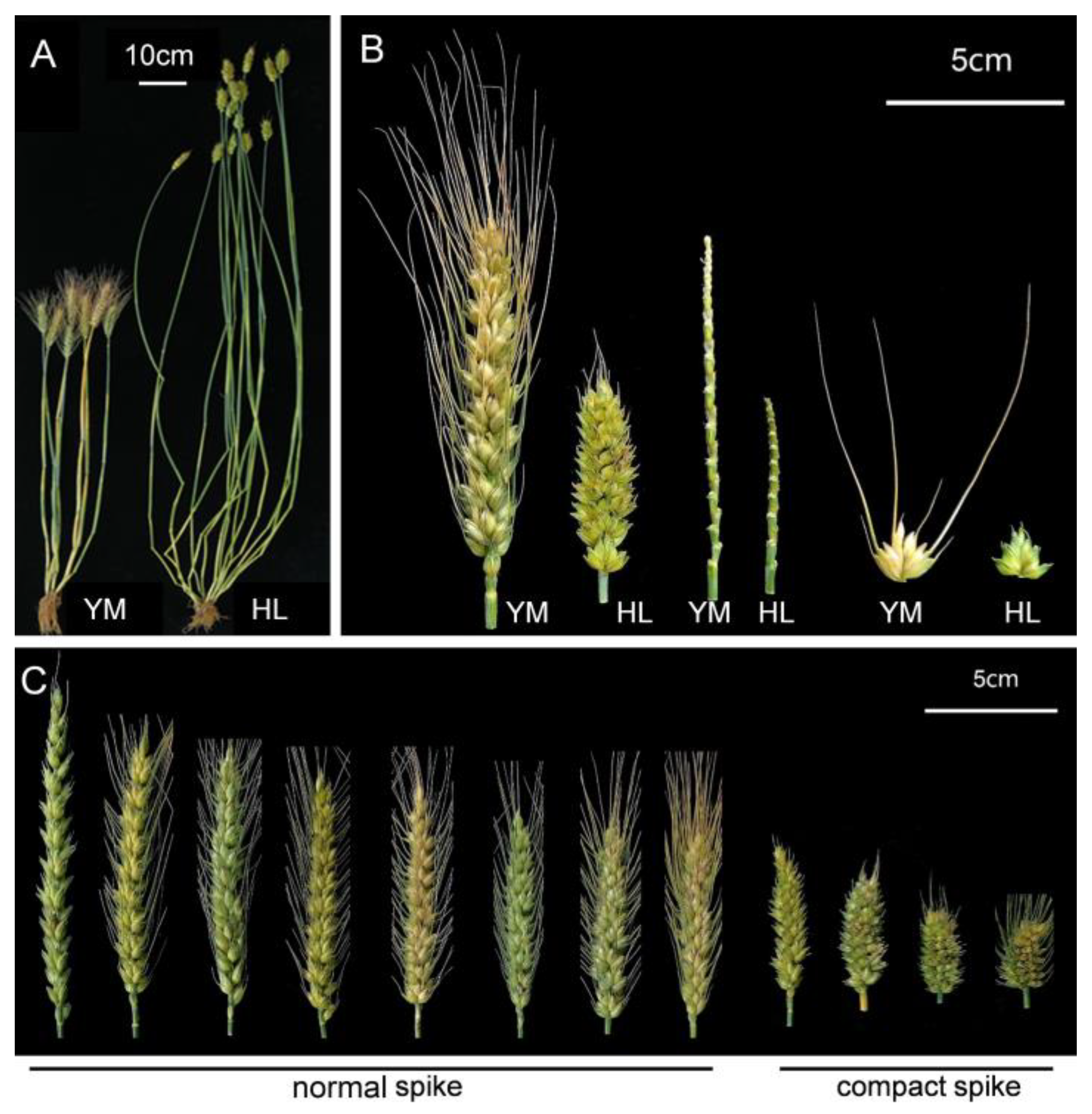
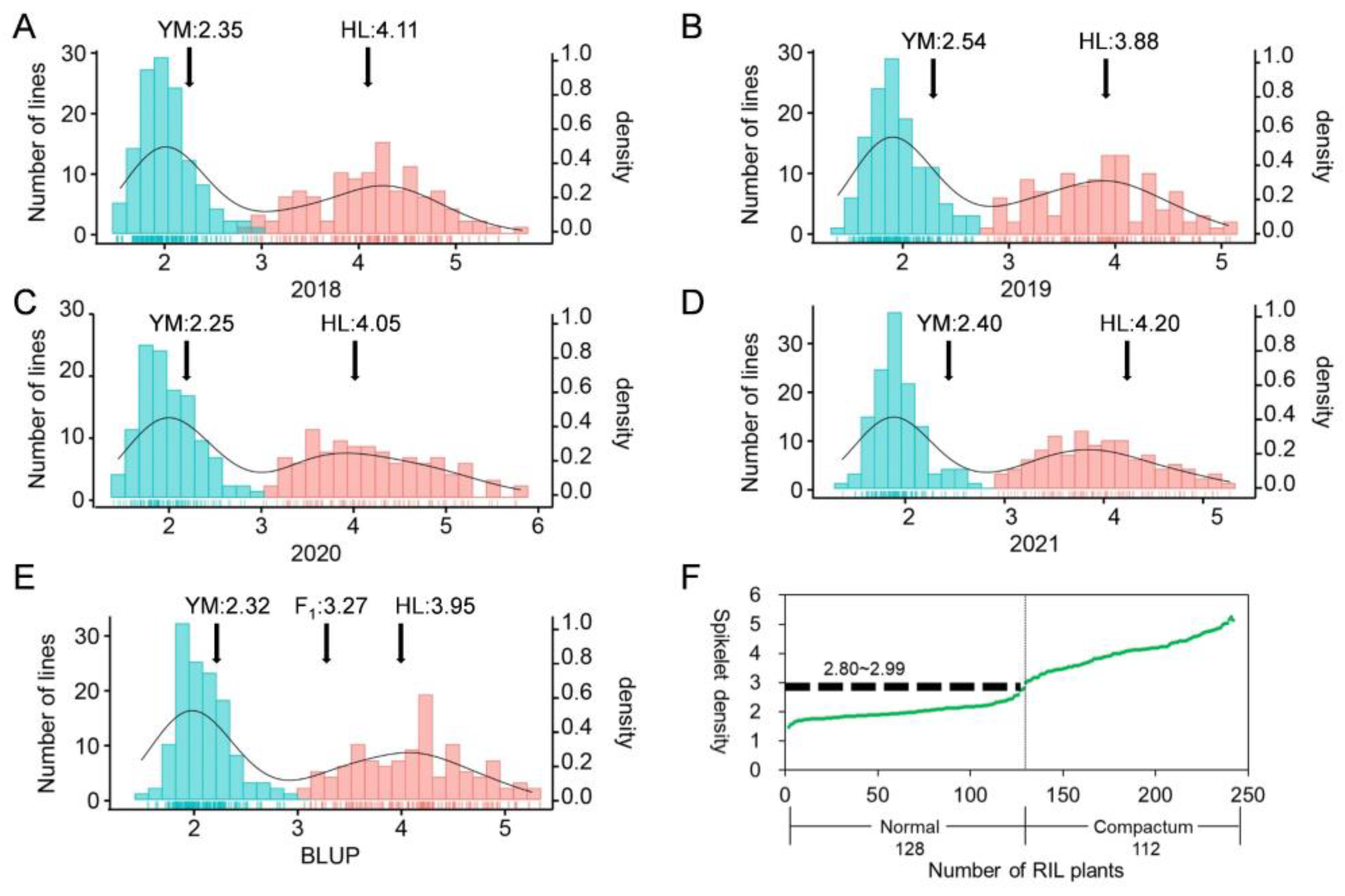
2.1. Phenotypic Analyses
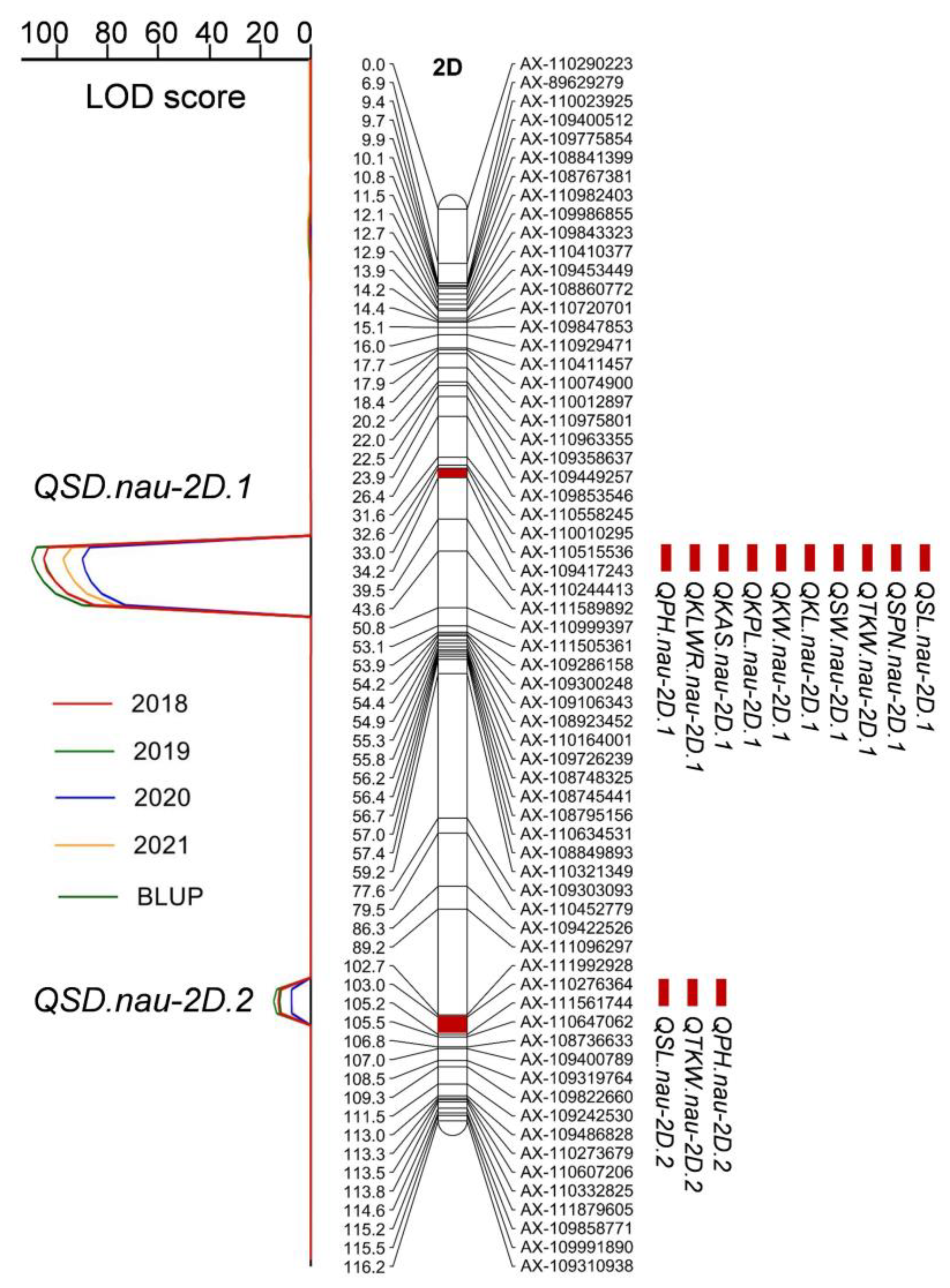
2.2. Linkage Groups Construction and QTL Detection for SD and Other Morphological Traits at C Locus
2.3. Mapping QTL for Five Agronomic Traits Located at Genome Regions Other Than the C Locus
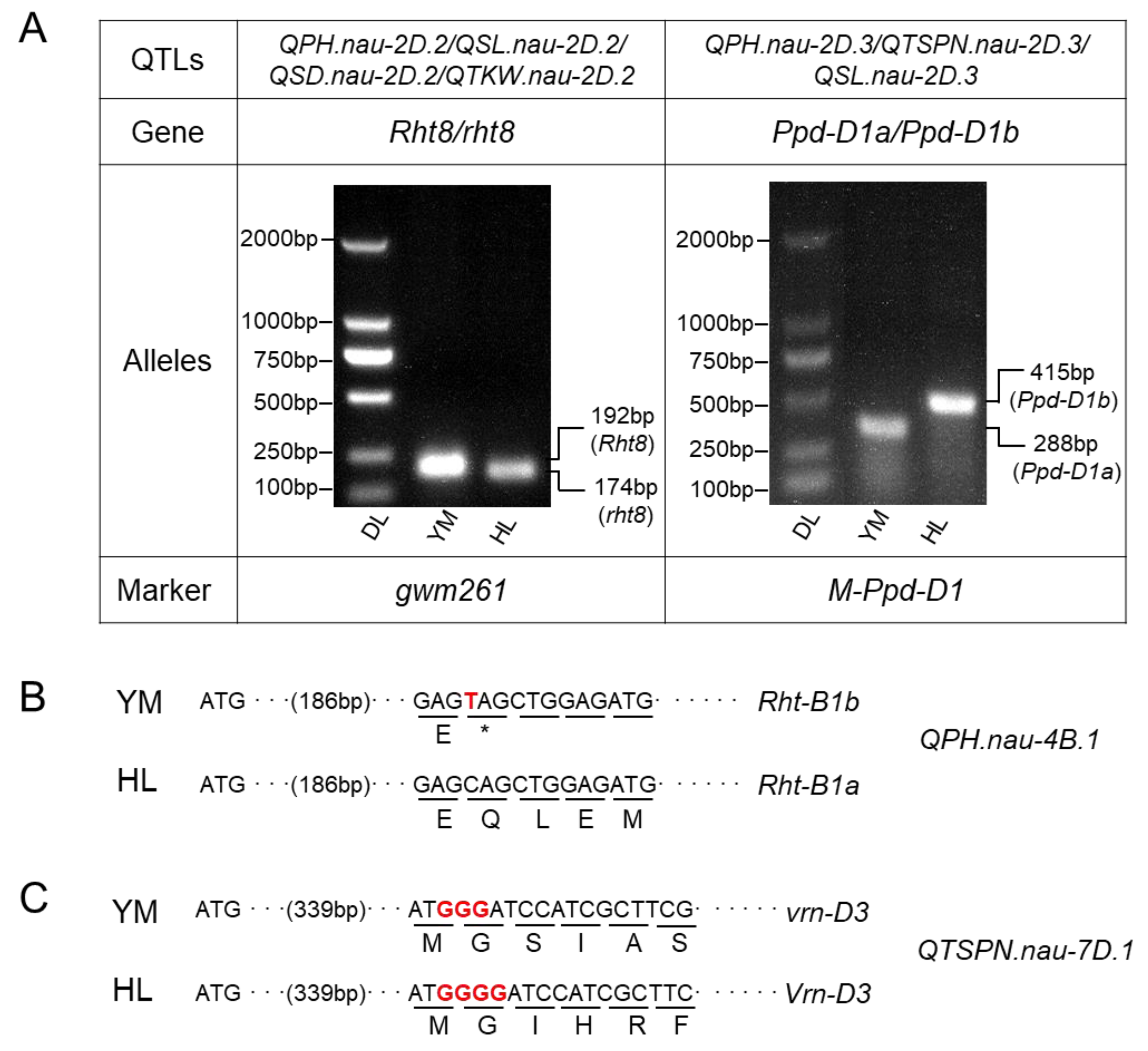
2.4. Validation of the Relationship of Mapped QTL with Known QTL or Genes
2.5. Genetic Effects of Allele Combinations between C and Other Five Known QTLs or Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotypic Evaluation and Statistical Analysis
4.3. Genetic Map Construction and QTL Mapping
4.4. Validation of Known Loci or Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hussain, W.; Baenziger, P.S.; Belamkar, V.; Guttieri, M.J.; Venegas, J.P.; Easterly, A.; Sallam, A.; Poland, J. Genotyping-by-sequencing derived high-density linkage map and its application to QTL mapping of flag leaf traits in bread wheat. Sci. Rep. 2017, 7, 16394. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Bai, G.; Carver, B.F.; Chao, S.; Wang, Z. Mapping quantitative trait loci for plant adaptation and morphology traits in wheat using single nucleotide polymorphisms. Euphytica 2016, 208, 299–312. [Google Scholar] [CrossRef]
- Cheng, X.; Xin, M.; Xu, R.; Chen, Z.; Cai, W.; Chai, L.; Xu, H.; Jia, L.; Feng, Z.; Wang, Z.; et al. A single amino acid substitution in STKc_GSK3 kinase conferring semispherical grains and its implications for the origin of Triticum sphaerococcum. Plant Cell 2020, 32, 923–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.B.; Nalam, V.J.; Zemetra, R.S.; Riera-Lizarazu, O. Mapping the compactum locus in wheat (Triticum aestivum L.) and its relationship to other spike morphology genes of the Triticeae. Euphytica 2008, 163, 193–201. [Google Scholar] [CrossRef]
- Faris, J.D.; Gill, B.S. Genomic targeting and high-resolution mapping of the domestication gene Q in wheat. Genome 2002, 45, 706–718. [Google Scholar] [CrossRef]
