
Identification of Candidate Gene-Based Markers for Girth Growth in Rubber Trees


 and
and
Abstract
:1. Introduction
2. Results
2.1. Development and Identification of ILP Markers
2.2. Population Structure and LD Analysis of ILP Markers
2.3. Association Mapping of ILP Markers
2.4. Validation of the Associated Markers in Rubber Tree Breeding Lines
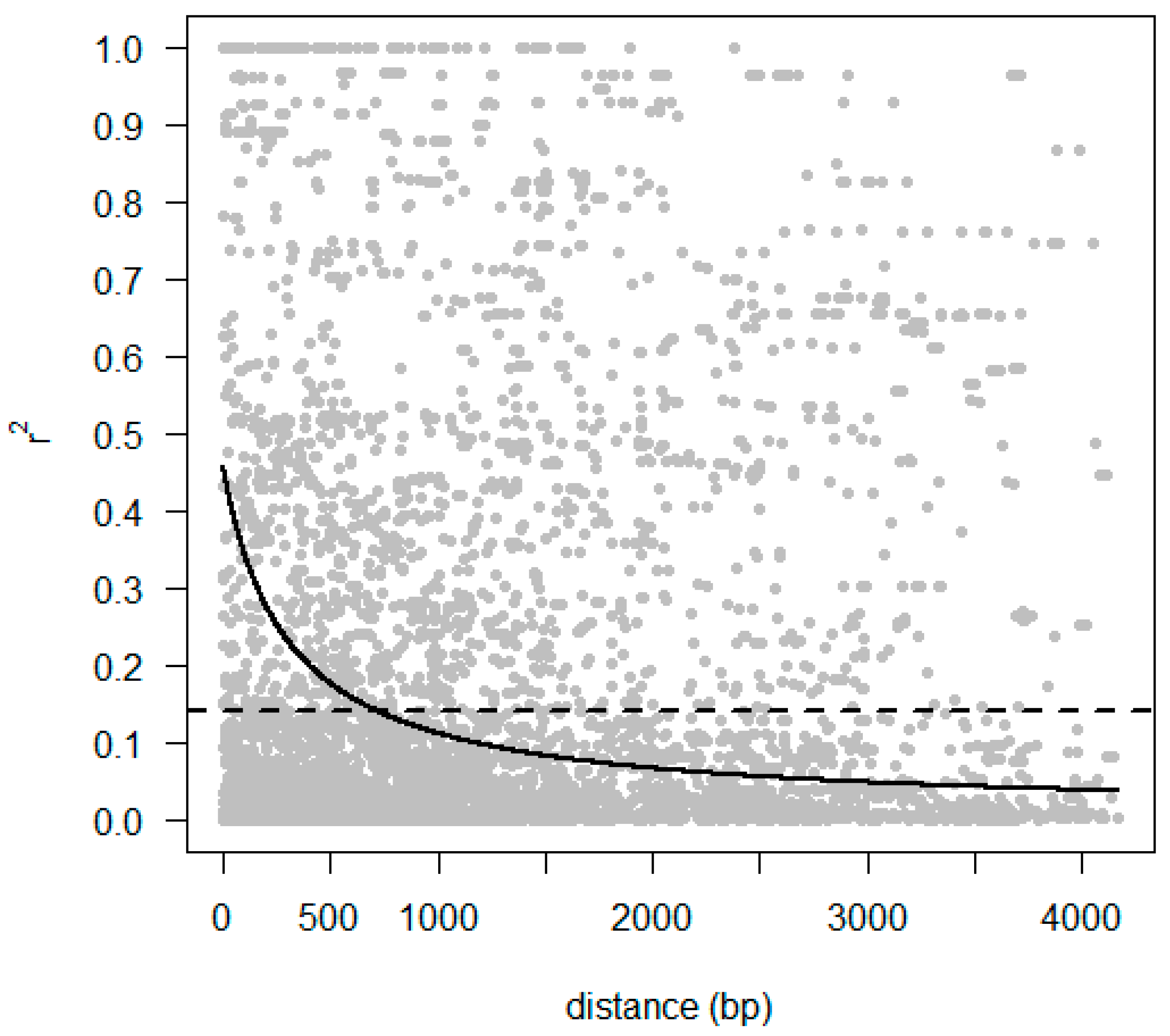
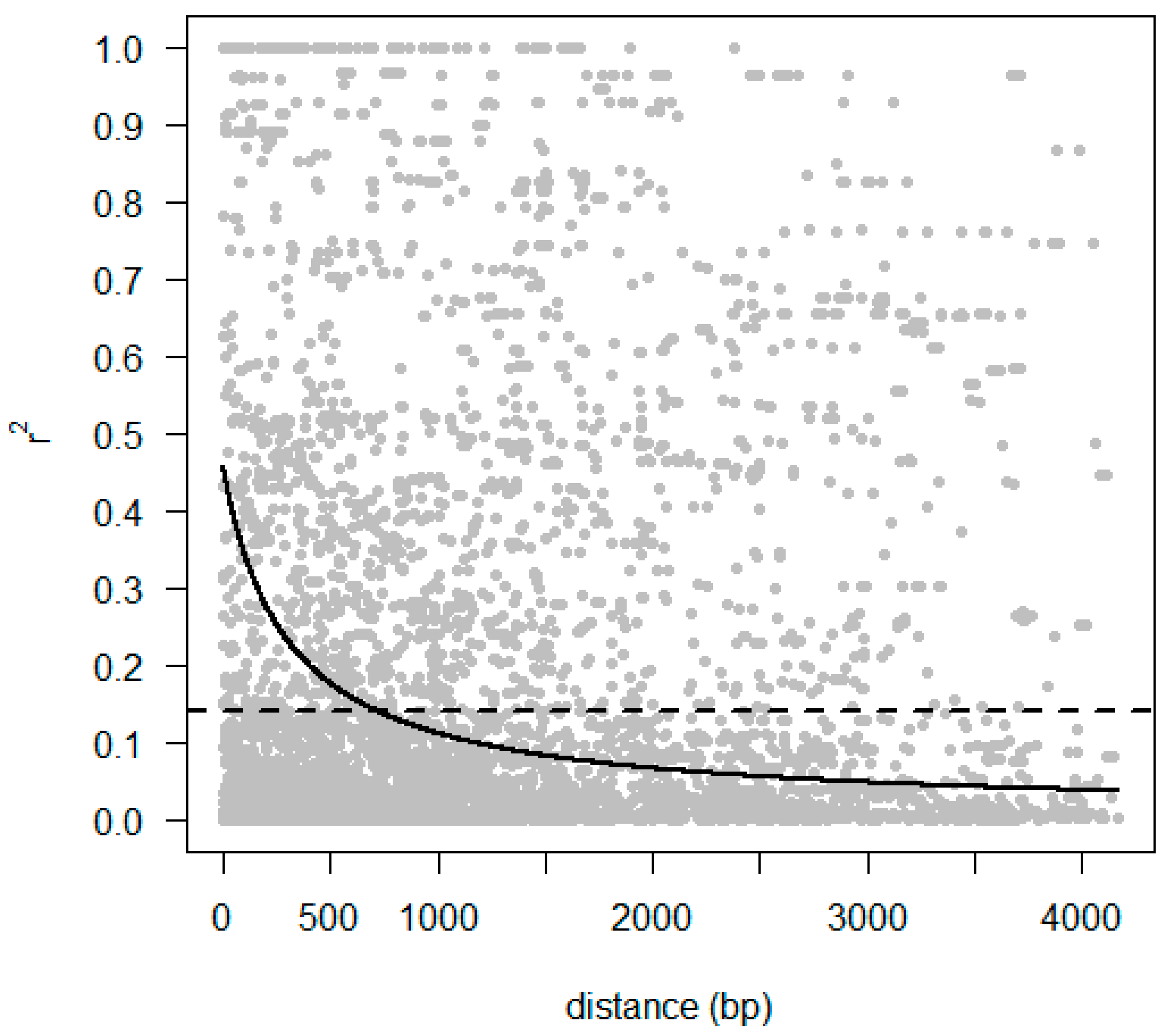
2.5. Identification of Polymorphism and Linkage Disequilibrium within COBL Gene
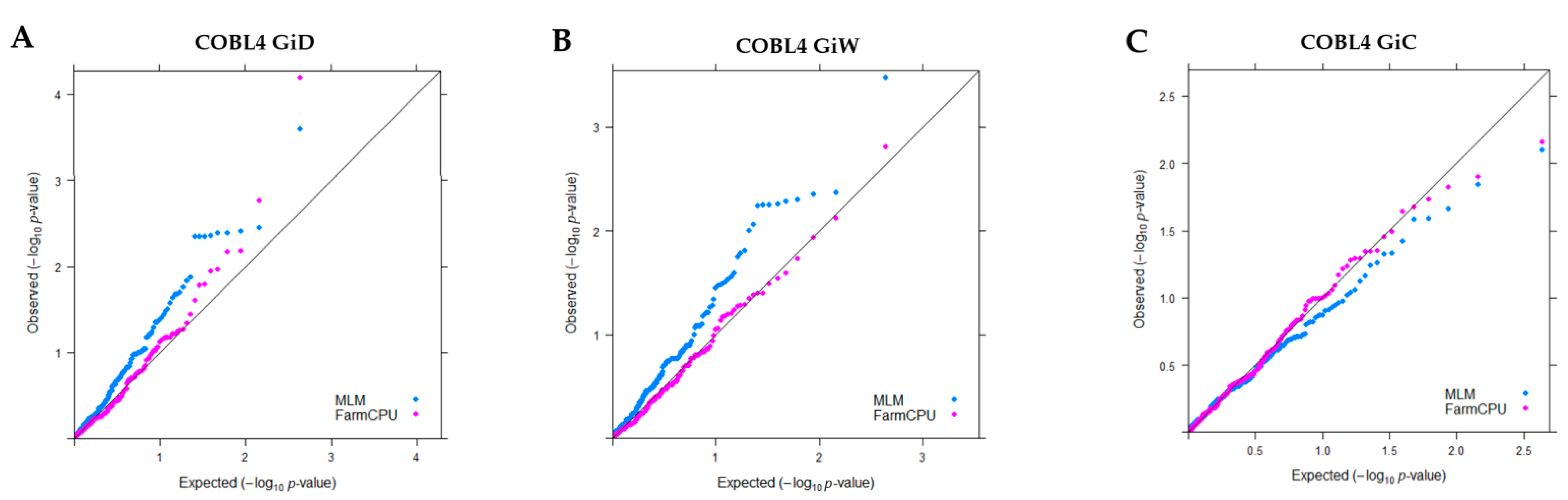
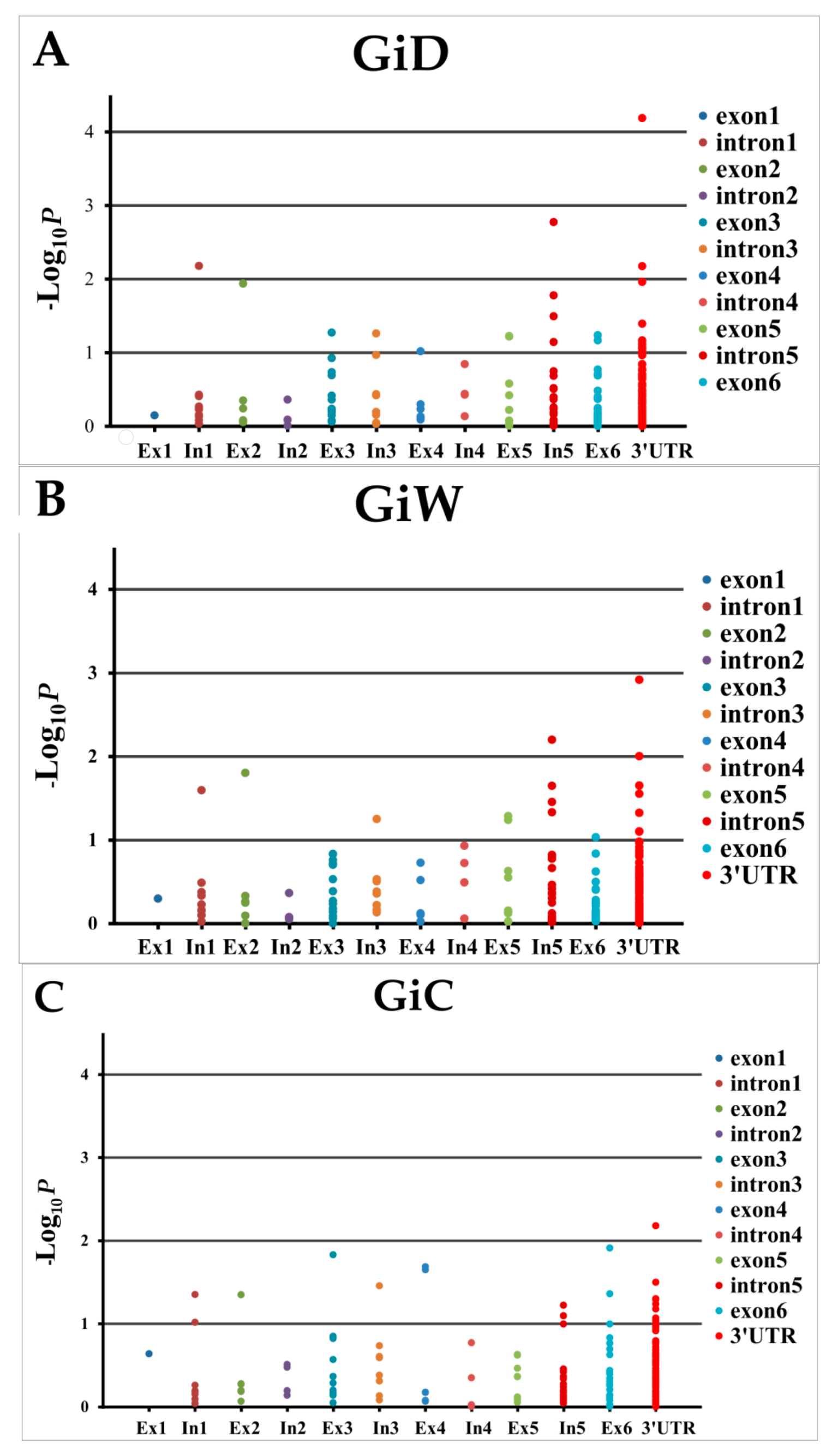
2.6. Association between SNP/Indel within COBL and Growth Girth
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Field Data
4.2. Candidate Gene Selection
4.3. ILP Marker Development
4.4. Population Structure and Linkage Disequilibrium Analysis of ILP Markers
4.5. Association Analysis of ILP Markers
4.6. Validation of the Identified ILP Markers
4.7. Library Preparation and SMRT Sequencing of the Candidate Gene
4.8. Linkage Disequilibrium and Candidate Gene Association Mapping of COBL4
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chanroj, V.; Rattanawong, R.; Phumichai, T.; Tangphasornruang, S.; Ukoskit, K. Genome-wide association mapping of latex yield and girth in Amazonian accessions of Hevea brasiliensis grown in a suboptimal climate zone. Genomics 2017, 109, 475–484. [Google Scholar] [CrossRef] [PubMed]
- De Souza, L.M.; Dos Santos, L.H.B.; Rosa, J.R.B.F.; Da Silva, C.C.; Mantello, C.C.; Conson, A.R.O.; Scaloppi, E.J., Jr.; Fialho, J.D.F.; De Moraes, M.L.T.; Gonçalves, P.D.S.; et al. Linkage disequilibrium and population structure in wild and cultivated populations of rubber tree (Hevea brasiliensis). Front. Plant Sci. 2018, 9, 815. [Google Scholar] [CrossRef] [Green Version]
- Ehrenreich, I.; Hanzawa, Y.; Chou, L.; Roe, J.; Kover, P.; Purugganan, M. Candidate Gene Association Mapping of Arabidopsis Flowering Time. Genetics 2009, 183, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alomari, D.Z.; Eggert, K.; Von Wiren, N.; Alqudah, A.M.; Polley, A.; Plieske, J.; Ganal, M.W.; Pillen, K.; Röder, M.S. Identifying Candidate Genes for Enhancing Grain Zn Concentration in Wheat. Front. Plant Sci. 2018, 9, 1313. [Google Scholar] [CrossRef]
