
Complete Genome Sequence of an Isolate of Passiflora chlorosis virus from Passion Fruit (Passiflora edulis Sims)



Abstract
1. Introduction
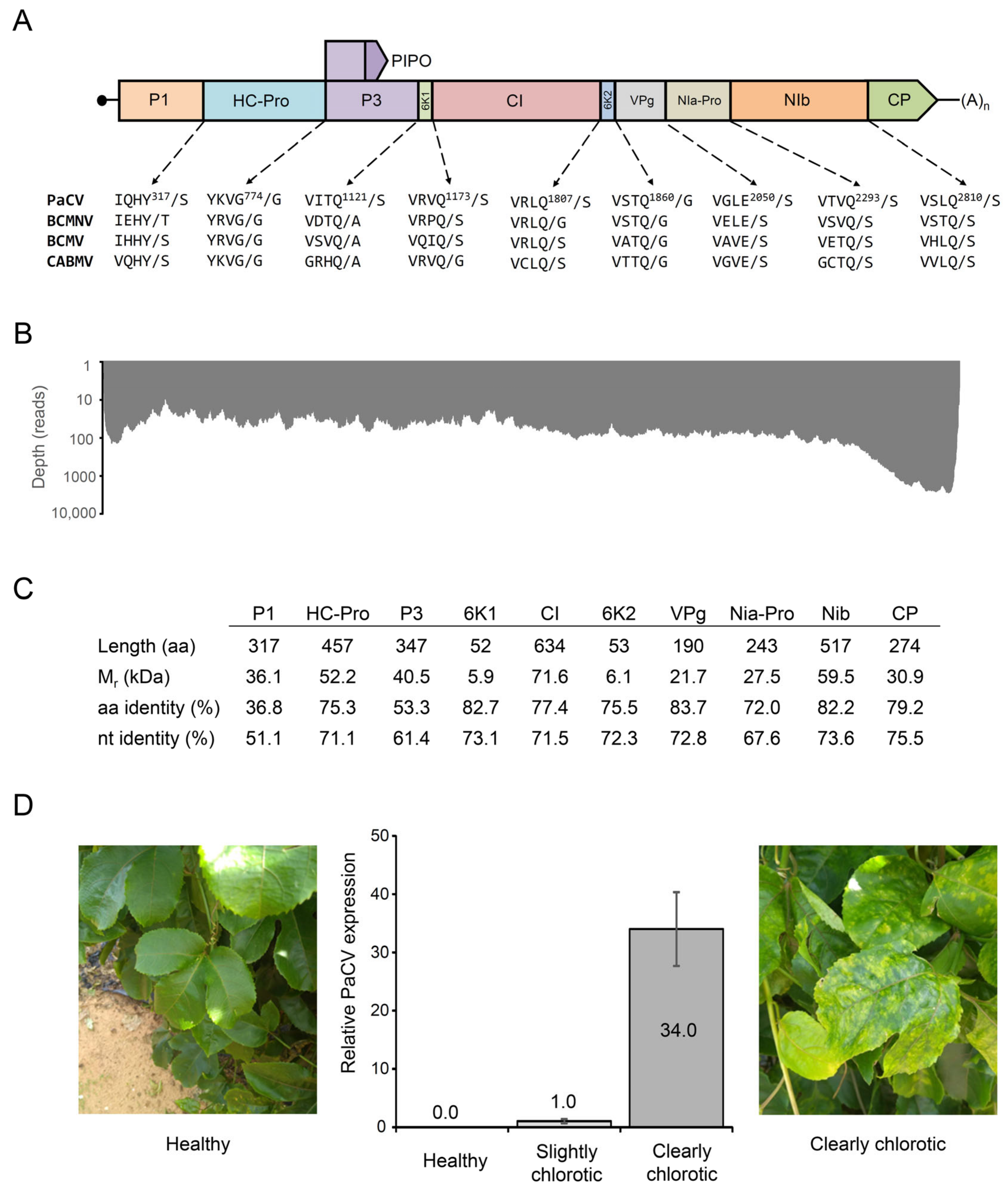
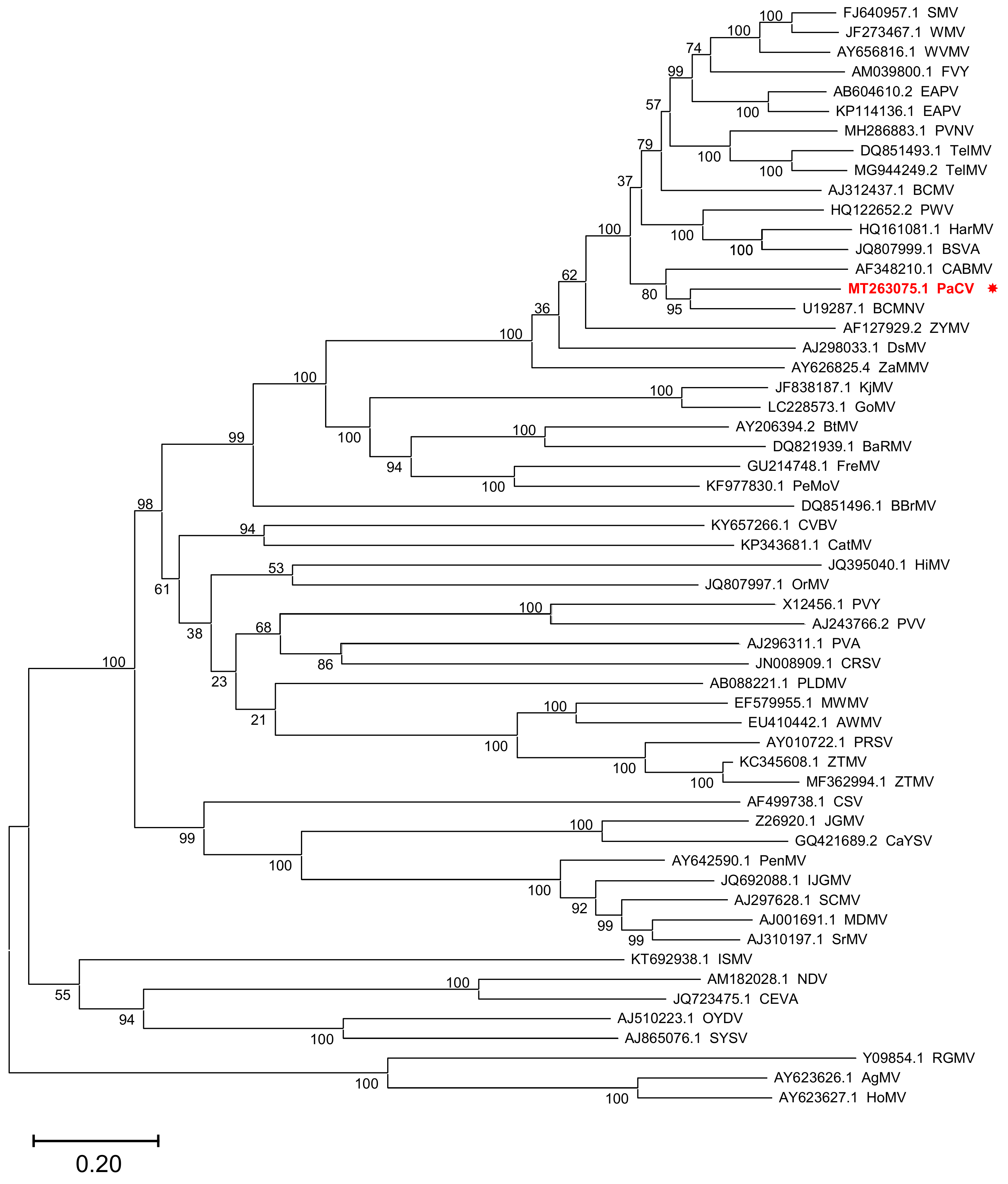
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. Plant Material
4.2. Genome Assembly and Annotation
4.3. 5′ Rapid Amplification of cDNA Ends (5′-RACE)
4.4. Sequence Alignment and Phylogenetic Analysis
4.5. Detection of PaCV Using Quantitative RT-PCR
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iwai, H.; Yamashita, Y.; Nishi, N.; Nakamura, M. The potyvirus associated with the dappled fruit of Passiflora edulis in Kagoshima prefecture, Japan is the third strain of the proposed new species East Asian Passiflora virus (EAPV) phylogenetically distinguished from strains of Passion fruit woodiness virus. Arch. Virol. 2006, 151, 811–818. [Google Scholar] [PubMed]
- Jover-Gil, S.; Beeri, A.; Fresnillo, P.; Samach, A.; Candela, H. Complete genome sequence of a novel virus, classifiable within the Potyviridae family, which infects passion fruit (Passiflora edulis). Arch. Virol. 2018, 163, 3191–3194. [Google Scholar] [CrossRef] [PubMed]
- Ochwo-Ssemakula, M.; Sengooba, T.; Hakiza, J.J.; Adipala, E.; Edema, R.; Redinbaugh, M.G.; Aritua, V.; Winter, S. Characterization and distribution of a Potyvirus associated with passion fruit woodiness disease in Uganda. Plant Dis. 2012, 96, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.A.; Jones, L. A new potyvirus found in Passiflora incence in Florida. Plant Dis. 2007, 91, 227. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baker, C.A.; Jeyaprakash, A.; Webster, C.G.; Adkins, S. Viruses Infecting Passiflora Species in Florida; Florida Department of Agriculture and Consumer Services: Tallahassee, FL, USA, 2014. [Google Scholar]