- Xu, B.J.; Chen, Q.; Zheng, T.; Jiang, Y.F.; Qiao, Y.Y.; Guo, Z.R.; Cao, Y.L.; Wang, Y.; Zhang, Y.Z.; Zong, L.J.; et al. An overexpressed Q allele leads to increased spike density and improved processing quality in common wheat (Triticum aestivum). G3-Genes Genom Genet. 2018, 8, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Xiao, J.; Liu, Y.; Chen, S.; Yuan, C.; Cao, A.; You, F.M.; Yang, D.; An, S.; Wang, H.; et al. Rht23 (5Dq′) likely encodes a Q homeologue with pleiotropic effects on plant height and spike compactness. Theor. Appl. Genet. 2018, 131, 1825–1834. [Google Scholar] [CrossRef]
- Kosuge, K.; Watanabe, N.; Melnik, V.M.; Laikova, L.I.; Goncharov, N.P. New sources of compact spike morphology determined by the genes on chromosome 5A in hexaploid wheat. Genet. Resour. Crop. Ev. 2012, 59, 1115–1124. [Google Scholar] [CrossRef]
- Kosuge, K.; Watanabe, N.; Kuboyama, T.; Melnik, V.M.; Yanchenko, V.I.; Rosova, M.A.; Goncharov, N.P. Cytological and microsatellite mapping of mutant genes for spherical grain and compact spikes in durum wheat. Euphytica 2008, 159, 289–296. [Google Scholar] [CrossRef]
- Pearce, S.; Saville, R.; Vaughan, S.P.; Chandler, P.M.; Wilhelm, E.P.; Sparks, C.A.; Al-Kaff, N.; Korolev, A.; Boulton, M.I.; Phillips, A.L.; et al. Molecular characterization of Rht-1 dwarfing genes in hexaploid wheat. Plant Physiol. 2011, 157, 1820–1831. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.S.; Du, Y.Y.; Yang, Z.Y.; Chen, L.; Condon, A.G.; Hu, Y.G. Comparing the effects of GA-responsive dwarfing genes Rht13 and Rht8 on plant height and some agronomic traits in common wheat. Field Crop. Res. 2015, 179, 35–43. [Google Scholar] [CrossRef]
- Gasperini, D.; Greenland, A.; Hedden, P.; Dreos, R.; Harwood, W.; Griffiths, S. Genetic and physiological analysis of Rht8 in bread wheat: An alternative source of semi-dwarfism with a reduced sensitivity to brassinosteroids. J. Exp. Bot. 2012, 63, 4419–4436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.Q.; Zhao, D.M.; Zhang, C.Q.; Zhang, Z.Z.; Xue, S.L.; Lin, F.; Kong, Z.X.; Tian, D.G.; Luo, Q.Y. Molecular genetic analysis of five spike-related traits in wheat using RIL and immortalized F-2 populations. Mol. Genet. Genom. 