- Sehgal, D.; Mondal, S.; Guzman, C.; Garcia Barrios, G.; Franco, C.; Singh, R.; Dreisigacker, S. Validation of Candidate Gene-Based Markers and Identification of Novel Loci for Thousand-Grain Weight in Spring Bread Wheat. Front. Plant Sci. 2019, 10, 1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, M.A.; Shaw, M.; Cooper, R.D.; Frew, T.J.; Butler, R.C.; Murray, S.R.; Moya, L.; Coyne, C.J.; Timmerman-Vaughan, G.M. Association mapping of starch chain length distribution and amylose content in pea (Pisum sativum L.) using carbohydrate metabolism candidate genes. BMC Plant Biol. 2017, 17, 132. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, M.F.; Angarita, M.; Delgado, M.C.; García, C.; Jiménez-Gomez, J.; Gebhardt, C.; Mosquera, T. Identification of Novel Associations of Candidate Genes with Resistance to Late Blight in Solanum tuberosum Group Phureja. Front. Plant Sci. 2017, 8, 1040. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Pijut, P.M.; Byrne, S.; Asp, T.; Bai, G.; Jiang, Y. Candidate gene association mapping for winter survival and spring regrowth in perennial ryegrass. Plant Sci. 2015, 235, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Palle, S.R.; Seeve, C.M.; Eckert, A.J.; Cumbie, W.P.; Goldfarb, B.; Loopstra, C.A. Natural variation in expression of genes involved in xylem development in loblolly pine (Pinus taeda L.). Tree Genet. Genomes 2011, 7, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Freeman, J.S.; Potts, B.M.; Downes, G.M.; Pilbeam, D.; Thavamanikumar, S.; Vaillancourt, R.E. Stability of quantitative trait loci for growth and wood properties across multiple pedigrees and environments in Eucalyptus globulus. New Phytol. 2013, 198, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Harakava, R. Genes encoding enzymes of the lignin biosynthesis pathway in Eucalyptus. Genet. Mol. Biol. 2005, 28 (Suppl. S3), 601–607. [Google Scholar] [CrossRef] [Green Version]