- Carstens, E.B.; Ball, L.A. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2008). Arch. Virol. 2009, 154, 1181–1188. [Google Scholar] [CrossRef]
- Cardin, L.; Moury, B. First Report of Passiflora chlorosis virus in Bituminaria bituminosa in Europe. Plant Dis. 2009, 93, 196. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Fauquet, C.M. Molecular criteria for genus and species discrimination within the family Potyviridae. Arch. Virol. 2005, 150, 459–479. [Google Scholar] [CrossRef]
- Wylie, S.J.; Adams, A.; Chalam, C.; Kreuze, J.; López-Moya, J.J.; Ohshima, K.; Praveen, S.; Rabenstein, F.; Stenger, D.; Wang, A.; et al. ICTV virus taxonomy profile: Potyviridae. J. Gen. Virol. 2017, 98, 352–354. [Google Scholar] [CrossRef]
- Fang, G.W.; Allison, R.F.; Zambolim, E.M.; Maxwell, D.P.; Gilbertson, R.L. The complete nucleotide sequence and genome organization of bean common mosaic virus (NL3 strain). Virus Res. 1995, 39, 13–23. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Beaudoin, F. Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Mol. Plant Pathol. 2005, 6, 471–487. [Google Scholar] [CrossRef]
- Goh, C.J.; Hahn, Y. Analysis of proteolytic processing sites in potyvirus polyproteins revealed differential amino acid preferences of NIa-Pro protease in each of seven cleavage sites. PLoS ONE 2021, 16, e0245853. [Google Scholar] [CrossRef] [PubMed]
- Atreya, P.L.; Atreya, C.D.; Pirone, T.P. Amino acid substitutions in the coat protein result in loss of insect transmissibility of a plant virus. Proc. Natl. Acad. Sci. USA 1991, 88, 7887–7891. [Google Scholar] [CrossRef] [PubMed]