2007, 277, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Phillips, A.L.; Condon, A.G.; Parry, M.A.; Hu, Y.G. GA-responsive dwarfing gene Rht12 affects the developmental and agronomic traits in common bread wheat. PLoS ONE 2013, 8, e62285. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.P.; Conway, B.; Miller, D.; Marshall, D.; Cooper, A.; Murphy, P.; Chao, S.M.; Brown-Guedira, G.; Costa, J. Quantitative trait loci mapping for spike characteristics in hexaploid wheat. Plant Genome 2017, 10, plantgenome2016-10. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Ma, J.; Zhou, X.H.; Sun, M.; Kong, X.C.; Wei, Y.M.; Jiang, Y.F.; Qi, P.F.; Jiang, Q.T.; Liu, Y.X.; et al. Identification of quantitative trait loci controlling agronomic traits indicates breeding potential of tibetan semiwild wheat (Triticum aestivum ssp tibetanum). Crop. Sci. 2016, 56, 2410–2420. [Google Scholar] [CrossRef]
- Faris, J.D.; Zhang, Q.; Chao, S.; Zhang, Z.; Xu, S.S. Analysis of agronomic and domestication traits in a durum x cultivated emmer wheat population using a high-density single nucleotide polymorphism-based linkage map. Theor. Appl. Genet. 2014, 127, 2333–2348. [Google Scholar] [CrossRef]
- Marza, F.; Bai, G.H.; Carver, B.F.; Zhou, W.C. Quantitative trait loci for yield and related traits in the wheat population Ning7840 x Clark. Theor. Appl. Genet. 2006, 112, 688–698. [Google Scholar] [CrossRef]
- Sourdille, P.; Cadalen, T.; Guyomarc’h, H.; Snape, J.W.; Perretant, M.R.; Charmet, G.; Boeuf, C.; Bernard, S.; Bernard, M. An update of the Courtot × Chinese Spring intervarietal molecular marker linkage map for the QTL detection of agronomic traits in wheat. Theor. Appl. Genet. 2003, 106, 530–538. [Google Scholar] [CrossRef]
- Sourdille, P.; Tixier, M.H.; Charmet, G.; Gay, G.; Cadalen, T.; Bernard, S.; Bernard, M. Location of genes involved in ear compactness in wheat (Triticum aestivum) by means of molecular markers. Mol. Breed. 2000, 6, 247–255. [Google Scholar] [CrossRef]