- Kirst, M.; Myburg, A.A.; De Leon, J.P.; Kirst, M.E.; Scott, J.; Sederoff, R. Coordinated genetic regulation of growth and lignin revealed by quantitative trait locus analysis of cDNA microarray data in an interspecific backcross of eucalyptus. Plant Physiol. 2004, 135, 2368–2378. [Google Scholar] [CrossRef] [Green Version]
- Southerton, S.G.; MacMillan, C.P.; Bell, J.C.; Bhuiyan, N.; Dowries, G.; Ravenwood, I.C.; Joyce, K.R.; Williams, D.; Thumma, B.R. Association of allelic variation in xylem genes with wood properties in Eucalyptus nitens. Aust. For. 2010, 73, 259–264. [Google Scholar] [CrossRef]
- Liu, Y.; He, Z.; Appels, R.; Xia, X. Functional markers in wheat: Current status and future prospects. Theor. Appl. Genet. 2012, 125, 10–14. [Google Scholar] [CrossRef]
- Badoni, S.; Das, S.; Sayal, Y.K.; Gopalakrishnan, S.; Singh, A.K.; Rao, A.R.; Agarwal, P.; Parida, S.K.; Tyagi, A.K. Genome-wide generation and use of informative intron-spanning and intron-length polymorphism markers for high-throughput genetic analysis in rice. Sci. Rep. 2016, 6, 23765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C.; Wu, S.; Niu, E.; Cheng, C.; Guo, W. Identification of genes related to salt stress tolerance using intron-length polymorphic markers, association mapping and virus-induced gene silencing in cotton. Sci. Rep. 2017, 7, 528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Bansal, R.; Gopalakrishna, T. Development of intron length polymorphism markers in cowpea [Vigna unguiculata (L.) Walp.] and their transferability to other Vigna species. Mol. Breed. 2012, 30, 1363–1370. [Google Scholar] [CrossRef]
- Jayaswall, K.; Sharma, H.; Bhandawat, A.; Sagar, R.; Yadav, V.K.; Sharma, V.; Mahajan, V.; Roy, J.; Singh, M. Development of intron length polymorphic (ILP) markers in onion (Allium cepa L.), and their cross-species transferability in garlic (A. sativum L.) and wild relatives. Genet. Resour. Crop Evol. 2019, 66, 1379–1388. [Google Scholar] [CrossRef]
- Li, D.; Xia, Z.; Deng, Z.; Liu, X.; Dong, J.; Feng, F. Development and characterization of intron-flanking EST-PCR markers in rubber tree (Hevea brasiliensis Muell. Arg.). Mol. Biotechnol. 2012, 51, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Saisug, W.; Ukoskit, K. Comparative analysis of EST-derived markers for allelic variation in Jatropha curcas L. and cross transferability among economically important species of Euphorbiaceae. Genes Genom. 2013, 35, 1–12. [Google Scholar] [CrossRef]