- Maia, I.G.; Haenni, A.L.; Bernardi, F. Potyviral HC-Pro: A multifunctional protein. J. Gen. Virol. 1996, 77, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Worrall, E.A.; Wamonje, F.O.; Mukeshimana, G.; Harvey, J.J.; Carr, J.P.; Mitter, N. Bean common mosaic virus and Bean common mosaic necrosis virus: Relationships, biology, and prospects for control. Adv. Virus Res. 2015, 93, 1–46. [Google Scholar]
- Desbiez, C.; Girard, M.; Lecoq, H. A novel natural mutation in HC-Pro responsible for mild symptomatology of Zucchini yellow mosaic virus (ZYMV, Potyvirus) in cucurbits. Arch. Virol. 2010, 155, 397–401. [Google Scholar] [CrossRef]
- Olspert, A.; Chung, B.Y.; Atkins, J.F.; Carr, J.P.; Firth, A.E. Transcriptional slippage in the positive-sense RNA virus family Potyviridae. EMBO Rep. 2015, 16, 995–1004. [Google Scholar] [CrossRef]
- Rodamilans, B.; Valli, A.; Mingot, A.; San León, D.; Baulcombe, D.; López-Moya, J.J.; García, J.A. RNA polymerase slippage as a mechanism for the production of frameshift gene products in plant viruses of the Potyviridae family. J. Virol. 2015, 89, 6965–6967. [Google Scholar] [CrossRef]
- Revers, F.; Le Gall, O.; Candresse, T.; Le Romancer, M.; Dunez, J. Frequent occurrence of recombinant potyvirus isolates. J. Gen. Virol. 1996, 77, 1953–1965. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Meth. 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Scorza, L.C.T.; Hernandes-Lopes, J.; Melo-de-Pinna, G.F.A.; Dornelas, M.C. Expression patterns of Passiflora edulis APETALA1/FRUITFULL homologues shed light onto tendril and corona identities. Evodevo 2017, 8, 3. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Pfam | Name | Location (aa) | Peptides | E-Value |