- Liu, H.; Ma, J.; Tu, Y.; Zhu, J.; Ding, P.; Liu, J.; Li, T.; Zou, Y.; Habib, A.; Mu, Y.; et al. Several stably expressed QTL for spike density of common wheat (Triticum aestivum) in multiple environments. Plant Breed. 2020, 139, 284–294. [Google Scholar] [CrossRef]
- Jantasuriyarat, C.; Vales, M.I.; Watson, C.J.; Riera-Lizarazu, O. Identification and mapping of genetic loci affecting the free-threshing habit and spike compactness in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2004, 108, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Mac Key, J. The taxonomy of hexaploid wheat. Sven. Bot. Tidskr. 1954, 26, 579–590. [Google Scholar]
- Gul, A.; Allan, R.E. Relation of the club gene with yield and yield components of near-isogenic wheat lines. Crop. Sci. 1972, 12, 297–301. [Google Scholar] [CrossRef]
- Zwer, P.K.; Sombrero, A.; Rickman, R.W.; Klepper, B. Club and common wheat yield component and spike development in the Pacific-Northwest. Crop. Sci. 1995, 35, 1590–1597. [Google Scholar] [CrossRef]
- Rao, M. Mapping of the compactum gene C on chromosome 2D of wheat. Wheat Inf. Serv. 1972, 35, 9. [Google Scholar]
- Dvorak, J.; Luo, M.C.; Yang, Z.L.; Zhang, H.B. The structure of the Aegilops tauschii genepool and the evolution of hexaploid wheat. Theor. Appl. Genet. 1998, 97, 657–670. [Google Scholar] [CrossRef]
- Salina, E.; Borner, A.; Leonova, I.; Korzun, V.; Laikova, L.; Maystrenko, O.; Roder, M.S. Microsatellite mapping of the induced sphaerococcoid mutation genes in Triticum aestivum. Theor. Appl. Genet. 2000, 100, 686–689. [Google Scholar] [CrossRef]
- Ellis, M.H.; Rebetzke, G.J.; Azanza, F.; Richards, R.A.; Spielmeyer, W. Molecular mapping of gibberellin-responsive dwarfing genes in bread wheat. Theor. Appl. Genet. 2005, 111, 423–430. [Google Scholar] [CrossRef]
- Xiong, H.; Zhou, C.; Fu, M.; Guo, H.; Xie, Y.; Zhao, L.; Gu, J.; Zhao, S.; Ding, Y.; Li, Y.; et al. Cloning and functional characterization of Rht8, a “Green Revolution” replacement gene in wheat. Mol. Plant 2022, 15, 373–376. [Google Scholar] [CrossRef]