- Yan, W.; Zhao, H.; Yu, K.; Wang, T.; Khattak, A.N.; Tian, E. Development of a multiparent advanced generation intercross (MAGIC) population for genetic exploitation of complex traits in Brassica juncea: Glucosinolate content as an example. Plant Breed. 2020, 139, 779–789. [Google Scholar] [CrossRef]
- Parra, G.; Bradnam, K.; Rose, A.B.; Korf, I. Comparative and functional analysis of intron-mediated enhancement signals reveals conserved features among plants. Nucleic Acids Res. 2011, 39, 5328–5337. [Google Scholar] [CrossRef] [Green Version]
- Pootakham, W.; Sonthirod, C.; Naktang, C.; Ruang-Areerate, P.; Yoocha, T.; Sangsrakru, D.; Theerawattanask, K.; Rattanawong, R.; Lekawipat, N.; Tangphatsornruang, S. De novo hybrid assembly of the rubber tree genome reveals evidence of paleotetraploidy in Hevea species. Sci. Rep. 2017, 7, 41457. [Google Scholar] [CrossRef] [Green Version]
- Shearman, J.; Pootakham, W.; Tangphatsornruang, S. The BPM 24 Rubber Tree Genome, Organellar Genomes and Synteny within the Family Euphorbiaceae. In Compendium of Plant Genomes: The Rubber Genome; Springer International Publishing: New York, NY, USA, 2020; pp. 67–79. [Google Scholar]
- Feng, S.P.; Li, W.G.; Huang, H.S.; Wang, J.Y.; Wu, Y.T. Development, characterization and cross-species/genera transferability of EST-SSR markers for rubber tree (Hevea brasiliensis). Mol. Breed. 2009, 23, 85–97. [Google Scholar] [CrossRef]
- Li, D.; Xia, Z.; Deng, Z.; Liu, X.; Feng, F. Development, characterization, genetic diversity and cross-species/genera transferability of ILP markers in rubber tree (Hevea brasiliensis). Genes Genom. 2013, 35, 719–731. [Google Scholar] [CrossRef]