|---|---|---|---|---|
| PF01577 | Potyvirus P1 protease | 79–316 | P1 | 2.1 × 10−43 |
| PF00851 | Helper component proteinase | 341–774 | HC-Pro | 3.2 × 10−111 |
| PF13608 | Protein P3 of Potyviral polyprotein | 787–1224 | P3, 6K1, CI | 5.7 × 10−57 |
| PF07652 | Flavivirus DEAD domain | 1272–1387 | CI | 6.8 × 10−7 |
| PF00271 | Helicase conserved C-terminal domain | 1418–1532 | CI | 4.2 × 10−10 |
| PF08440 | Potyviridae polyprotein | 1558–1828 | CI, 6K2 | 9.0 × 10−63 |
| PF00863 | Peptidase family C4 | 2050–2282 | NIa-Pro | 2.5 × 10−62 |
| PF00680 | Viral RNA-dependent RNA polymerase | 2340–2751 | NIb | 8.8 × 10−74 |
| PF00767 | Potyvirus coat protein | 2850–3080 | CP | 1.2 × 10−87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fresnillo, P.; Jover-Gil, S.; Samach, A.; Candela, H. Complete Genome Sequence of an Isolate of Passiflora chlorosis virus from Passion Fruit (Passiflora edulis Sims). Plants 2022, 11, 1838. https://doi.org/10.3390/plants11141838
Fresnillo P, Jover-Gil S, Samach A, Candela H. Complete Genome Sequence of an Isolate of Passiflora chlorosis virus from Passion Fruit (Passiflora edulis Sims). Plants. 2022; 11(14):1838. https://doi.org/10.3390/plants11141838
Chicago/Turabian StyleFresnillo, Patricia, Sara Jover-Gil, Alon Samach, and Héctor Candela. 2022. "Complete Genome Sequence of an Isolate of Passiflora chlorosis virus from Passion Fruit (Passiflora edulis Sims)" Plants 11, no. 14: 1838. https://doi.org/10.3390/plants11141838
APA StyleFresnillo, P., Jover-Gil, S., Samach, A., & Candela, H. (2022). Complete Genome Sequence of an Isolate of Passiflora chlorosis virus from Passion Fruit (Passiflora edulis Sims). Plants, 11(14), 1838. https://doi.org/10.3390/plants11141838