- Chai, L.; Xin, M.; Dong, C.; Chen, Z.; Zhai, H.; Zhuang, J.; Cheng, X.; Wang, N.; Geng, J.; Wang, X.; et al. A natural variation in Ribonuclease H-like gene underlies Rht8 to confer “Green Revolution” trait in wheat. Mol. Plant 2022, 15, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Mokanu, N.V.; Fayt, V.I. Differences in the effects of alleles of the genes Vrn1 and Ppd-D1 with respect to winter hardiness, frost tolerance and yield in winter wheat. Cytol. Genet. 2008, 42, 384–390. [Google Scholar] [CrossRef]
- Yan, L.; Fu, D.; Li, C.; Blechl, A.; Tranquilli, G.; Bonafede, M.; Sanchez, A.; Valarik, M.; Yasuda, S.; Dubcovsky, J. The wheat and barley vernalization gene VRN3 is an orthologue of FT. Proc. Natl. Acad. Sci. USA 2006, 103, 19581–19586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, D.L.; Szucs, P.; Yan, L.L.; Helguera, M.; Skinner, J.S.; von Zitzewitz, J.; Hayes, P.M.; Dubcovsky, J. Large deletions within the first intron in VRN-1 are associated with spring growth habit in barley and wheat. Mol. Genet. Genom. 2005, 273, 54–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worland, A.J.; Korzun, V.; Roder, M.S.; Ganal, M.W.; Law, C.N. Genetic analysis of the dwarfing gene Rht8 in wheat. Part II. The distribution and adaptive significance of allelic variants at the Rht8 locus of wheat as revealed by microsatellite screening. Appl. Genet. 1998, 96, 1110–1120. [Google Scholar] [CrossRef]
- Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; Poland, J.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [Green Version]
- Peterson, C.J., Jr.; Morris, C.F.; Line, R.F.; Donaldson, E.; Jones, S.S.; Allan, R.E. Registration of ‘Hiller’ Wheat. Crop. Sci. 1999, 39, 1531–1532. [Google Scholar] [CrossRef]
- Smith, S.E.; Kuehl, R.O.; Ray, I.M.; Hui, R.; Soleri, D. Evaluation of simple methods for estimating broad-sense heritability in stands of randomly planted genotypes. Crop. Sci. 1998, 38, 1125–1129. [Google Scholar] [CrossRef]