- De Souza, L.M.; Guen, V.L.; Cerqueira-Silva, C.B.M.; Silva, C.C.; Mantello, C.C.; Conson, A.R.O.; Vianna, J.P.G.; Zucchi, M.I.; Scaloppi Junior, E.J.; Fialho, J.D.F.; et al. Genetic diversity strategy for the management and use of rubber genetic resources: More than 1000 wild and cultivated accessions in a 100-genotype core collection. PLoS ONE 2015, 10, e0134607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Guen, V.; Doare, F.; Weber, C.; Seguin, M. Genetic structure of Amazonian populations of Hevea brasiliensis is shaped by hydrographical network and isolation by distance. Tree Genet. Genomes 2009, 5, 673–683. [Google Scholar] [CrossRef]
- Pootakham, W.; Shearman, J.; Tangphatsornruang, S. Development of Molecular Markers in Hevea brasiliensis for Marker-assisted Breeding. In Compendium of Plant Genomes: The Rubber Genome; Springer International Publishing: New York, NY, USA, 2020; pp. 55–66. [Google Scholar]
- Brady, S.M.; Song, S.; Dhugga, K.S.; Rafalski, J.A.; Benfey, P.N. Combining expression and comparative evolutionary analysis. Plant Physiol. 2007, 143, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Thumma, B.R.; Matheson, B.A.; Zhang, D.; Meeske, C.; Meder, R.; Downes, G.M.; Southerton, S.G. Identification of a Cis-acting regulatory polymorphism in a Eucalypt COBRA-like gene affecting cellulose content. Genetics 2009, 183, 1153–1164. [Google Scholar] [CrossRef] [Green Version]
- Chamary, J.V.; Hurst, L.D. The price of silent mutations. Sci. Am. 2009, 300, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Hunt, R.; Sauna, Z.E.; Ambudkar, S.V.; Gottesman, M.M.; Kimchi-Sarfaty, C. Silent (synonymous) SNPs: Should we care about them? Methods Mol. Biol. 2009, 578, 23–39. [Google Scholar] [PubMed]
- Ingvarsson, P.K.; Garcia, M.V.; Luquez, V.; Hall, D.; Jansson, S. Nucleotide polymorphism and phenotypic associations within and around the phytochrome B2 Locus in European aspen (Populus tremula, Salicaceae). Genetics 2008, 178, 2217–2226. [Google Scholar] [CrossRef] [Green Version]
- Larsen, P.A.; Heilman, A.M.; Yoder, A.D. The utility of PacBio circular consensus sequencing for characterizing complex gene families in non-model organisms. BMC Genom. 2014, 15, 720. [Google Scholar] [CrossRef] [Green Version]
- Travers, K.J.; Chin, C.-S.; Rank, D.R.; Eid, J.S.; Turner, S.W. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010, 38, e159. [Google Scholar] [CrossRef] [PubMed]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Shearman, J.R.; Tangphatsornruang, S. Nanopore sequencing in agricultural and food applications. In Handbook of Nanotechnology Applications; Elsevier: Oxford, UK, 2021; pp. 443–459. [Google Scholar]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh, B.I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unifed mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Kaler, A.S.; Gillman, J.D.; Beissinger, T.; Purcell, L.C. Comparing different statistical models and multiple testing corrections for association mapping in Soybean and Maize. Front. Plant Sci. 2020, 10, 1794. [Google Scholar] [CrossRef]
- Zhao, W.; Pollack, J.L.; Blagev, D.P.; Zaitlen, N.; McManus, M.T.; Erle, D.J. Massively parallel functional annotation of 3′ untranslated regions. Nat. Biotechnol. 2014, 32, 387–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin biosynthesis and structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.; Choi, H.; An, G. Roles of lignin biosynthesis and regulatory genes in plant development. J. Integr. Plant Biol. 2015, 57, 902–912. [Google Scholar] [CrossRef]
- Rahman, A.Y.A.; Usharraj, A.O.; Misra, B.B.; Thottathil, G.P.; Jayasekaran, K.; Feng, Y.; Hou, S.; Ong, S.Y.; Ng, F.L.; Lee, L.S.; et al. Draft genome sequence of the rubber tree Hevea brasiliensis. BMC Genom. 2013, 14, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozen, S.; Skaletsky, H.J. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [PubMed] [Green Version]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker: An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef] [Green Version]
- Rohlf, F.J. NTSYSpc Numerical Taxonomy and Multivariate Analysis System Version 2.0 User Guide, 2nd ed.; Applied Biostatistics Inc.: New York, NY, USA, 1998. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association mapping in structured population. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Bernardo, R. Genomewide markers as cofactors for precision mapping of quantitative trait loci. Theor. Appl. Genet. 2013, 126, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.; Price, A.L.; Reich, D. Population Structure and Eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Trait 1 | Marker 2 | p Value | Phenotypic Variation (%) |