- Beales, J.; Turner, A.; GriYths, S.; Snape, J.W.; Laurie, D.A. A Pseudo-Response Regulator is misexpressed in the photoperiod insensitive Ppd-D1a mutant of wheat (Triticum aestivum L.). Appl. Genet. 2007, 115, 721–733. [Google Scholar] [CrossRef]
- Peng, J.R.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. ’Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef]
- Bonnin, I.; Rousset, M.; Madur, D.; Sourdille, P.; Dupuits, C.; Brunel, D.; Goldringer, I. FT genome A and D polymorphisms are associated with the variation of earliness components in hexaploid wheat. Appl. Genet. 2008, 116, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Carver, B.F.; Wang, S.W.; Cao, S.H.; Yan, L.L. Genetic regulation of developmental phases in winter wheat. Mol. Breed. 2010, 26, 573–582. [Google Scholar] [CrossRef]
- Wilhelm, E.P.; Mackay, I.J.; Saville, R.J.; Korolev, A.V.; Balfourier, F.; Greenland, A.J.; Boulton, M.I.; Powell, W. Haplotype dictionary for the Rht-1 loci in wheat. Theor. Appl. Genet. 2013, 126, 1733–1747. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits * | Parents | RILs | ||||
|---|---|---|---|---|---|---|
| YM | HL | p-Value | Normal Spike | Compact Spike | p-Value | |
| PH (cm) | 84.99 ± 4.34 | 130.58 ± 5.72 | 3.36 × 1027 *** | 119.01 ± 14.50 | 114.89 ± 13.80 | 0.026 * |
| TN | 4.05 ± 1.50 | 5.75 ± 2.69 | 0.018 * | 6.98 ± 0.19 | 7.01 ± 0.19 | 0.249 |
| SL (cm) | 8.50 ± 0.59 | 4.46 ± 0.39 | 1.32 × 1025 *** | 10.72 ± 1.49 | 5.23 ± 0.87 | 6.53 × 1094 *** |
| SD | 2.32 ± 0.14 | 3.95 ± 0.32 | 2.30 × 1022 *** | 2.02 ± 0.25 | 4.04 ± 0.54 | 0 *** |
| SW (g) | 2.51 ± 0.40 | 1.55 ± 0.41 | 5.19 × 1009 *** | 2.38 ± 0.26 | 2.19 ± 0.28 | 6.37 × 1008 *** |
| SPN | 19.65 ± 0.75 | 17.50 ± 1.05 | 5.89 × 1009 *** | 20.89 ± 1.39 | 20.32 ± 1.23 | 0.001 ** |
| FSPN | 19.35 ± 0.99 | 17.15 ± 1.35 | 8.18 × 1007 *** | 19.84 ± 1.18 | 19.69 ± 1.06 | 0.312 |
| SSPN | 1.00 ± 0.79 | 0.50 ± 0.76 | 0.049 * | 1.05 ± 0.51 | 0.63 ± 0.37 | 4.66 × 1012 *** |
| KNS | 53.70 ± 7.36 | 45.45 ± 8.85 | 0.003 ** | 55.32 ± 4.72 | 57.32 ± 5.66 | 0.003 ** |
| KL (mm) | 6.50 ± 0.12 | 5.83 ± 0.07 | 2.86 × 1007 *** | 6.55 ± 0.31 | 6.07 ± 0.29 | 8.97 × 1027 *** |