|---|---|---|---|
| GiD | COBL064_1 * | 2.0 × 10−7 | 14.0 |
| PIN227_1 * | 1.0 × 10−5 | 10.2 | |
| PIN220 * | 1.1 × 10−4 | 7.7 | |
| CaS108 | 9.2 × 10−4 | 4.9 | |
| GiW | COBL064_1 * | 4.32 × 10−7 | 14.6 |
| PIN227_1 * | 4.82 × 10−5 | 10.0 | |
| PIN220 | 4.7 × 10−4 | 7.3 | |
| CaS108 | 1.8 × 10−3 | 4.9 | |
| GiC | CAD076 | 1.6 × 10−3 | 8.9 |
| Marker | Size (bp) | GiD | GiW | GiC | |||
|---|---|---|---|---|---|---|---|
| p-Value | Effect | p-Value | Effect | p-Value | Effect | ||
| COBL4 | 380 | 0.033 | −1.52 | 0.044 | −1.68 | ns | −0.08 |
| PIN227 | 261 | 0.01 | 1.22 | 0.026 | 1.16 | ns | −0.20 |
| 289 | 0.005 | −1.66 | 0.01 | −1.74 | ns | −0.29 | |
| 298 | 0.013 | −2.02 | 0.027 | −2.17 | ns | −0.41 | |
| PIN220 | 461 | 0.025 | 3.98 | 0.023 | 4.70 | ns | 3.32 |
| CAD76 | 263 | ns | −0.93 | ns | −1.07 | 0.017 | −1.49 |
| 312 | ns | 0.41 | ns | 0.49 | 0.043 | 1.25 | |
| Trait 1 | Marker | Position 2 | p-Value 3 | Phenotypic Variation (%) |
|---|---|---|---|---|
| GiD | 3’ UTR | 5589 | 6.50 × 10−5 * | 11.1 |
| intron5 | 3221 | 1.69 × 10−3 | 7.0 | |
| intron1 | 1475 | 6.63 × 10−3 | 6.1 | |
| 3’ UTR | 4406 | 6.69 × 10−3 | 10.1 | |
| GiW | 3’ UTR | 5589 | 1.56 × 10−3 | 9.8 |
| intron5 | 3221 | 7.64 × 10−3 | 6.4 | |
| GiC | 3’ UTR | 5526 | 6.89 × 10−3 | 3.6 |
| Functional Class | Gene | Full Name | No. of ESTs | References | |
|---|---|---|---|---|---|
| Cellulose | 1 | CesA2 | Cellulose synthase2 | 1 | [9,13] |
| synthesis | 2 | CesA3 | Cellulose synthase3 | 1 | [9,13] |
| 3 | COBL4 | COBRA-like protein 4 | 1 | [10,13] | |
| 4 | CaS | Callose synthase 10 | 3 | [9] | |
| Lignin synthesis | 5 | 4CL | 4-coumarate:CoA ligase | 1 | [9,11,12,13] |
| 6 | CAD | Cinnamyl alcohol dehydrogenase | 3 | [9,10,11,12,13] | |
| 7 | COMT1 | Caffeic O-methylransferase1 | 2 | [9,11,13] | |
| 8 | CCoAMT | Caffeoyl-CoA O-methyltransferase | 3 | [9,10,13] | |
| 9 | CCR | Cinnamoyl-CoA reductase | 2 | [9,10,11,12] | |
| 10 | C4H1 | Cinnamate 4-hydroxylase 1 | 2 | [10,11] | |
| 11 | PAL | Phenylalanine ammonia-lyase | 1 | [9,11] | |