| KW (mm) | 3.37 ± 0.10 | 3.00 ± 0.04 | 5.33 × 1006 *** | 3.31 ± 0.13 | 3.22 ± 0.15 | 2.27 × 1006 *** |
| KLWR | 1.95 ± 0.05 | 2.00 ± 0.02 | 0.054 | 2.01 ± 0.07 | 1.93 ± 0.08 | 1.91 × 1014 *** |
| KPL (mm) | 17.11 ± 0.31 | 15.29 ± 0.15 | 1.58 × 1007 *** | 16.89 ± 0.71 | 15.79 ± 0.68 | 4.71 × 1027 *** |
| KAS (mm2) | 16.29 ± 0.73 | 12.27 ± 0.33 | 2.25 × 1007 *** | 16.10 ± 1.30 | 14.22 ± 1.23 | 1.40 × 1024 *** |
| TKW (g) | 47.47 ± 9.57 | 34.89 ± 10.10 | 2.50 × 1004 *** | 43.93 ± 4.19 | 37.75 ± 3.61 | 9.15 × 1027 *** |
| Trials | QTL | LOD Score | Marker Interval | Physical Distance (Mb) | Additive Effect | Contribution (%) |
|---|---|---|---|---|---|---|
| 2018 | QSD.nau-2D.1 | 116.53 | AX-110515536-AX-109417243 | 370.1–406.3 | −1.09 | 81.61 |
| 2018 | QSD.nau-2D.2 | 17.15 | AX-110276364-AX-111561744 | 23.4–24.9 | 0.25 | 4.42 |
| 2019 | QSD.nau-2D.1 | 114.17 | AX-110515536-AX-109417243 | 370.1–406.3 | −0.98 | 80.57 |
| 2019 | QSD.nau-2D.2 | 18.12 | AX-110276364-AX-111561744 | 23.4–24.9 | 0.24 | 5.03 |
| 2020 | QSD.nau-2D.1 | 93.71 | AX-110515536-AX-109417243 | 370.1–406.3 | −1.11 | 77.82 |
| 2020 | QSD.nau-2D.2 | 9.16 | AX-110276364-AX-111561744 | 23.4–24.9 | 0.22 | 3.02 |
| 2021 | QSD.nau-2D.1 | 101.44 | AX-110515536-AX-109417243 | 370.1–406.3 | −1.04 | 79.68 |
| 2021 | QSD.nau-2D.2 | 9.10 | AX-110276364-AX-111561744 | 23.4–24.9 | 0.20 | 2.83 |
| BLUP | QSD.nau-2D.1 | 119.61 | AX-110515536-AX-109417243 | 370.1–406.3 | −1.04 | 82.35 |
| BLUP | QSD.nau-2D.2 | 15.60 | AX-110276364-AX-111561744 | 23.4–24.9 | 0.23 | 3.96 |
| Trait * | QTL | LOD Score | Marker Interval | Physical Distance (Mb) | Additive Effect | Contribution (%) |
|---|---|---|---|---|---|---|
| SPN | QSPN.nau-2D.1 | 11.37 | AX-109417243-AX-110515536 | 370.1–406.3 | 0.57 | 11.00 |
| SL | QSL.nau-2D.1 | 106.61 | AX-109417243-AX-110515536 | 370.1–406.3 | 2.89 | 80.08 |
| TKW | QTKW.nau-2D.1 | 37.91 | AX-109417243-AX-110515536 | 370.1–406.3 | 3.64 | 30.46 |
| SW | QSW.nau-2D.1 | 8.58 | AX-109417243-AX-110515536 | 370.1–406.3 | 0.12 | 9.51 |