| 12 | Peroxidase2 | Peroxidase 2 | 1 | [9] | |
| Transcription | 13 | LIM | LIM gene for LIM transcription factor | 2 | [9,13] |
| factors | 14 | MYB1 | MYB1 gene for MYB transcription factor 1 | 5 | [9,10,13] |
| 15 | BTF3 | Transcription factor BTF3 homolog 4 | 2 | [13] | |
| 16 | HD-zip | Homeobox-leucine zipper protein ANTHOCYANINLESS 2 | 1 | [13] | |
| 17 | NAC1 | NAC domain-containing protein 100-like | 4 | [13] | |
| 18 | APL | myb family transcription factor APL | 1 | [9] | |
| 19 | KNAT | Homeobox protein knotted-1-like | 1 | [9] | |
| 20 | NtLIM1 | Eglim1 gene for transcription factor lim1 | 2 | [10] | |
| 21 | MOR1 | Protein MOR1 | 1 | [9] | |
| 22 | PIN1 | Auxin efflux carrier component 1-like | 1 | [9] | |
| 23 | RIC1 | CRIB domain-containing protein RIC7 | 1 | [9] | |
| 24 | FRA2 | Katanin p60 ATPase-containing subunit A1 | 1 | [9] | |
| Cell expansion | 25 | COB | COBRA-like extracellular glycosyl-phosphatidyl inositol-anchored protein family | 1 | [9] |
| 26 | KORRIGAN | Korrigan | 1 | [9,10] | |
| Total | 26 | 45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhusudsawang, G.; Rattanawong, R.; Phumichai, T.; Pootakham, W.; Tangphatsornruang, S.; Ukoskit, K. Identification of Candidate Gene-Based Markers for Girth Growth in Rubber Trees. Plants 2021, 10, 1440. https://doi.org/10.3390/plants10071440
Bhusudsawang G, Rattanawong R, Phumichai T, Pootakham W, Tangphatsornruang S, Ukoskit K. Identification of Candidate Gene-Based Markers for Girth Growth in Rubber Trees. Plants. 2021; 10(7):1440. https://doi.org/10.3390/plants10071440
Chicago/Turabian StyleBhusudsawang, Gunlayarat, Ratchanee Rattanawong, Thitaporn Phumichai, Wirulda Pootakham, Sithichoke Tangphatsornruang, and Kittipat Ukoskit. 2021. "Identification of Candidate Gene-Based Markers for Girth Growth in Rubber Trees" Plants 10, no. 7: 1440. https://doi.org/10.3390/plants10071440
APA StyleBhusudsawang, G., Rattanawong, R., Phumichai, T., Pootakham, W., Tangphatsornruang, S., & Ukoskit, K. (2021). Identification of Candidate Gene-Based Markers for Girth Growth in Rubber Trees. Plants, 10(7), 1440. https://doi.org/10.3390/plants10071440