| KL | QKL.nau-2D.1 | 47.78 | AX-109417243-AX-110515536 | 370.1–406.3 | 0.28 | 37.76 |
| KW | QKW.nau-2D.1 | 10.61 | AX-109417243-AX-110515536 | 370.1–406.3 | 0.06 | 11.80 |
| KPL | QKPL.nau-2D.1 | 42.38 | AX-109417243-AX-110515536 | 370.1–406.3 | 0.61 | 38.80 |
| KAS | QKAS.nau-2D.1 | 37.70 | AX-109417243-AX-110515536 | 370.1–406.3 | 1.10 | 33.23 |
| KLWR | QKLWR.nau-2D.1 | 24.88 | AX-109417243-AX-110515536 | 370.1–406.3 | 0.05 | 19.68 |
| PH | QPH.nau-2D.1 | 8.09 | AX-109417243-AX-110515536 | 370.1–406.3 | 4.20 | 5.35 |
| SL | QSL.nau-2D.2 | 11.38 | AX-111561744-AX-110276364 | 23.4–24.9 | −0.52 | 2.27 |
| TKW | QTKW.nau-2D.2 | 32.28 | AX-111561744-AX-110276364 | 23.4–24.9 | −2.40 | 12.90 |
| PH | QPH.nau-2D.2 | 25.30 | AX-111561744-AX-110276364 | 23.4–24.9 | −6.11 | 13.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, M.; Su, J.; Jiao, C.; Zhang, X.; Xu, T.; Wang, T.; Liu, X.; Wang, Z.; Sun, L.; Yuan, C.; et al. Pleiotropic Effect of the compactum Gene and Its Combined Effects with Other Loci for Spike and Grain-Related Traits in Wheat. Plants 2022, 11, 1837. https://doi.org/10.3390/plants11141837
Wen M, Su J, Jiao C, Zhang X, Xu T, Wang T, Liu X, Wang Z, Sun L, Yuan C, et al. Pleiotropic Effect of the compactum Gene and Its Combined Effects with Other Loci for Spike and Grain-Related Traits in Wheat. Plants. 2022; 11(14):1837. https://doi.org/10.3390/plants11141837
Chicago/Turabian StyleWen, Mingxing, Jiaxuan Su, Chengzhi Jiao, Xu Zhang, Tao Xu, Tong Wang, Xiaoxue Liu, Zongkuan Wang, Li Sun, Chunxia Yuan, and et al. 2022. "Pleiotropic Effect of the compactum Gene and Its Combined Effects with Other Loci for Spike and Grain-Related Traits in Wheat" Plants 11, no. 14: 1837. https://doi.org/10.3390/plants11141837
APA StyleWen, M., Su, J., Jiao, C., Zhang, X., Xu, T., Wang, T., Liu, X., Wang, Z., Sun, L., Yuan, C., Wang, H., Wang, X., & Xiao, J. (2022). Pleiotropic Effect of the compactum Gene and Its Combined Effects with Other Loci for Spike and Grain-Related Traits in Wheat. Plants, 11(14), 1837. https://doi.org/10.3390/plants11141837